
Abstract
Group A Rotavirus, Human Astrovirus, and Norovirus (RVA, HAstV, and NoV) are recognized as the major causative agents of acute gastroenteritis in children and adults worldwide. The aim of this study was to determine the prevalence and molecular epidemiology of RVA, HAstV, and NoV in wastewater from three cities in Uruguay. Thirty-six samples from Bella Unión, Salto, and Fray Bentos cities were analyzed using quantitative and qualitative PCR. RVA was the most frequently detected virus (50%), followed by HAstV (39%), NoV GII (36%), and NoV GI (25%). RVA strains were characterized as P[8] and G3 based on the VP4 and VP7 genes, respectively. Among NoV-positive samples, genotypes GI.2, GI.3, GI.5, GI.6, GI.7, GII.2, GII.6, and GII.4 were detected, and only one HAstV genotype (MLB1) was found. Our wastewater-based epidemiological approach provides a snapshot of the overall genetic diversity of these viruses in three cities of the Uruguay River basin during 2017–2018. These findings reinforce the importance of this environmental surveillance tool for monitoring epidemiological trends of enteric viruses circulating in the population, which can be used to guide public health intervention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Enteric viruses are a group of viruses that infect and replicate in the gastrointestinal tract of the host, causing different diseases such as gastroenteritis, encephalitis, or viral hepatitis. Their mode of transmission is through the fecal–oral route, being therefore, commonly found in wastewater. (Banyai et al., 2018). These viruses in wastewater frequently contaminate rivers and lakes constituting a source of infection when people enter in contact with these recreational environments. Their transmission is very efficient since they are frequently excreted in high concentrations (reaching up to 1011 viral particles per gram of fecal matter) in the feces of infected people, persist for long periods of time in the environment, and present a low infective dose (Banyai et al., 2018). Viral acute gastroenteritis (VAGE) is a major cause of morbidity and mortality in infants less than 5‐years‐old mainly in developing countries (Ghssein et al., 2018; Gupta et al., 2017). Diarrheal disease is the second most important cause of death in children under five years old, with 1.7 billion cases and 525,000 deaths annually in children in this age group, worldwide (WHO, 2017).
Within the enteric viruses, Group A Rotavirus (RVA), Human Astrovirus (HAstV), and Norovirus (NoV) are recognized as the main cause of gastroenteritis in humans worldwide (Estes & Greenberg, 2013, Robilotti et al., 2015; Vu et al., 2017). RVA belongs to the family Reoviridae and possesses a genome composed of 11 segments of double-stranded RNA. The current classification of RVA genotypes is based on the sequence of the VP7 gene (which encodes the G-capsid protein) and VP4 gene (which encodes the P-capsid protein), and at least 36 G genotypes and 51 P genotypes have been identified (RCWG, 2018). Human Astroviruses belong to the Astroviridae family and are classified into classical HAstV, Melbourne (MLB) clade, and Virginia clade (VA) according to the International Committee on Taxonomy of Viruses (ICTV, 2018). Classical HAstVs consist in 1-8 genotypes (HAstV-1-8), while the MLB and VA clades are genetically distinct from classical HAstVs and present a near genetic relationship with animal astroviruses (Arowolo et al., 2020). Finally, NoV belongs to the Caliciviridae family and is subdivided into 10 genogroups (G), GI to GX, with GI and GII (which include human strains) further divided into 9 and 27 genotypes, respectively (Chhabra et al., 2019).
RVA during 2015–2016 in wastewater treatment plants (WWTP) in Catalonia, Spain was detected in the 90% of the samples demonstrating high prevalence of circulation in this population with the presence of genotypes G2, G3, G9, G12, P[4], P[8], P[9], and P[10] (Silva-Sales et al., 2020). Considering RVA molecular epidemiology in Iran, genotypes G10 and G1 were the predominating genotypes detected in treated and raw sewage of two WWTPs (Atabakhsh et al., 2020). Another study conducted by Kargar et al. (2013) in Shiraz Hospital, reported the predominance of genotype G1.
Recent studies in HAstVs suggested that they also affect the central nervous system. Type 1 VA-AstVs are strongly suggested to be associated with encephalitis in immunocompromised patients (Lum et al., 2016). Compared to other human enteric viruses such as noroviruses and rotaviruses, HAstVs are not deeply characterized (Hata et al., 2018). Types 1 and 2 MLB-AstVs were identified in wastewater samples collected in the United States, whereas all known genotypes of MLB-AstVs (types 1–3) were found from samples collected in Nepal (Hata et al., 2018). In children with acute gastroenteritis in Japan (2012–2013) MLB1 and MLB2 strains were the most predominant clade, followed by classical astrovirus and VA astrovirus (Khamrin et al., 2016).
Previous studies determined a high prevalence of NoV in several regions worldwide, such as Africa where GII.4 genotype dominates (Afework et al., 2022). Similarly, in Brazil, GII was responsible for 71% of NoV-positive VAGE cases, with GII.4 prevailing across all the regions of the country (Fioretti et al., 2011; Bitencurt et al., 2019). Notably, there have been at least six epidemic strains of GII.4 since 1995, namely US 1995/96, Farmington Hills 2002, Hunter 2004, Den Haag 2006b/Yerseke_2006a, New Orleans 2009, and Sydney 2012 (Bitencurt et al., 2019).
Studies of enteric viruses in Uruguay, conducted nearly a decade ago (2011–2012), analyzed both clinical and wastewater samples. These investigations revealed the presence of several viral strains reflecting a wide circulation in the local population. These findings provide evidence that wastewater is a significant source of contamination and poses a potential public health risk in this region (Lizasoain et al., 2015, Victoria et al., 2014, 2016, Tort et al., 2015a, 2015b).
Wastewater-based epidemiology (WBE) is a tool that provides a snapshot of the overall health of a community based on what is excreted in sewage (Mao et al., 2021). The WBE approach has multiple applications, including its use as an early alert of a viral circulation (Aguiar-Oliveira et al., 2020) and as an effective method to determine the viral molecular epidemiology (Barril et al., 2010, Hellmer et al. 2014, Prevost et al. 2015, Tort et al., 2015a, Guo et al., 2022, Yu et al., 2022).
The present study aim to shed light on the RVA, NoV and HAstV strains that circulated in three cities from the northwestern region of Uruguay by analyzing wastewater samples obtained from March 2017 to February 2018 through a WBE approach.
Materials and Methods
Wastewater Sampling
From March 2017 to February 2018, monthly collections of wastewater were performed in three cities from the northwestern region of Uruguay: Bella Unión, Salto, and Fray Bentos. Bella Union is located at the northern region of Uruguay with a population of 18,406 inhabitants (INE, 2011). Salto is located 143 km south from Bella Union with a population of 104,028 inhabitants (INE, 2011). Fray Bentos is located 241 km south from Salto with a population of 24,406 inhabitants (INE, 2011). All the cities are located on the riverside of the Uruguay River, the most important river of the country (Fig. 1). Samples from Salto and Fray Bentos city were collected in the single pipe of each city, which directly discharges untreated sewage from these cities into the Uruguay river. In Bella Union city, the sewage samples were collected into a stream where the treated wastewater is directly discharged from a two stabilization ponds system. This wastewater collection sites were determined in order to represent the population of each city.

Map of Uruguay showing the sampling points located in each city: Bella Union, Salto, and Fray Bentos
Viral Concentration, Nucleic Acid Extraction, and Reverse Transcription
Wastewater samples were concentrated by the ultracentrifugation method described by Pina et al. (1998). Briefly, 42 mL of sample were centrifuged at 100,000 × g for 1 h at 4 °C. The pellet was eluted in 3.5 mL of 0.25 N glycine buffer (pH 9.5) and incubated on ice for 30 min with shaking every 5 min. Then, 3.5 mL of PBS 2X (pH 7.2) was added, mixed, and centrifuged at 12,000 × g for 15 min. The supernatant was quickly transferred to an ultracentrifugation tube and the pellet was removed. Another centrifugation at 100,000 × g for 1 h at 4 °C was performed and the supernatant was quickly discarded. The pellet was resuspended in 200 µL of 1 × PBS and stored at − 80 °C for further analysis. The viral recovery rate of ultracentrifugation viral concentration method is about 12% for both PP7 and recombinant AdV from wastewater (Blanco Fernandez et al., 2017).
Viral RNA was extracted from 140 µL of the concentrated sample using the commercial QIAamp Viral RNA kit (Qiagen Inc., Hilden, Germany). The manufacturer’s protocol was followed, and the purified viral RNA was eluted in 30 µL of elution buffer and stored at − 80 °C until the reverse transcription (RT) procedure. Extracted RNA was reverse transcribed into cDNA using the SuperScript II Reverse Transcriptase and Random Hexamer Primers (both from Invitrogen™, USA), according to the manufacturer’s instructions. Ten microlitres of RNA and tenfold dilution of the RNA were used as a template for RT in order to remove enzymatic inhibitors presented in wastewater samples.
Quantitative PCR (qPCR)
For detection and quantification of RVA, NoV GI, GII, and HAstV, three qPCR were performed with TaqMan technology and Rotor-Gene Q instrument (Qiagen®). Undiluted and tenfold diluted samples were analyzed in duplicate using SensiFAST™ Probe No-ROX Kit (Bioline, London, UK) together with primers and probes for RVA (Zeng et al., 2008), HAstV (Dai et al., 2010), and a multiplex for NoV GI and GII (Pang et al, 2005; Schultz et al, 2011).
RVA standard curve was performed with nine points of serial dilutions of plasmid from 108 to 100 genomic copies/reaction (gc/r) that yield a slope of − 3.59 and a reaction efficiency of 0.90. The standards curve of NoV GII and NoV GI were performed with the same serial dilutions as the RVA quantification that yields a slope of − 3.43 and a PCR efficiency of 0.96 for NoV GII and − 3.02 and 1.14 for NoV GI. For HAstV, the standard curve was performed with the same dilutions as RVA, NoV GI, and NoV GII with a slope of − 3.31 and efficiency of the reaction of 1.01. The detection limits of the qPCR reactions for RVA, NoV GI, NoV GII, and HAstV were 5, 30, 5, and 5 genomic copies per qPCR reaction, respectively. Samples presenting no signal amplification (not detected) were considered below the detection limit of this technique.
Molecular Characterization
Nested PCR for RVA
Two nested PCR were used for G and P genotypes of the VP7 and VP4 genes, respectively (WHO, 2009). The primers used in the first round PCR for G and P, were 9con1/9con2 and 4con2/4con3, respectively. The second round PCR contains the internal primers VP7F/VP7Rdeg (881 bp) for G and VP4F/VP4R (663 bp) for P genotypes.
PCR for HAstV
Detection of emerging HAstVs was performed with primers SF0073 and SF0076 that target a highly conserved region of the ORF 1b (viral RNA polymerase) of astroviruses. The amplicon obtained is 409 bp long (Finkbeiner et al., 2009).
Semi-nested PCR for NoV
Two semi-nested PCRs (GI and GII) were used. For GI, primers COG1F/G1SKR and G1SKF/G1SKR were used for the first (380 bp) and second (330 bp) rounds, respectively. For GII, primers COG2F/G2SKR and G2SKF/G2SKR were used for the first (390 bp) and second (340 bp) round PCRs, respectively. These primers hybridize in the ORF1/ORF2 junction region and the 5' region of ORF2 (VP1 capsid protein) (Kitajima et al., 2010).
All the PCR products were run in a 1.5% agarose gel electrophoresis and the amplicons were purified with the commercial AxyPrep™ DNA Gel Extraction kit (AXYGEN®). DNA strands were sequenced by Sanger sequencing methodology at Macrogen Inc. (Seoul, South Korea) and sequences were deposited in GenBank with the following accession numbers: OQ324916, OQ346254, OQ346261, OQ346262, OQ414203, OQ414237, OQ414241, OQ414242, OQ421105, OQ421123 for NoV GI; OQ356350, OQ356465, OQ357263, OQ357880, OQ357897, OQ357900, OQ358122, OQ358123, OQ421257, OQ421437, OQ503416 for NoV GII. OQ513257 and OQ603360 for HAstV. OQ603361, OQ603362, OQ626714, and OQ626713 for RVA.
All the procedures were performed using nuclease-free water as negative control and viral strains of known genotypes for each virus as positive controls.
Phylogenetic Analysis
The molecular characterization of RVA, HAstV, and NoV strains was performed by Maximum Likelihood (ML) phylogenetic analysis of the sequences obtained from the amplicons of these viruses.
Assembly and editing of the sequences were performed with Seqman® version 7.0 (DNAStar Lasergene, Madison, WI, USA). All datasets were aligned using MAFFT v7.467 program and subjected to ML phylogenetic analysis.
The ML phylogenetic trees were inferred with IQ-TREE 1.6.1 software under the best-fit model of nucleotide substitution selected using the ModelFinder application (RVA VP4: TIM + F + G4, RVA VP7: HKY + F + I, NoV GI: TIM2e + I + G4, NoV GII: TIM3e + I + G4, and HAstV: TIM2 + F + I + G4). Branch support was assessed using the approximate likelihood-ratio test based on a Shimodaira–Hasegawa-like procedure (SH-aLRT) with 1,000 replicates. Final trees were visualized in FigTree v1.4.4.
Results
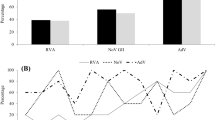
Between March 2017 and February 2018, monthly wastewater sampling was performed from three cities in the northwestern region of Uruguay, Bella Unión, Salto, and Fray Bentos. RVA was detected in 50% of the analyzed samples (18/36) through qPCR. Additionally, HAstV was detected in 39% of the wastewater samples, while NoV GII and GI were present in 36% and 25% of the analyzed samples, respectively (Fig. 2).

Frequency and concentration of RVA, NoV GI, NoV GII, and HAstV in wastewater samples monthly collected for one year in Bella Union, Salto, and Fray Bentos cities, Uruguay
In untreated wastewater from both Fray Bentos and Salto cities, the studied viruses were detected year-round except in July 2017 in Fray Bentos and October 2017 and January 2018 in Salto. On the other hand, in treated wastewater from Bella Union, these viruses were scarce detected over the analyzed period and NoV GII was not detected (Fig. 2).
Regarding untreated wastewater samples, RVA and NoV GII were the viruses with the highest frequency of detection in Fray Bentos, both detected in 67% of the samples. Additionally 5 out of 12 samples (42%) from Fray Bentos were positive for HAstV and 4 out of 12 (33%) were positive for NoV GI. In Salto, the virus with the highest frequency of detection was RVA, detected in 58% of the samples, followed by HAstV in 42%. NoV GII and NoV GI were detected in 33% and 25% of the samples analyzed from Salto, respectively.
In Bella Unión, RVA and HAstV were detected in 25% of the samples and NoV GI in 8.3% of them. It is important to highlight that in Bella Unión, treated wastewater samples were analyzed.
The co-circulation of different viruses in the same samples was observed as follows: in November 2017 in Fray Bentos RV and NoV GI; in November 2017 in Salto NoV GI and GII; in September 2017 in Salto NoV GII and HAstV (Fig. 2).
RVA was observed with a geometric mean concentration of 1.3E + 03 gc/L, NoV GI and GII presented a geometric mean concentration of 2.8E + 05 and 1.7E + 04 gc/L, respectively, and for HAstV the geometric mean concentration was 1.3E + 04 gc/L (Fig. 2).
In order to perform the molecular characterization of RVA, we analyzed VP4 and VP7 genes. For VP4, we obtained 2 amplicons that corresponded to samples collected in Salto in December 2017 (OQ603361) and January 2018 (OQ603362). Both samples were classified as genotype P[8] by the Rotavirus Genotyping web-based tool (Maes et al., 2009) and according to the phylogenetic analysis they were closely related (SH-aLRT = 95) to Argentinian and Brazilian strains detected in 2006, 2008, and 2009 (Fig. 3). The amplicons obtained for VP7 corresponded to samples collected in Fray Bentos in September 2017 (OQ626714) and November 2017 (OQ626713). These samples were classified by the Rotavirus Genotyping web-based tool as G3 genotype and according to the phylogenetic analysis they are closely related to strains detected in Brazil and Peru in 2010, 2011 and 2014 (SH-aLRT = 100) (Fig. 4).

Phylogenetic analysis of a partial region of the RVA VP4 gene (630 bp) for P[8] genotype. The statistical support for each branch is indicated by the SH-aLRT value. Samples sequenced in this study are highlighted in red, while those from previous studies conducted in Uruguay are shown in blue (Color figure online)

Phylogenetic analysis of a partial region of the RVA VP7 gene (809 bp) for G3 genotype. The statistical support for each branch is indicated by the SH-aLRT value. Samples sequenced in this study are highlighted in red, while those from previous studies conducted in Uruguay are shown in blue (Color figure online)
The phylogenetic analysis of the emerging HAstV strains revealed that strains collected in 2017 in August in Salto (OQ513257) and September in Fray Bentos (OQ603360) were classified as Mamastrovirus 6 of the MLB1 group. These strains were closely related to previously published Uruguayan strains collected in Treinta y Tres and Melo in 2012 and 2013 (SH-aLRT = 78) (Fig. 5).

Phylogenetic analysis of emerging HAstVs based on a partial region of the RNA polymerase region (394 bp). The statistical support for each branch is indicated by the SH-aLRT value. Samples sequenced in this study are highlighted in red, while those from previous studies conducted in Uruguay are shown in blue (Color figure online)
The phylogenetic analysis of the NoV GI strains characterized in this study showed that the isolates detected belonged to genotypes GI.2 (n = 1), GI.3 (n = 2), GI.5 (n = 2), GI.6 (n = 1), and GI.7 (n = 4). Our GI.2 strain from Bella Unión sampled in 2017 (OQ414203) was closely related (SH-aLRT = 99.9) to previously detected GI.2 strains sampled in Brazil in the period 2015–2016. The GI.3 sample collected from Salto in March 2017 (OQ324916) clustered together with Brazilian strains from 2012 and 2015 and an Uruguayan strain from Treinta y Tres collected in 2011 (SH-aLRT = 87). The GI.3 sample collected in Salto in May 2017 (OQ421105) represents a second independent introduction of this genotype to this city and was closely related (SH-aLRT = 83) to samples from Brazil collected in 2013, 2014, and 2016. The GI.5 strains detected in January and February 2018 in Fray Bentos, grouped together (SH-aLRT = 99), probably representing a unique introduction of this genotype to this Uruguayan city. Additionally, the phylogenetic analysis suggests that these GI.5 strains were more closely related (SH-aLRT = 92) to a GI.5 Brazilian strain from 2015 than to an Uruguayan GI.5 strain from Paysandu detected in 2012. Similarly, the GI.6 strain collected in Fray Bentos in 2017 (OQ414241) was more closely related to a Brazilian GI.6 strain collected in 2014 (SH-aLRT = 84) than to an Uruguayan GI.6 strain from Treinta y Tres detected in 2013. The four GI.7 strains from Salto and Fray Bentos collected between October and December 2017 segregated in a highly supported (SH-aLRT = 99.5) monophyletic group, raising the possibility of a single event of introduction probably from Brazil with a subsequent regional dispersion (Fig. 6).

Phylogenetic analysis of a partial region of the VP1 gene (291 bp) of NoV GI. The statistical support for each branch is indicated by the SH-aLRT value. Samples sequenced in this study are highlighted in red, while those from previous studies conducted in Uruguay are shown in blue (Color figure online)
The phylogenetic analysis of the NoV GII strains characterized in this study showed that the isolates detected belonged to genotypes GII.4 (n = 3), GII.2 (n = 5), GII.6 (n = 3). The samples A732 from March 2017 Fray Bentos (OQ357263), A759 September 2017 (OQ357900), and A761 October 2017 (OQ357880) both from Bella Unión, samples A758 collected from September 2017 (OQ357897) and A741 from May 2017 (OQ421437) from Salto were classified as GII.2. Samples A732 and A741 were closely related to a strain observed in Chile 2016. Samples A761, A758, and A759 were similar to Brazilian strains from 2003. The Uruguayan strains observed in 2011–2012, classified as GII.2 were not closely related to the sequences obtained in this study. Samples A745 from June 2017 (OQ358122) and A766 (OQ358123) from November 2017, both from Salto, and sample A754 (OQ421257) collected in August 2017 in Fray Bentos, grouped together in GII.6 and were closely related to a strain detected in Chile 2016. Concerning GII.4 strains, samples A777 (OQ356350) from February 2018 (Salto) and A778 (OQ503416) from the same date (Fray Bentos) were similar and closely related with a strain described in Chile 2018. The sample A771 (OQ356465) collected in December 2017 in Bella Unión was closely related to a strain isolated in Chile 2018. The Uruguayan GII.4 strains isolated in 2017–2018 were classified as New Orleans 2009 and Sydney 2012 variants by the Norovirus genotyping tool. Similar to the results of the GII.2 Uruguayan strains, the Uruguayan GII.4 strains isolated in 2011–2012 were divergent when compared with the Uruguayan strains detected in 2017–2018. The SH-aLRT support values were > 80% for all the genotypes of NoV GII (Fig. 7).

Phylogenetic analysis of a partial region (302 bp) of NoV GII VP1 gene. The SH-aLRT support values were > 80% for all the genotypes. Samples sequenced in this study are highlighted in red, while those from previous studies conducted in Uruguay are shown in blue (Color figure online)
Discussion
In this study, a molecular characterization of three gastroenteric viruses, RVA, NoV, and HAstV was performed using a WBE approach. In comparison to surveillance based on clinical diagnostic testing, monitoring enteric viruses in a WBE approach present a major advantage of comprehensive population-based surveillance of circulating virus by capturing persons who did not have diagnostic testing performed during gastroenteritis as well as including patients with asymptomatic infection (Lin et al., 2021). The objective of this work was to obtain a snapshot of the circulating strains in the population of three Uruguayan cities throughout a year (2017–2018). Analyzing wastewater samples provides an estimate of the epidemiology of viruses infecting the populations served by the sanitation network and enables the observation of gastroenteritis outbreaks caused by the studied viruses. Unfortunately, no epidemiological information from clinical samples was available in our country in order to compare the presence of these viruses in wastewater with gastroenteritis outbreaks in the population. Therefore, it is not possible to know the significant impact of the strains detected in wastewater on human health.
Considering the frequency and distribution of all the untreated wastewater analyzed samples, the prevalence of RVA was higher when compared with NoV GI, NoV GII, and HAstV. Moreover, RVA was detected throughout the year, indicating its continuous circulation in these areas which discharge their wastewater into the Uruguay River. These findings are consistent with the previous studies from southern Brazil based on clinical samples, where RVA was also detected year-round (Gutierrez et al., 2020). The ability of rotaviruses to remain infectious in harsh environments and wastewater treatment process may contribute to their prevalence as one of the most common enteric viruses (Tavakoli Nick et al., 2020). However, despite these findings, previous studies from Brazil, Uruguay, and Iran reported a marked RVA seasonality during colder months in both environmental and clinical samples (Atabakhsh et al., 2020; Bortagaray et al., 2019; Gutierrez et al., 2020). Further studies with longer periods and higher frequency of sample collection are necessary in order to confirm the absence of seasonality as observed in this work.
Considering the distribution of NoV GI and GII throughout the study period of this work in all the untreated wastewater analyzed samples, no clear seasonality pattern was observed. However, previous studies suggested that NoV has greater incidence and persistence in cold environments and its spread may be affected by environmental factors such as relative humidity and latitude, as well as demographic characteristics. In our study, although NoV GI and GII were detected in both cold and warm seasons, the virus was more frequently distributed during the warm season. This contrast with numerous clinical studies where NoV VAGE cases and outbreaks tend to occur during winter or cooler months (Eftim et al., 2017; Shamkhali Chenar & Deng, 2017; Farkas et al., 2018; van Beek et al. 2018; Huang et al., 2022). Additionally, marked seasonality of NoV has been observed in China where the majority of NoV disease peaks occur from October to March during the cold season (Wei et al., 2021). In our study, NoV and HAstV were also detected in all the seasons throughout the year, without any significant seasonality. In the literature there is no information about Astrovirus seasonality in the environment.
It is important to highlight that the sample collection site in Bella Union was a stream where the treated wastewater was discharged, then, the reduced frequency of viral detection compared to the other sites was expected. Virus removal in wastewater by stabilization ponds treatment is highly variable among several studies (reviewed in Verbyla & Mihelcic, 2015). This remotion combined with the possibility of a lower viral recovery due to the high concentration of algae (compared with untreated wastewater) could explain the low viral frequency in this site.
Regarding the molecular characterization of RVA, we found strains that clustered with genotypes P[8] and G3. These genotypes have been previously reported in other studies, with G3P[8] being the most prevalent in southern Brazil and corresponding to an equine-like genotype (Gutierrez et al., 2020). However, in our study, we did not observed a high genetic diversity in RVA strains, since only one VP4 and one VP7 genotype were detected. This limited diversity could be due to the small sample size of our study, which included only 36 samples or maybe because of the predominance of this two genotypes. It has been observed that both the P[8] and G3 genotype continue to circulate in our country (Tort et al., 2015a, 2015b). Unfortunately, several samples detected by qPCR were not detected by nested PCR since the quantitative approach presents a higher sensitivity (Zeng et al., 2008).
Our study found a higher frequency of NoV GII compared to GI, which is in agreement with the previous studies based on clinical samples reporting a higher frequency of gastroenteritis outbreaks caused by GII than GI (Matthews et al., 2012; Parikh et al., 2020).
Regarding the molecular characterization of NoV GI, we observed a high genetic variability detecting genotypes GI.2, GI.5, GI.6, GI.3, and GI.7. Specifically, we detected GI.5 in Fray Bentos during January and February 2018, which suggests the occurrence of at least one outbreak in this city during that period. Similarly, we observed the circulation of GI.7 in Salto during October to December 2017. In contrast, the detection of NoV GII revealed the presence of genotypes GII.2, GII.5, GII.6, and GII.4, with the predominance of GII.4, variants New Orleans 2009 and Sydney 2012, which is consistent with a study conducted in Brazil (Hernandez et al., 2018). Several studies, including Huang et al. (2022) reported the detection of NoV GI.2, GI.5, and GI.3, with the more recent detection of genotype GI.6 at a high frequency since 2014. In our study, we also observed the presence of genotypes GI.2, GI.5, GI.3, and GI.6, but with the additional detection of genotype GI.7. For NoV GII, the frequently detected genotypes include GII.4, GII.17, GII.2, GII.3, and GII.13, while in our study we detected the presence of genotypes GII.2, GII.5, GII.6, and GII.4, with the latter two being detected in both studies. Since 2012, the GII.4 Sydney 2012 variant has been the most commonly detected strain in wastewater and has been associated with VAGE outbreaks worldwide. These findings are in agreement with recent molecular epidemiological analyses of NoV over the past 5 years (Zhou et al., 2020; Cannon et al., 2021; Utsumi et al. 2021; Huang et al., 2022).
Comparing our study with a previous one performed by our team in 2011–2012 in our country, it is observed that NoV GI.8, GI.6, GI.5, and GI.7 were previously circulating but they were not detected in the present study. NoV GI.3 continued circulating and GI.7 and GI.2 currently circulate but were absent in 2011–2012. Regarding NoV GII, GII.3, GII.13, GII.17, and GII.1 no longer circulate compared to the previously work. GII.4, GII.2, and GII.6 continue to circulate while other genotypes were not detected (Lizasoain et al., 2015; Victoria et al. 2016).
In addition, co-circulation of different NoV genotypes within a defined human population during the same period provides opportunities for genetic recombination between strains (Wei et al., 2021). Our study detected the presence of both NoV GI and GII strains in the same sample collected in February 2018 and August 2017 in Fray Bentos, as well as in May 2017 in Salto, suggesting the co-circulation of these two genotypes in the same city at the same time.
By analyzing the HAstV stains, we observed only the presence of MLB1 genotype. In a previous study performed in Uruguay the presence of MLB1 was also observed (Lizasoain et al., 2015). On the other hand, in a study with wastewater in Japan, MLB2, VA1, and VA2 genotypes were identified (Hata et al., 2015). Moreover, in the United States, only types 1 and 2 MLB-AstVs were identified from the wastewater samples collected, whereas all known genotypes of MLB-AstVs (types 1–3) were found from the samples collected in Nepal (Hata et al., 2018). These findings suggest the wide distribution of until recently called emergent HAstV.
Conclusions
Our study revealed the circulation of RVA, NoV GI, NoV GII, and HAstV in wastewater reflecting their circulation in the population served by the sewerage network present in the analyzed cities. Only a single genotype for each analyzed gene (VP4 and VP7) of RVA was observed. A high genetic diversity was detected for NoV, with the co-circulation of several genotypes in both Genogroup I and Genogroup II. HAstV showed low variability, with only one genotype detected. We observed that several genotypes that were circulating in 2011–2012 were also present in 2017–2018 but others were only present in one of the analyzed period. It is important to highlight that, in a general way, the strains detected in this study were more similar to contemporary strains circulating in neighboring countries than strains detected in our country in 2011–2012.
References
Afework, D. T., Shumie, M. K., Endalew, G. F., Adugna, A. G., & Tarekegn, B. G. (2022). Pooled prevalence and genetic diversity of norovirus in Africa: A systematic review and meta-analysis. Virology Journal, 19(1), 115. https://doi.org/10.1186/s12985-022-01835-w
Aguiar-Oliveira, M. L., Campos, A., Matos, A. R., Rigotto, C., Sotero-Martins, A., Teixeira, P. F. P., & Siqueira, M. M. (2020). Wastewater-based epidemiology (WBE) and viral detection in polluted surface water: A valuable tool for COVID-19 surveillance—A brief review. International Journal of Environmental Research and Public Health, 17(24), 9251. https://doi.org/10.3390/ijerph17249251
Arowolo, K. O., Ayolabi, C. I., Adeleye, I. A., Lapinski, B., Santos, J. S., & Raboni, S. M. (2020). Molecular epidemiology of astrovirus in children with gastroenteritis in southwestern Nigeria. Archives of Virology, 165(11), 2461–2469. https://doi.org/10.1007/s00705-020-04741-0
Atabakhsh, P., Kargar, M., & Doosti, A. (2020). Molecular detection and genotyping of group A rotavirus in two wastewater treatment plants, Iran. Brazilian Journal of Microbiology, 51(1), 197–203. https://doi.org/10.1007/s42770-019-00131-0
Barril, P. A., Giordano, M. O., Isa, M. B., Masachessi, G., Ferreyra, L. J., Castello, A. A., Glikmann, G., & Nates, S. V. (2010). Correlation between rotavirus A genotypes detected in hospitalized children and sewage samples in 2006, Córdoba, Argentina. Journal of Medical Virology, 82(7), 1277–1281. https://doi.org/10.1002/jmv.21800
van Beek, J., de Graaf, M., Al-Hello, H., Allen, D. J., Ambert-Balay, K., Botteldoorn, N., Brytting, M., Buesa, J., Cabrerizo, M., Chan, M., Cloak, F., Di Bartolo, I., Guix, S., Hewitt, J., Iritani, N., Jin, M., Johne, R., Lederer, I., Mans, J., … Koopmans, M. P. G. (2018). NoroNet. Molecular surveillance of norovirus, 2005–2016: an epidemiological analysis of data collected from the NoroNet network. The Lancet Infectious Diseases, 18(5), 545–553. https://doi.org/10.1016/S1473-3099(18)30059-8
Bitencurt, E. L. R., Siqueira, J. A. M., Medeiros, T. B., Bandeira, R. D. S., de Souza Oliveira, D., de Paula Souza E Guimarães, R. J., da Silva Soares, L., Macarenhas, J. D. P., Teixeira, D. M., Silva, R. S. U., Loureiro, E. C. B., de Moraes Silva, M. C., da Silva, L. D., & Gabbay, Y. B. (2019). Epidemiological and molecular investigation of norovirus and astrovirus infections in Rio Branco, Acre, Northern Brazil: A retrospective study. Journal of Medical Virology, 91(6), 997–1007. https://doi.org/10.1002/jmv.25395
Blanco Fernández, M. D., Barrios, M. E., Cammarata, R. V., Torres, C., Taboga, O. A., & Mbayed, V. A. (2017). Comparison of internal process control viruses for detection of food and waterborne viruses. Applied Microbiology and Biotechnology, 101(10), 4289–4298. https://doi.org/10.1007/s00253-017-8244-2
Bortagaray, V., Lizasoain, A., Piccini, C., Gillman, L., Berois, M., Pou, S., Díaz, M. D. P., Tort, F. L., Colina, R., & Victoria, M. (2019). Microbial source tracking analysis using viral indicators in Santa Lucía and Uruguay rivers, Uruguay. Food and Environmental Virology, 11(3), 259–267. https://doi.org/10.1007/s12560-019-09384-2
Bányai, K., Estes, M. K., Martella, V., & Parashar, U. D. (2018). Viral gastroenteritis. The Lancet, 392(10142), 175–186. https://doi.org/10.1016/S0140-6736(18)31128-0
Cannon, J. L., Bonifacio, J., Bucardo, F., Buesa, J., Bruggink, L., Chan, M. C., Fumian, T. M., Giri, S., Gonzalez, M. D., Hewitt, J., Lin, J. H., Mans, J., Muñoz, C., Pan, C. Y., Pang, X. L., Pietsch, C., Rahman, M., Sakon, N., Selvarangan, R., … Vinjé, J. (2021). Global trends in norovirus genotype distribution among children with acute gastroenteritis. Emerging Infectious Diseases, 27(5), 1438–1445. https://doi.org/10.3201/eid2705.204756
Chhabra, P., de Graaf, M., Parra, G. I., Chan, M. C., Green, K., Martella, V., Wang, Q., White, P. A., Katayama, K., Vennema, H., Koopmans, M. P. G., & Vinjé, J. (2019). Updated classification of norovirus genogroups and genotypes. The Journal of General Virology, 100(10), 1393–1406. https://doi.org/10.1099/jgv.0.001318. Erratum in: The Journal of General Virology 2020;101(8), 893.
Dai, Y. C., Xu, Q. H., Wu, X. B., Hu, G. F., Tang, Y. L., Li, J. D., Chen, Q., & Nie, J. (2010). Development of real-time and nested RT PCR to detect astrovirus and one-year survey of astrovirus in Jiangmen City, China. Archives of Virology, 155(6), 977–982.
Eftim, S. E., Hong, T., Soller, J., Boehm, A., Warren, I., Ichida, A., & Nappier, S. P. (2017). Occurrence of norovirus in raw sewage: A systematic literature review and meta-analysis. Water Research, 111, 366–374. https://doi.org/10.1016/j.watres.2017.01.017
Estes, M. K., & Greenberg, H. B. (2013) Rotaviruses. In: Fields virology, 6th edn. Philadelphia: Lippincott Williams & Wilkins.
Farkas, K., Marshall, M., Cooper, D., McDonald, J. E., Malham, S. K., Peters, D. E., Maloney, J. D., & Jones, D. L. (2018). Seasonal and diurnal surveillance of treated and untreated wastewater for human enteric viruses. Environmental Science and Pollution Research, 25(33), 33391–33401. https://doi.org/10.1007/s11356-018-3261-y
Finkbeiner, S. R., Holtz, L. R., Jiang, Y., Rajendran, P., Franz, C. J., Zhao, G., Kang, G., & Wang, D. (2009). Human stool contains a previously unrecognized diversity of novel astroviruses. Virology Journal, 6, 161.
Fioretti, J. M., Ferreira, M. S. R., Victoria, M., et al. (2011). Genetic diversity of noroviruses in Brazil. Memórias do Instituto Oswaldo Cruz, 106, 942–947.
Ghssein, G., Salami, A., Salloum, L., Chedid, P., Joumaa, W. H., & Fakih, H. (2018). Surveillance study of acute gastroenteritis etiologies in hospitalized children in South Lebanon (SAGE study). Pediatric Gastroenterology, Hepatology & Nutrition, 21(3), 176–183.
Guo, Y., Li, J., O’Brien, J., Sivakumar, M., & Jiang, G. (2022). Back-estimation of norovirus infections through wastewater-based epidemiology: A systematic review and parameter sensitivity. Water Research, 219, 118610. https://doi.org/10.1016/j.watres.2022.118610
Gupta, S., Krishnan, A., Sharma, S., Kumar, P., Aneja, S., & Ray, P. (2017). Changing pattern of prevalence, genetic diversity, and mixed infections of viruses associated with acute gastroenteritis in pediatric patients in New Delhi, India. Journal of Medical Virology, 90(3), 469–476.
Gutierrez, M. B., Fialho, A. M., Maranhão, A. G., Malta, F. C., Andrade, J. D. S. R., Assis, R. M. S., Mouta, S. D. S. E., Miagostovich, M. P., Leite, J. P. G., & Machado, F. T. (2020). Rotavirus A in Brazil: Molecular epidemiology and surveillance during 2018–2019. Pathogens, 9(7), 515. https://doi.org/10.3390/pathogens9070515
Hata, H., Katayama, M., & Kitajima, H. (2015). Furumai, Wastewater analysis indicates that genetically diverse astroviruses, including strains belonging to novel clades MLB and VA, are circulating within Japanese populations. Applied and Environmental Microbiology, 81, 4932–4939. https://doi.org/10.1128/AEM.00563-15
Hata, A., Kitajima, M., Haramoto, E., Lee, S., Ihara, M., Gerba, C. P., & Tanaka, H. (2018). Next-generation amplicon sequencing identifies genetically diverse human astroviruses, including recombinant strains, in environmental waters. Scientific Reports, 8(1), 11837. https://doi.org/10.1038/s41598-018-30217-y
Hellmér, M., Paxéus, N., Magnius, L., Enache, L., Arnholm, B., Johansson, A., Bergström, T., & Norder, H. (2014). Detection of pathogenic viruses in sewage provided early warnings of hepatitis A virus and norovirus outbreaks. Applied and Environmental Microbiology, 80(21), 6771–6781. https://doi.org/10.1128/AEM.01981-14
Hernandez, J. M., Silva, L. D., Junior, E. C. S., Bandeira, R. S., Rodrigues, E. A. M., Lucena, M. S. S., Costa, S. T. P., & Gabbay, Y. B. (2018). Molecular epidemiology and temporal evolution of norovirus associated with acute gastroenteritis in Amazonas state, Brazil. BMC Infectious Diseases, 18(1), 147. https://doi.org/10.1186/s12879-018-3068-y
Huang, Y., Zhou, N., Zhang, S., Yi, Y., Han, Y., Liu, M., Han, Y., Shi, N., Yang, L., Wang, Q., Cui, T., & Jin, H. (2022). Norovirus detection in wastewater and its correlation with human gastroenteritis: A systematic review and meta-analysis. Environmental Science and Pollution Research, 29(16), 22829–22842. https://doi.org/10.1007/s11356-021-18202-x
ICTV. (2018). International Committee on Taxonomy of Viruses. Updated in July 2018 and available at https://talk.ictvonline.org/taxonomy/
I.N.E. Instituto Nacional de Estadıstica. (2011). Censos2011. http://www5.ine.gub.uy/censos2011/index.html. Accessed March 12, 2023.
Kargar, M., Javdani, N., Najafi, A., & Tahamtan, Y. (2013). First molecular detection of group A rotavirus in urban and hospital sewage systems by nested-RT PCR in Shiraz, Iran. Journal of Environmental Health Science and Engineering, 11(1), 4. https://doi.org/10.1186/2052-336X-11-4
Khamrin, P., Thongprachum, A., Okitsu, S., Hayakawa, S., Maneekarn, N., & Ushijima, H. (2016). Multiple astrovirus MLB1, MLB2, VA2 clades, and classic human astrovirus in children with acute gastroenteritis in Japan. Journal of Medical Virology, 88(2), 356–360. https://doi.org/10.1002/jmv.24337
Kitajima, M., Oka, T., Haramoto, E., Takeda, N., Katayama, K., & Katayama, H. (2010). Seasonal distribution and genetic diversity of genogroups I, II, and IV noroviruses in the Tamagawa River, Japan. Environmental Science and Technology, 44, 7116–7122.
Lin, X., Zou, R., Liu, Y., Ji, F., Tao, Z., & Xu, A. (2021). Continuous detection of norovirus and astrovirus in wastewater in a coastal city of China in 2014–2016. Letters in Applied Microbiology, 73(4), 418–425. https://doi.org/10.1111/lam.13530
Lizasoain, A., Tort, L. F., García, M., Gómez, M. M., Cristina, J., Leite, J. P., Miagostovich, M. P., Victoria, M., & Colina, R. (2015). Environmental assessment of classical human astrovirus in Uruguay. Food and Environmental Virology. https://doi.org/10.1007/s12560-015-9186-4
Lum, S., et al. (2016). An emerging opportunistic infection: Fatal astrovirus (VA1/HMO-C) encephalitis in a pediatric stem cell transplant recipient. Transplant Infectious Disease, 18(6), 960–964.
Maes, P., Matthijnssens, J., Rahman, M., & Van Ranst, M. (2009). RotaC: A web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiology, 9, 1–4.
Mao, K., Zhang, H., Pan, Y., & Yang, Z. (2021). Biosensors for wastewater-based epidemiology for monitoring public health. Water Research, 191, 116787. https://doi.org/10.1016/j.watres.2020.116787
Matthews, J. E., Dickey, B. W., Miller, R. D., Felzer, J. R., Dawson, B. P., Lee, A. S., Rocks, J. J., Kiel, J., Montes, J. S., Moe, C. L., Eisenberg, J. N., & Leon, J. S. (2012). The epidemiology of published norovirus outbreaks: A review of risk factors associated with attack rate and genogroup. Epidemiology & Infection, 140(7), 1161–1172. https://doi.org/10.1017/S0950268812000234
Pang, X. L., Preiksaitis, J. K., & Lee, B. (2005). Multiplex real time RT-PCR for the detection and quantitation of norovirus genogroups I and II in patients with acute gastroenteritis. Journal of Clinical Virology, 33(2), 168–171.
Parikh, M. P., Vandekar, S., Moore, C., Thomas, L., Britt, N., Piya, B., Stewart, L. S., Batarseh, E., Hamdan, L., Cavallo, S. J., Swing, A. M., Garman, K. N., Constantine-Renna, L., Chappell, J., Payne, D. C., Vinjé, J., Hall, A. J., Dunn, J. R., & Halasa, N. (2020). Temporal and genotypic associations of sporadic norovirus gastroenteritis and reported norovirus outbreaks in middle Tennessee, 2012–2016. Clinical Infectious Diseases, 71(9), 2398–2404. https://doi.org/10.1093/cid/ciz1106
Pina, S., Puig, M., Lucena, F., Jofre, J., & Girones, R. (1998). Viral pollution in the environment and in shellfish: Human adenovirus detection by PCR as an index of human viruses. Applied and Environmental Microbiology, 64, 3376–3382.
Prevost, B., Lucas, F. S., Ambert-Balay, K., Pothier, P., Moulin, L., & Wurtzer, S. (2015). Deciphering the diversities of astroviruses and noroviruses in wastewater treatment plant effluents by a high-throughput sequencing method. Applied and Environmental Microbiology, 81(20), 7215–7222. https://doi.org/10.1128/AEM.02076-15
RCWG. 2018. Web site: Rotavirus Classification Working Group (RCWG). Newly assigned genotypes, List of accepted genotypes (Updated: June-30–2017). https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/rcwg
Robilotti, E., Deresinski, S., & Pinsky, B. A. (2015). Norovirus. Clinical Microbiology Reviews, 28(1), 134–164. https://doi.org/10.1128/CMR.00075-14
Schultz, A. C., Vega, E., Dalsgaard, A., Christensen, L. S., Nørrung, B., Hoorfar, J., & Vinjé, J. (2011). Development and evaluation of novel one-step TaqMan realtime RT-PCR assays for the detection and direct genotyping of genogroup I and II noroviruses. Journal of Clinical Virology, 50(3), 230–234. https://doi.org/10.1016/j.jcv.2010.12.001
Shamkhali Chenar, S., & Deng, Z. (2017). Environmental indicators for human norovirus outbreaks. International Journal of Environmental Health Research, 27(1), 40–51. https://doi.org/10.1080/09603123.2016.1257705
Silva-Sales, M., Martínez-Puchol, S., Gonzales-Gustavson, E., Hundesa, A., & Gironès, R. (2020). High prevalence of rotavirus A in raw sewage samples from northeast Spain. Viruses, 12(3), 318. https://doi.org/10.3390/v12030318
Tavakoli Nick, S., Mohebbi, S. R., Hosseini, S. M., Mirjalali, H., & Alebouyeh, M. (2020). Monitoring of rotavirus in treated wastewater in Tehran with a monthly interval, in 2017–2018. Journal of Water and Health, 18(6), 1065–1072. https://doi.org/10.2166/wh.2020.112
Tort, L. F., Victoria, M., Lizasoain, A. A., Castells, M., Maya, L., Gómez, M. M., Arreseigor, E., López, P., Cristina, J., Leite, J. P., & Colina, R. (2015a). Molecular epidemiology of group A rotavirus among children admitted to hospital in Salto, Uruguay, 2011–2012: First detection of the emerging genotype G12. Journal of Medical Virology, 87(5), 754–763. https://doi.org/10.1002/jmv.24123
Tort, L. F., Victoria, M., Lizasoain, A., García, M., Berois, M., Cristina, J., Leite, J. P., Gómez, M. M., Miagostovich, M. P., & Colina, R. (2015b). Detection of common, emerging and uncommon VP4, and VP7 human group A rotavirus genotypes from urban sewage samples in Uruguay. Food and Environmental Virology, 7(4), 342–353. https://doi.org/10.1007/s12560-015-9213-5
Utsumi, T., Lusida, M. I., Dinana, Z., Wahyuni, R. M., Soegijanto, S., Athiyyah, A. F., Sudarmo, S. M., Ranuh, R. G., Darma, A., Yamani, L. N., Doan, Y. H., Shimizu, H., Ishii, K., Matsui, C., Deng, L., Abe, T., Katayama, K., & Shoji, I. (2021). Molecular epidemiology and genetic diversity of norovirus infection in children hospitalized with acute gastroenteritis in East Java, Indonesia in 2015–2019. Infection, Genetics and Evolution, 88, 104703. https://doi.org/10.1016/j.meegid.2020.104703
Verbyla, M. E., & Mihelcic, J. R. (2015). A review of virus removal in wastewater treatment pond systems. Water Research, 71, 107–124. https://doi.org/10.1016/j.watres.2014.12.031
Victoria, M., Tort, L. F., García, M., Lizasoain, A., Maya, L., Leite, J. P., Miagostovich, M. P., Cristina, J., & Colina, R. (2014). Assessment of gastroenteric viruses from wastewater directly discharged into Uruguay River, Uruguay. Food and Environmental Virology, 6(2), 116–124. https://doi.org/10.1007/s12560-014-9143-7
Victoria, M., Tort, L. F., Lizasoain, A., García, M., Castells, M., Berois, M., Divizia, M., Leite, J. P., Miagostovich, M. P., Cristina, J., & Colina, R. (2016). Norovirus molecular detection in Uruguayan sewage samples reveals a high genetic diversity and GII.4 variant replacement along time. Journal of Applied Microbiology, 120(5), 1427–35. https://doi.org/10.1111/jam.13058
Vu, D. L., Bosch, A., Pintó, R. M., & Guix, S. (2017). Epidemiology of classic and novel human astrovirus: Gastroenteritis and beyond. Viruses, 9(2), 33. https://doi.org/10.3390/v9020033
WHO. (2017). Diarrhoeal disease. Accessed July 25th, 2023 in https://www.who.int/news-room/fact-sheets/detail/diarrhoeal-disease
WHO. (2009). Manual of rotavirus detection and characterization methods. Immunization, Vaccines and Biologicals, WHO/IVB/08.17. www.who.int/vaccines-documents/
Wei, N., Ge, J., Tan, C., Song, Y., Wang, S., Bao, M., & Li, J. (2021). Epidemiology and evolution of Norovirus in China. Human Vaccines & Immunotherapeutics, 17(11), 4553–4566. https://doi.org/10.1080/21645515.2021.1961465
Yu, F., Jiang, B., Guo, X., Hou, L., Tian, Y., Zhang, J., Li, Q., Jia, L., Yang, P., Wang, Q., Pang, X., & Gao, Z. (2022). Norovirus outbreaks in China, 2000–2018: A systematic review. Reviews in Medical Virology, 32(6), e2382. https://doi.org/10.1002/rmv.2382
Zeng, S. Q., Halkosalo, A., Salminen, M., et al. (2008). One-step quantitative RT-PCR for the detection of rotavirus in acute gastroenteritis. Journal of Virological Methods, 153(2), 238–2340.
Zhou, H., Wang, S., von Seidlein, L., & Wang, X. (2020). The epidemiology of norovirus gastroenteritis in China: Disease burden and distribution of genotypes. Frontiers of Medicine, 14(1), 1–7. https://doi.org/10.1007/s11684-019-0733-5
Acknowledgements
We would like to thank the financial support for this project by Fondo Clemente Estable (FCE_1_2017_1_136092) of the National Agency of Research and Innovation (ANII), Uruguay. We also want to thank Dr. Moller and Dr. Thomson from Biological Chemistry Institute, School of Sciences, UdelaR, for their technical assistance with ultracentrifugation procedure.
Author information
Authors and Affiliations
Contributions
Conceived the study: MV and RC. Wrote the manuscript: VB. Performed the laboratory experiments: VB and MS. Performed the bioinfomatic analysis: DM and VB. Supervised data, collection and laboratory work: MV and RC. Critical reviewed the manuscript: VB, MS, DM, RC and MV. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Viviana, B., Matias, S., Daiana, M. et al. Molecular Characterization of Gastroenteric Viruses in Wastewater from Cities in Uruguay. Food Environ Virol 15, 318–330 (2023). https://doi.org/10.1007/s12560-023-09567-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12560-023-09567-y