
Abstract
Viruses remain the leading cause of acute gastroenteritis (AGE) worldwide. Recently, we reported the abundance of AGE viruses in raw sewage water (SW) during the COVID-19 pandemic, when viral AGE patients decreased dramatically in clinics. Since clinical samples were not reflecting the actual state, it remained important to determine the circulating strains in the SW for preparedness against impending outbreaks. Raw SW was collected from a sewage treatment plant in Japan from August 2018 to March 2022, concentrated by polyethylene-glycol-precipitation method, and investigated for major gastroenteritis viruses by RT-PCR. Genotypes and evolutionary relationships were evaluated through sequence-based analyses. Major AGE viruses like rotavirus A (RVA), norovirus (NoV) GI and GII, and astrovirus (AstV) increased sharply (10–20%) in SW during the COVID-19 pandemic, though some AGE viruses like sapovirus (SV), adenovirus (AdV), and enterovirus (EV) decreased slightly (3–10%). The prevalence remained top in the winter. Importantly, several strains, including G1 and G3 of RVA, GI.1 and GII.2 of NoV, GI.1 of SV, MLB1 of AstV, and F41 of AdV, either emerged or increased amid the pandemic, suggesting that the normal phenomenon of genotype changing remained active over this time. This study crucially presents the molecular characteristics of circulating AGE viruses, explaining the importance of SW investigation during the pandemic when a clinical investigation may not produce the complete scenario.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute gastroenteritis (AGE) remains a major global health problem: causing significant morbidity and mortality, though mainly in developing countries, it poses a substantial economic burden in both developing and developed countries around the world (Chow et al., 2010; Hoque et al., 2019a, 2019b). Viruses including rotavirus (RV), norovirus (NoV), adenovirus (AdV), astrovirus (AstV), sapovirus (SV), and enteroviruses (EV) remain the leading causes of AGE, particularly, in children which represent about 70% of total AGE (Chow et al., 2010; Maria & Hrishikesh, 2018). Being a developed and industrialized nation, Japan still struggles against AGE-associated morbidity and economic losses throughout the year (Kawata et al., 2020). To prevent severe RV infections, Japan has introduced RV vaccines as voluntary vaccines since 2011 and included these in the national immunization program from October 2020, yet the diversity in RV genotypes and increasing trends of other diarrheal viruses have been found in post-RV-vaccination era worldwide including Japan (Gikonyo et al., 2020; Hoque et al., 2022a, 2022b, 2022c; Tsugawa et al., 2021). However, after the emergence of the pandemic coronavirus disease-2019 (COVID-19), the implementation of non-pharmaceutical interventions (NPIs) like social distancing, mask-wearing, increased hand hygiene, school closures, and working from home have proven effective to reduce COVID-19 along with many other infectious diseases including AGE (Eigner et al., 2021; Zhang et al., 2022). It was thought that the precautious lifestyle adopted during the COVID-19 pandemic may have decreased viral AGE. However, more recently, we detected an abundance of diarrheal viruses in raw sewage during the COVID-19 pandemic, suggesting that infections in the population may not have decreased with the introduction of NPIs (Hoque et al., 2022a, 2022b, 2022c). The number of cases declined, likely due to reduced transmission because of school closures, activity restrictions, as well as patients’ disinterest in seeking medical care during the pandemic, resulting in fewer reports (Armistead et al., 2022). Since the infections are widespread in the community, there is a constant risk that a new outbreak strain may emerge in a short time. Therefore, it remains urgent to investigate the circulating strains in the wastewater, exceptionally, when clinical samples are no more efficient to provide the complete scenario of the infections. In fact, wastewater-based epidemiology (WBE), filling the gap of individual testing, provides an early warning of the emergence of infectious agents in the community and its subsequent progression toward an outbreak. WBE is especially important for AGE viruses, as diarrhea is usually caused by contaminated food and/or water that may be adulterated from the environment (Hafliger et al., 2000; Laine et al., 2011). Again, enteric viruses may remain asymptomatic (Okitsu et al., 2020), which could be missed in clinical diagnosis but identified by WBE.
In this study, we collected raw sewage water (SW) regularly from a wastewater treatment plant in Japan well before the pandemic and examined the molecular characteristics of circulating strains of the major diarrheal viruses before and after the emergence of the COVID-19 pandemic, which remain critical for understanding the evolutionary relationship among genotypes during the pandemic.
Materials and Methods
Design of the Study
A total of 39 raw SWs collected almost monthly from August 2018 to March 2022 from a sewage treatment plant in the Kansai region of Japan were concentrated and analyzed for the presence of group A RV (RVA), NoV GI, NoV GII, AdV, AstV, SV, EV, and in addition nCoV (novel coronavirus 2019) after detection of spiked internal control murine norovirus (MNV) by reverse transcription-polymerase chain reaction (RT-PCR). In Japan, although the first COVID-19 case was identified on January 14, 2020, a series of containment measures, including suspension of large gatherings, campaigning for hygiene, and nationwide school closures were implemented from early March 2020 (Imai et al., 2022). Thus, 16 samples collected from August 2018 to February 2020 were considered pre-restriction samples, while the remaining 23 samples were collected thereafter during the restriction period of COVID-19.
Sewage Water (SW) Collection and Concentration
About half-liter of the inlet raw SW was collected in a clean PET bottle and kept frozen until further processing for concentration. About 50 ml of raw SW was first mixed with MNV as internal process control and centrifuged at 3000 rpm (RCF 1000×g) for 10 min to remove large particles from the supernatant. The clear supernatant was then mixed with 4 g of polyethylene glycol (PEG)-6000 (Kanto Chemical Co., Inc., Tokyo, Japan) and 1.2 g of NaCl (Sigma Aldrich, St. Louis, MO) and stirred at room temperature for 4 h. The suspension was then centrifuged at 10,000 rpm (RCF 11,900×g) for 30 min and the supernatant was discarded without disturbing the pellet. Finally, the pellet was suspended in 0.5 ml of nuclease-free water (QIAGEN, Hilden, Germany), aliquoted at 200 µl/tube for storage at − 30 °C.
Extraction of Viral RNA and Reverse Transcription (RT)
Viral RNA was extracted from 140 µl of concentrated SW using QIAamp Viral RNA mini kit (QIAGEN) following the manufacturer’s instructions and stored at − 30 °C until use. For cDNA synthesis, 5 µl of RNA was mixed with 0.5 µl of 50% DMSO and denatured at 95 °C for 5 min, followed by immediate cooling on ice. The RT was performed using ReverTra Ace reverse transcriptase (Toyobo, Osaka, Japan) and random primers (Takara, Shiga, Japan) through a single thermal cycle of 30 °C for 10 min, followed by 42 °C for 40 min, and 95 °C for 5 min before cooling or storage at − 30 °C.
Detection of Viruses and Molecular Analyses
Target viruses were detected after the RT by monoplex polymerase chain reaction (PCR) using GoTaq Green Master mixes (Promega, Madison, WI) and specific primers as described previously (Thongprachum et al., 2018). Namely, the partial regions of VP7 of RVA (primer pair: sBeg9-VP7-1’), capsid of NoV GI (G1SKF-G1SKR), GII (1st PCR: COG2F-G2SKR, nested PCR: G2SKF-G2SKR) and SV (SLV5317-SLV5749), hexon of AdV (Ad1-Ad2), ORF-1b of AstV (SF0073-SF0076), and 5’UTR of EV (F1-R1) were amplified and sequenced for screening and genotyping, respectively (Thongprachum et al., 2018). The MNV was confirmed by a nested RT-PCR (Kitajima et al., 2009). The PCR was maintained: initial denaturation at 94 °C for 3 min, followed by 37 cycles of denaturation, annealing, and elongation, each for 1 min at 94 °C, 50 °C, and 72 °C, respectively, and the final extension at 72 °C for 7 min. The first PCR product (1 μl) was re-amplified using the same specific primers if the first PCR remained negative or faint. In addition, RV-vaccine strain-specific RT-qPCR was done to assess Rotarix (NSP2 gene) and RotaTeq (VP6 gene) strains in all RVA-positive samples as described previously (Gautam et al., 2014; Hoque et al., 2022a, 2022b, 2022c). To detect nCoV, both nested and real-time RT-PCRs were performed using primer sets of ORF1a (Shirato et al., 2020) and CDC 2019-nCoV_N2 (Kitamura et al., 2021), respectively.
PCR amplicons were purified and sequenced through the Sanger sequencing method. The sequences were analyzed using the Chromas (ver 2.6.6) and Bioedit Sequence Alignment Editor (ver 7.2.6.1). The genotypes were determined by the identity (nucleotide identity) with reference sequences obtained from the NCBI GenBank database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). In addition, the major G-genotypes including G1, G2, human typical G3, equine-like G3 (G3e), G4, G8, G9, and G12 were determined in RVA-positive samples by nested PCR using primers as described by Fujii et al., (2019) with some modifications: the 1st PCR products were 30-fold diluted and 1 μl of it was used in 2nd monoplex PCR ran for 30 cycles.
The final multiple sequence alignments and the construction of the phylogenetic trees were performed using MEGA7 software using the Neighbor-Joining method with the Kimura 2-parameter model and statistical significance testing by 1000 bootstrapping replicates.
Nucleotide Accession Number
A total of 76 nucleotide sequences determined in this study were deposited in the GenBank database for the accession numbers: RVA (OP696914-OP696931), NoV GI (OP692718-OP692724), NoV GII (OP692737- OP692751), SV (OP696906-OP696913), AdV (OP696942-OP696949), AstV (OP696932-OP696941), and EV (OP696950-OP696959).
Results
An Abundance of Enteric Viruses Before and After the Emergence of Pandemic
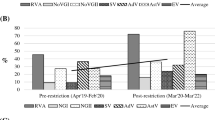
Among the 16 pre-restriction samples, SV was detected in 81.2% (13 of 16) of the samples, whereas other viruses were detected in nearly 50% (7 or 8 of 16) of the samples (Table 1). In contrast, RVA, NoV GI, and SV were positively detected in about 70% (16 of 23) of samples, followed by 65.2% AstV, 61% NoV GII, 47.8% EV, and 43.5% AdV among 23 samples collected during the restriction period of COVID-19 from March 2020 to March 2022.
The average detection rates in the pre- (53.6%) and post-restriction (61%) periods did not differ significantly (P = 0.26 of t-test), suggesting that AGE did not decrease in the community during the pandemic. However, nCoV was not detected in any of these samples.
Seasonal Distributions
Although these viruses remain abundant in all four seasons found in Japan including the spring (March to May), summer (June to August), autumn (September to November), and winter (December to February), the average detection rate remained highest in the winter (74.6%), followed by spring (61.4%), autumn (52.3%), and summer (42.8%) (Table 2). Namely, except EV, all other six diarrheal viruses were detected most frequently in the winter and lowest in the summer, while EV was detected vice versa (Table 2). Seasonal distribution of these viruses appeared similar before and after the pandemic (Table 2).
Genotype Diversity of RVA
As shown in Table 1, all major genotypes of RVA including G1, G2, G3, G3e, G8, and G9 increased sharply during the pandemic. The most common genotype was G3 (85%), followed by G3e, G8, G9 (each remained 55%), G1 (40%), G2 (30%), and G6, G10, G11 (each remained 5%) over the study period.
Since RVAs are found in both humans and animals, we did a sequence analysis of the VP7 gene to differentiate the animal strains and human strains of RVAs. Phylogenetic analysis of RVA revealed that this raw sewage contained RVA from both human and animal sources: R41, R48, R50, R51, and R60 strains remained genetically close (93–100% identity) to animal strains, while R40, R49, R52, R61, and R63 exhibited almost similar identities (98.2%) with both human and animal strains (Fig. 1). R25 remained 100% interchangeable with Tokyo 18–25 strain (LC477366) detected in the same year (2018) from human stool and characterized as G3e bearing DS-1-like backbone with P[8] genes (Fujii et al., 2020). Both G2 genotypes (R29 and R35) belonged to the IV-A1 sub-lineage and showed 98.2% nucleotide identity with Sewage_4 strain (KX931935) detected previously from the same sewage plant in 2015 (Thongprachum et al., 2018) and akin to the epidemic strains of 2014/2015 and 2016/2017 seasons in Japan (Fujii et al., 2020; Thongprachum et al., 2018). RotaTeq, but not Rotarix, vaccine strains were detected in only 2 (5.2%) SW samples by RT-qPCR: one in March 2020 (20 copies/ml), and another in February 2022 (2.4 × 103 copies/ml).

Phylogenetic analysis of RVA based on partial VP7 region. The tree was constructed by the Neighbor-Joining method and drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The tree is rooted by the G18 strain and constructed with the study strains (bold, underlined with sampling time and accession number in the first bracket) and the reference strains obtained from the GenBank database. Bootstrap values ≥ 75% are shown at the branch nodes
Genetic Diversity of NoVs and SVs
Both NoV GI and GII increased sharply in SWs during the pandemic (Table 1). Among NoV GI-positive samples, the detection rate of the GI.3 genotype was increased from 4.3 to 13% during the pandemic. In addition, GI.1 (8.7%) and GI.6 (4.3%) were detected only during the pandemic. As shown in Fig. 2A, NGI50 and NGI60 showed 100% identities with a strain (LC521521) detected from asymptomatic/less symptomatic people in Thailand in 2018 (Phattanawiboon et al., 2020), whereas NGI30 and NGI61 remained 100% indistinguishable from epidemic strains (MN922737, MT491999), as well as environmental strains (MN752674, MN495013), detected in Taiwan, Spain, Thailand, and South Korea in 2018–2019 (Chiu et al., 2020). Both NGI62 and NGI63 were related (> 99.5% identity) to GI.1 clinical (MT492066) and environmental (MZ021974) strains. NGI49 showed 100% similarity with GI.6 strains detected in the wastewater in South Korea (MN494556) and Pakistan (OK326849) in 2018 and later reported in the patients in Tokyo in 2021 (LC646339).



Phylogenetic analysis of NoV GI (a), NoV GII (b), and SV (c) based on partial capsid region. The trees were constructed by the Neighbor-Joining method and drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic trees. The strains detected in this study are shown with the collection time and accession number in bold and underlined. Bootstrap values ≥ 75% are shown at the branch nodes. Asterisks represent the prototypes
Among NoV GII genotypes, GII.2 (45.5%) remained the major that was mainly detected during the pandemic (Table 1). Other dominating NoV GII genotypes include GII.17 (13.6%), GII.1 (4.5%), and GII.6 (4.5%). Phylogenetic analysis demonstrated that, except the NGII39 and NGII63, other GII.2 strains remained close to the circulating GII.P16-GII.2 strain that was isolated from both sporadic cases and outbreaks in many countries including Japan (Honjo et al., 2022; Nagasawa et al., 2018), and is thought to be derived from the epidemic GII.2 strains of 2010–2012 (Fig. 2(B)). NGII37, NGII52, and NGII62 remained in a cluster with strain (LC169560) characterized as sub-lineage IIIc of GII.17 and has the similarity with outbreak strain reported in central Japan in 2015 (Thongprachum et al., 2018).
In contrast, the detection rate of SVs decreased slightly (81.2–69.6%) during the pandemic, yet remained comparative to other viruses. S60, S61, and S62 strains, detected in the winter of 2021–2022, showed 100% identities with clinical strains of the GI.1 genotype that were reported from different parts of the world including China (2013–2017), Japan (2010, 2017), Russia (2012), Brazil (2010) and USA (2014) (Fig. 2(C)) (Okitsu et al., 2021). S39 and 41 remained similar to GI.2 strains detected in China in 2016–2017, while S33, S34, and S40 were clustered in the GII.3 genotype with both environmental and clinical strains collected from China and Russia earlier.
The Predominance of HAdV Type F41 and the Genetic Diversity of AstVs and EVs
The detection rate of AdV and EV decreased slightly (3–8%), whereas AstV increased by > 15% during the pandemic (Table 1). Genotyping was possible for 44.4% (8 of 18) of AdV which all showed 100% identity with the F41 genotype and remained largely divided into two groups (Fig. 3A). Importantly, most strains detected during the pandemic (AD38, AD40, AD60, AD61, and AD63) remained in a group with strains of lineage 1, while AD25, 26, and 37 strains detected earlier were clustered into the alternate group with strains of lineage 2 and 3 of F41 (Jasper Götting, 2022), indicating the emergence of different F41 strains during the pandemic.



Phylogenetic analysis of AdV (a), AstV (b), and EV (c) based on partial hexon, ORF-1b, and 5’UTR regions, respectively. The trees were constructed by the Neighbor-Joining method and drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic trees. The strains detected in this study are shown with the isolation time in bold, underlined. Bootstrap values ≥ 75% are shown at the branch nodes. The name of the disease other than AGE has been mentioned if available
To determine the evolutionary relationships among AstV-genotypes, the partial ORF-1b region of 10 AstV-positive samples were sequenced. Half of them pertained to MLB (Melbourne) clade including MLB1 (AS37, AS38, AS60, and AS61) and MLB3 (AS39) showing 99–100% identities with clinical strains detected worldwide in various countries including Japan (LC694998 and LC695000) (Fig. 3B). The remaining half was related to canine AstV (CAstV), close to the strain (KY271995) characterized earlier as China-type group-2 CAstV (Zhou et al., 2017). However, both classical (HAstV) and Virginia (VA) types AstVs were not detected in this study.
To find out the genetic diversity of EVs, the 5’UTR region of 10 EV-positive samples were sequenced. Half (5/10, 50%) of them remained attached to human EV-A species of CVA2, CVA4, CVA6, CVA10, and CVA16 serotypes, while 30% (3/10) strains matched with human EV-B of CVB4, E11, and E30 genotypes, and remaining 20% (2/10) fitted with human EV-C of CVA19 and CVA24 types. Among these, EV32 (CVA16), EV25 (CVA10), and EV41 (CVA19) remained relevant to strains responsible for HFMD in China, Australia, and Malaysia (Wang et al., 2022; Yi et al., 2022; Zhou et al., 2011), while EV59 (CVA4) was akin to strain (MN964082) caused herpangina in China in 2018 (Guo et al., 2020) (Fig. 3C). EV30 strain showed 98.4% identity with the outbreak strain (MZ171089) caused hemorrhagic conjunctivitis in China in 2007 (Li et al., 2022). Since HFMD, herpangina, and hemorrhagic conjunctivitis remain summer diseases (Ghaznavi et al., 2022), it explains the reason for the high detection rate of EVs in the summer (Table 2).
Discussion
Recently, we wrote a letter to the editor pointing out the abundance of AGE viruses in raw SW during the COVID-19 pandemic, when AGE patients in clinics were drastically reduced (Hoque et al., 2022a, 2022b, 2022c). However, the molecular characteristics of circulating strains in SW during the pandemic are not yet known. Because the emergence or reemergence of predominant genotypes through evolutionary change is a common phenomenon in AGE viruses, understanding the molecular changes in genotypes over time is absolutely essential to be prepared for impending outbreaks caused by emerging variants.
SW remained the best means of determining the circulating strains, especially when the number of clinical specimens declined during the pandemic. The SWs collected in the present study remained a mixture of black (wastewater from toilets) and gray waters (all domestic wastewater except toilets) from approximately 245,000 inhabitants, coming from homes, schools, markets, farms, and hospitals (after initial treatment), together with environmental waste from rainwater.
We observed that although the detection rates of SV, AdV, and EV decreased slightly with the introduction of NPIs, most enteric viruses such as RVA, NoV GI, GII, and AstV were detected more frequently at the time of the pandemic (Table 1). The SWs of this SW treatment plant were also studied in 2015–2016 (Thongprachum et al., 2018). As expected, several similarities and dissimilarities in genotypes were found between past and present data. For example, the G3-genotype of RVA remained high in both the pre- and post-pandemic periods in the present study, while the major RVA genotype was G2 in the earlier study (Thongprachum et al., 2018). Several clinical data revealed that the G2 genotype dominated in many places in Japan during 2014–2017 (Hoque et al., 2020; Khandoker et al., 2018), whereas the G3 genotype emerged in the post-vaccine era with an equine-like G3 (eG3) containing the DS-1-like genotype constellation that was firstly reported to cause an outbreak in Japan in Hokkaido in 2016 and then gradually increased (Akane et al., 2021; Okitsu et al., 2022). Importantly, RVA was detected frequently even after the introduction of RV vaccines in the national immunization program, although the RV-vaccine strains were detected in only 2 SW samples. Previously, we have shown that RV vaccines can reduce the severity of disease but cannot prevent the occurrence of RV infection (Hoque et al., 2019a, 2019b; Kawata et al., 2021; Ushijima et al., 2021), which is consistent with the present results. A similar interpretation has also been documented by other groups (Araki et al., 2016; Braeckman et al., 2012; Fujii et al., 2017; Kozawa et al., 2022; Markkula et al., 2017, 2020; Payne et al., 2019; Santos et al., 2016). Indeed, RV vaccines were originally developed to protect against serious diseases caused by commonly circulating strains (Hoque et al., 2018a, 2018b). The introduction of RV vaccination has succeeded in nearly halving the number of global RV deaths from 453,000 in 2008 to 215,000 in 2013 (Moyo et al., 2014; Tate et al., 2012). Indeed, RV vaccines continue to be successful in minimizing disease severity as well as hospitalizations and thus reducing mortality from RV infection, but the infection nevertheless remains ineradicable, showing multiple outbreaks and greater diversity of circulating RV genotypes in the post-vaccination period documented in our previous studies (Hoque et al., 2020; Okitsu et al., 2022) as well as by other groups (Gibory et al., 2022; Gikonyo et al., 2020; Tsugawa et al., 2021). A similar diversity of RV genotypes was observed in the present study at SW (Table 1). RV vaccines provide poor protection from some emerging strains like G8P[8] (Hoque et al., 2018a, 2018b). Again, since the target population for RV vaccination is infants (≤ 6 months), there is a possibility that the vaccines failed to protect older children (≥ 3 years) from infection due to not receiving the vaccine or that the effectiveness of the vaccine may have declined. RV vaccine strains were detected low in SW in earlier studies as well (Ito et al., 2021), probably because the infants typically use diapers.
In agreement with the prior study (Thongprachum et al., 2018), AdV-F41, Ast-MLB1, and different species of EVs predominated in the present study (Table 1). However, globally dominant NoV GII.4 was not detected in the present study; instead, NoV GII.2 has emerged in this population. Indeed, NoV GII.2 (particularly GII.P16-GII.2) has emerged abruptly in the last 6–7 years and remained associated with both sporadic cases and outbreaks in many countries, e.g., the USA, Australia, Thailand, China, and Japan, including this community (Gao et al., 2019; Nagasawa et al., 2018; Supadej et al., 2019).
Importantly, herein, we detected a high genotype diversity of AGE viruses in raw sewage in both pre-and post-pandemic eras. Although some stains looked like the animal strains (e.g., RVA from bovine/dog/cat/porcine/equine, and canine AstV) and some remained associated with different diseases (e.g., HFMD, herpangina, and hemorrhagic conjunctivitis), yet most of these viruses remained closely related to human enteric viruses. In addition, most of the strains like G1, G3 of RVA, GI.1 of NoV GI, GII.2 of NoV GII, GI.1 of SV, and MLB1 of AstV either increased or re-emerged amid the pandemic, suggesting that the normal phenomena of genotype changes remained active during the pandemic. Although these types of molecular data are deemed evidence of viral fragments, and not the infectious virus directly, the risk of impending outbreaks through exposure cannot be ruled out. Several extensive AGE outbreaks have been reported due to contamination with SW (Hafliger et al., 2000; Laine et al., 2011).
Of note, nCoV was not detected in the present study by both conventional and real-time RT-PCR, suggesting that COVID-19 infection remained low in the community which was difficult to detect from the wastewater. According to WHO, the absence of nCoV in wastewater does not indicate a lack of the virus in the community (WHO, 2022). However, the possibility of inefficient recovery of RNA from enveloped viruses like nCoV cannot be ruled out. Recent evidence suggests that, unlike non-enveloped viruses, the detection of enveloped nCoV from the sewage is much more difficult, particularly from the effluent (Hata et al., 2021; Kitamura et al., 2021). Very recently, Adachi Katayama et al. (2023) demonstrated a novel highly sensitive COPMAN (Coagulation and Proteolysis method using Magnetic beads for detection of Nucleic acids) method to detect nCoV from wastewater. Since our chief objective was to determine non-enveloped diarrheal viruses from the SW, we used non-enveloped MNV as the process control and the PEG precipitation (ppt) method for the concentration of the virus, which remained the most popular and widely used method for the concentration of non-enveloped diarrheal viruses from wastewater. Nonetheless, in agreement with recent evidence (Adachi Katayama et al., 2023), the weakness of the PEG ppt method for the detection of nCoV from SW has been found in the present study.
Rather than quantitative analyses, the goal of this study was to gain insight into molecular characteristics and changes in genotypes of diarrheal viruses in the community. However, samples that yielded faint and/or multiple bands were not sequenced for genotyping. Thus, some genotypes remained unidentified. One major limitation of our sequenced-based genotyping method was only a single dominating strain could be identified from a single sample of a month which could not reflect the prevalence of the infection in the community. In addition, this study did not include the examination of clinical samples of the same area. Therefore, the exact correlation between human infection and SW remained unknown. Nevertheless, a considerable length of time before and after the emergence of COVID-19, plus the genotyping results of the majority of samples with evolutionary relationships, remained sufficient to understand the overall trend of infections over this period.
In conclusion, this study provides the first crucial information on the circulating strains in the community during the COVID-19 pandemic. The data reveals not only the high frequency of infections in the community but also the existence of robust genetic evolutionary features among genotypes during the pandemic. Concurrently, the present study also explains the importance of WBE during the pandemic when clinical data fail to delineate the actual scenario. WBE should be strengthened further for regular monitoring of the trend of infection and early interventions before any emerging variant attends an epidemic.
References
Adachi Katayama, Y., Hayase, S., Ando, Y., Kuroita, T., Okada, K., Iwamoto, R., et al. (2023). COPMAN: a novel high-throughput and highly sensitive method to detect viral nucleic acids including SARS-CoV-2 RNA in wastewater. Science of the Total Environment, 856(Pt 1), 158966. https://doi.org/10.1016/j.scitotenv.2022.158966
Akane, Y., Tsugawa, T., Fujii, Y., Honjo, S., Kondo, K., Nakata, S., et al. (2021). Molecular and clinical characterization of the equine-like G3 rotavirus that caused the first outbreak in Japan, 2016. Journal of General Virology. https://doi.org/10.1099/jgv.0.001548
Araki, K., Hara, M., Sakanishi, Y., Shimanoe, C., Nishida, Y., Matsuo, M., & Tanaka, K. (2016). Estimating rotavirus vaccine effectiveness in Japan using a screening method. Human Vaccines & Immunotherapeutics, 12(5), 1244–1249. https://doi.org/10.1080/21645515.2015.1121337
Armistead, I., Tran, A., White, A. E., Wilson, E., & Scallan Walter, E. J. (2022). Trends in outpatient medical-care seeking for acute gastroenteritis during the COVID-19 Pandemic, 2020. Foodborne Pathogens and Disease, 19(4), 290–292. https://doi.org/10.1089/fpd.2021.0099
Braeckman, T., Van Herck, K., Meyer, N., Pircon, J. Y., Soriano-Gabarro, M., Heylen, E., et al. (2012). Effectiveness of rotavirus vaccination in prevention of hospital admissions for rotavirus gastroenteritis among young children in Belgium: Case-control study. BMJ, 345, e4752. https://doi.org/10.1136/bmj.e4752
Chiu, S. C., Hsu, J. K., Hu, S. C., Wu, C. Y., Wang, Y. C., & Lin, J. H. (2020). Molecular Epidemiology of GI.3 Norovirus Outbreaks from Acute Gastroenteritis Surveillance System in Taiwan, 2015–2019. BioMed Research International, 2020, 4707538. https://doi.org/10.1155/2020/4707538
Chow, C. M., Leung, A. K., & Hon, K. L. (2010). Acute gastroenteritis: From guidelines to real life. Clinical and Experimental Gastroenterology, 3, 97–112.
Eigner, U., Verstraeten, T., & Weil, J. (2021). Decrease in norovirus infections in Germany following COVID-19 containment measures. Journal of Infection, 82(6), 276–316. https://doi.org/10.1016/j.jinf.2021.02.012
Fujii, Y., Doan, Y. H., Wahyuni, R. M., Lusida, M. I., Utsumi, T., Shoji, I., & Katayama, K. (2019). Improvement of rotavirus genotyping method by using the semi-nested multiplex-PCR with new primer set. Frontiers in Microbiology, 10, 647. https://doi.org/10.3389/fmicb.2019.00647
Fujii, Y., Noguchi, A., Miura, S., Ishii, H., Nakagomi, T., Nakagomi, O., & Takahashi, T. (2017). Effectiveness of rotavirus vaccines against hospitalisations in Japan. BMC Pediatrics, 17(1), 156. https://doi.org/10.1186/s12887-017-0916-7
Fujii, Y., Oda, M., Somura, Y., & Shinkai, T. (2020). Molecular Characteristics of Novel Mono-Reassortant G9P[8] Rotavirus A Strains Possessing the NSP4 Gene of the E2 Genotype Detected in Tokyo, Japan. Japanese Journal of Infectious Diseases, 73(1), 26–35. https://doi.org/10.7883/yoken.JJID.2019.211
Gao, Z., Liu, B., Yan, H., Li, W., Jia, L., Tian, Y., et al. (2019). Norovirus outbreaks in Beijing, China, from 2014 to 2017. Journal of Infection, 79(2), 159–166. https://doi.org/10.1016/j.jinf.2019.05.019
Gautam, R., Esona, M. D., Mijatovic-Rustempasic, S., Ian Tam, K., Gentsch, J. R., & Bowen, M. D. (2014). Real-time RT-PCR assays to differentiate wild-type group A rotavirus strains from Rotarix((R)) and RotaTeq((R)) vaccine strains in stool samples. Human Vaccines & Immunotherapeutics, 10(3), 767–777. https://doi.org/10.4161/hv.27388
Ghaznavi, C., Sakamoto, H., Kawashima, T., Horiuchi, S., Ishikane, M., Abe, S. K., et al. (2022). Decreased incidence followed by comeback of pediatric infections during the COVID-19 pandemic in Japan. World Journal of Pediatrics, 18(8), 564–567. https://doi.org/10.1007/s12519-022-00575-9
Gibory, M., Bruun, T., Flem, E., Dembinski, J. L., Haltbakk, I., Stordal, K., et al. (2022). Genetic diversity of rotavirus strains circulating in Norway before and after the introduction of rotavirus vaccination in children. Journal of Medical Virology, 94(6), 2624–2631. https://doi.org/10.1002/jmv.27484
Gikonyo, J. N., Mbatia, B., Okanya, P. W., Obiero, G. F. O., Sang, C., Steele, D., & Nyangao, J. (2020). Post-vaccine rotavirus genotype distribution in Nairobi County, Kenya. International Journal of Infectious Diseases, 100, 434–440. https://doi.org/10.1016/j.ijid.2020.09.005
Guo, W. P., Chen, G. Q., Xie, G. C., Du, L. Y., & Tang, Q. (2020). Mosaic genome of Human Coxsackievirus A4 associated with herpangina and HFMD in Yancheng, China, 2016 and 2018. International Journal of Infectious Diseases, 96, 538–540. https://doi.org/10.1016/j.ijid.2020.05.057
Hafliger, D., Hubner, P., & Luthy, J. (2000). Outbreak of viral gastroenteritis due to sewage-contaminated drinking water. International Journal of Food Microbiology, 54(1–2), 123–126. https://doi.org/10.1016/s0168-1605(99)00176-2
Hata, A., Hara-Yamamura, H., Meuchi, Y., Imai, S., & Honda, R. (2021). Detection of SARS-CoV-2 in wastewater in Japan during a COVID-19 outbreak. Science of the Total Environment, 758, 143578. https://doi.org/10.1016/j.scitotenv.2020.143578
Honjo, S., Kuronuma, K., Fujiya, Y., Nakae, M., Ukae, S., Nihira, H., et al. (2022). Genotypes and transmission routes of noroviruses causing sporadic acute gastroenteritis among adults and children, Japan, 2015–2019. Infection, Genetics and Evolution, 104, 105348. https://doi.org/10.1016/j.meegid.2022.105348
Hoque, S. A., Iizuka, I., Kobayashi, M., Takanashi, S., Anwar, K. S., Islam, M. T., et al. (2019a). Determining effectiveness of rotavirus vaccine by immunochromatography and reverse transcriptase polymerase chain reaction: A comparison. Vaccine, 37(39), 5886–5890. https://doi.org/10.1016/j.vaccine.2019.07.091
Hoque, S. A., Islam, M. T., Kobayashi, M., Takanashi, S., Anwar, K. S., Watanabe, T., et al. (2018a). Our response to the letter to the editor. Vaccine, 36(34), 5110–5111. https://doi.org/10.1016/j.vaccine.2018.05.004
Hoque, S. A., Khandoker, N., Thongprachum, A., Khamrin, P., Takanashi, S., Okitsu, S., et al. (2020). Distribution of rotavirus genotypes in Japan from 2015 to 2018: Diversity in genotypes before and after introduction of rotavirus vaccines. Vaccine, 38(23), 3980–3986. https://doi.org/10.1016/j.vaccine.2020.03.061
Hoque, S. A., Kobayashi, M., Takanashi, S., Anwar, K. S., Watanabe, T., Khamrin, P., et al. (2018b). Role of rotavirus vaccination on an emerging G8P[8] rotavirus strain causing an outbreak in central Japan. Vaccine, 36(1), 43–49. https://doi.org/10.1016/j.vaccine.2017.11.056
Hoque, S. A., Kotaki, T., Pham, N. T. K., Onda, Y., Okitsu, S., Sato, S., et al. (2022a). Abundance of Viral Gastroenteritis before and after the Emergence of COVID-19: Molecular Evidence on Wastewater. Journal of Infection. https://doi.org/10.1016/j.jinf.2022.11.007
Hoque, S. A., Nishimura, K., Thongprachum, A., Khamrin, P., Thi Kin Pham, N., Islam, M. T., et al. (2022b). An increasing trend of human sapovirus infection in Japan, 2009 to 2019: An emerging public health concern. Journal of Infection and Public Health, 15(3), 315–320. https://doi.org/10.1016/j.jiph.2022.01.019
Hoque, S. A., Thongprachum, A., Takanashi, S., Mostafa, S. M., Saito, H., Anwar, K. S., et al. (2019b). Alarming situation of spreading enteric viruses through sewage water in Dhaka city: Molecular nces. Food and Environmental Virology, 11(1), 65–75. https://doi.org/10.1007/s12560-018-09363-z
Hoque, S. A., Wakana, A., Shimizu, H., Takanashi, S., Okitsu, S., Anwar, K. S., et al. (2022c). Detection of rotavirus strains in freshwater clams in Japan. Food Environ Virol, 14(1), 94–100. https://doi.org/10.1007/s12560-021-09505-w
Imai, N., Gaythorpe, K. A. M., Bhatia, S., Mangal, T. D., Cuomo-Dannenburg, G., Unwin, H. J. T., et al. (2022). COVID-19 in Japan, January-March 2020: Insights from the first three months of the epidemic. BMC Infectious Diseases, 22(1), 493. https://doi.org/10.1186/s12879-022-07469-1
Jasper Götting, A. K. C., Steinbrück, L., & Heim, A. (2022). Molecular Phylogeny of human adenovirus type 41 lineages. bioRxiv. https://doi.org/10.1101/2022.05.30.493978
Kawata, K., Hikita, T., Takanashi, S., Hikita, H., Ogita, K., Okitsu, S., et al. (2020). Diagnosis of acute gastroenteritis with immunochromatography and effectiveness of rotavirus vaccine in a Japanese Clinic. Access Microbiology, 2(3), acmi000085. https://doi.org/10.1099/acmi.0.000085
Kawata, K., Hoque, S. A., Nishimura, S., Yagyu, F., Islam, M. T., Sharmin, L. S., et al. (2021). Role of rotavirus vaccination on G9P[8] rotavirus strain during a seasonal outbreak in Japan. Human Vaccines and Immunotherapeutics. https://doi.org/10.1080/21645515.2021.1925060
Khandoker, N., Thongprachum, A., Takanashi, S., Okitsu, S., Nishimura, S., Kikuta, H., et al. (2018). Molecular epidemiology of rotavirus gastroenteritis in Japan during 2014–2015: Characterization of re-emerging G2P[4] after rotavirus vaccine introduction. Journal of Medical Virology, 90(6), 1040–1046. https://doi.org/10.1002/jmv.25067
Kitajima, M., Oka, T., Tohya, Y., Katayama, H., Takeda, N., & Katayama, K. (2009). Development of a broadly reactive nested reverse transcription-PCR assay to detect murine noroviruses, and investigation of the prevalence of murine noroviruses in laboratory mice in Japan. Microbiology and Immunology, 53(9), 531–534. https://doi.org/10.1111/j.1348-0421.2009.00152.x
Kitamura, K., Sadamasu, K., Muramatsu, M., & Yoshida, H. (2021). Efficient detection of SARS-CoV-2 RNA in the solid fraction of wastewater. Science of the Total Environmen, 763, 144587. https://doi.org/10.1016/j.scitotenv.2020.144587
Kozawa, K., Higashimoto, Y., Kawamura, Y., Miura, H., Negishi, T., Hattori, F., et al. (2022). Rotavirus genotypes and clinical outcome of natural infection based on vaccination status in the post-vaccine era. Human Vaccines & Immunotherapeutics, 18(1), 2037983. https://doi.org/10.1080/21645515.2022.2037983
Laine, J., Huovinen, E., Virtanen, M. J., Snellman, M., Lumio, J., Ruutu, P., et al. (2011). An extensive gastroenteritis outbreak after drinking-water contamination by sewage effluent, Finland. Epidemiology and Infection, 139(7), 1105–1113. https://doi.org/10.1017/S0950268810002141
Li, J., Huang, F., Zhang, Y., Ji, T., Zhu, S., Wang, D., et al. (2022). Molecular analysis of Coxsackievirus A24 variant isolates from three outbreaks of acute hemorrhagic conjunctivitis in 1988, 1994 and 2007 in Beijing, China. Virologica Sinica, 37(2), 168–176. https://doi.org/10.1016/j.virs.2022.01.024
Maria, C., & Hrishikesh, S. (2018). Diarrhea. StatPearls Publishing.
Markkula, J., Hemming-Harlo, M., Salminen, M. T., Savolainen-Kopra, C., Pirhonen, J., Al-Hello, H., & Vesikari, T. (2017). Rotavirus epidemiology 5–6 years after universal rotavirus vaccination: Persistent rotavirus activity in older children and elderly. Infectious Diseases (london), 49(5), 388–395. https://doi.org/10.1080/23744235.2016.1275773
Markkula, J., Hemming-Harlo, M., Savolainen-Kopra, C., Al-Hello, H., & Vesikari, T. (2020). Continuing rotavirus circulation in children and adults despite high coverage rotavirus vaccination in Finland. Journal of Infection, 80(1), 76–83. https://doi.org/10.1016/j.jinf.2019.09.009
Nagasawa, K., Matsushima, Y., Motoya, T., Mizukoshi, F., Ueki, Y., Sakon, N., et al. (2018). Phylogeny and immunoreactivity of norovirus GII.P16-GII.2, Japan, winter 2016–17. Emerging Infectious Diseases, 24(1), 144–148. https://doi.org/10.3201/eid2401.170284
Okitsu, S., Khamrin, P., Hikita, T., Thongprachum, A., Pham, N. T. K., Hoque, S. A., et al. (2022). Changing distribution of rotavirus A genotypes circulating in Japanese children with acute gastroenteritis in outpatient clinic, 2014–2020. Journal of Infection and Public Health, 15(7), 816–825. https://doi.org/10.1016/j.jiph.2022.06.009
Okitsu, S., Khamrin, P., Takanashi, S., Thongprachum, A., Hoque, S. A., Takeuchi, H., et al. (2020). Molecular detection of enteric viruses in the stool samples of children without diarrhea in Bangladesh. Infection, Genetics and Evolution, 77, 104055. https://doi.org/10.1016/j.meegid.2019.104055
Okitsu, S., Khamrin, P., Thongprachum, A., Hikita, T., Kumthip, K., Pham, N. T. K., et al. (2021). Diversity of human sapovirus genotypes detected in Japanese pediatric patients with acute gastroenteritis, 2014–2017. Journal of Medical Virology, 93(8), 4865–4874. https://doi.org/10.1002/jmv.26934
Payne, D. C., Englund, J. A., Weinberg, G. A., Halasa, N. B., Boom, J. A., Staat, M. A., et al. (2019). Association of rotavirus vaccination with inpatient and emergency department visits among children seeking care for acute gastroenteritis, 2010–2016. JAMA Network Open, 2(9), e1912242. https://doi.org/10.1001/jamanetworkopen.2019.12242
Phattanawiboon, B., Nonthabenjawan, N., Boonyos, P., Jetsukontorn, C., Towayunanta, W., Chuntrakool, K., et al. (2020). Norovirus transmission mediated by asymptomatic family members in households. PLoS ONE, 15(7), e0236502. https://doi.org/10.1371/journal.pone.0236502
Santos, V. S., Marques, D. P., Martins-Filho, P. R., Cuevas, L. E., & Gurgel, R. Q. (2016). Effectiveness of rotavirus vaccines against rotavirus infection and hospitalization in Latin America: Systematic review and meta-analysis. Infectious Diseases of Poverty, 5(1), 83. https://doi.org/10.1186/s40249-016-0173-2
Shirato, K., Nao, N., Katano, H., Takayama, I., Saito, S., Kato, F., et al. (2020). Development of Genetic Diagnostic Methods for Detection for Novel Coronavirus 2019(nCoV-2019) in Japan. Japanese Journal of Infectious Diseases, 73(4), 304–307. https://doi.org/10.7883/yoken.JJID.2020.061
Supadej, K., Khamrin, P., Kumthip, K., Malasao, R., Chaimongkol, N., Saito, M., et al. (2019). Distribution of norovirus and sapovirus genotypes with emergence of NoV GII.P16/GII.2 recombinant strains in Chiang Mai, Thailand. Journal of Medical Virology, 91(2), 215–224. https://doi.org/10.1002/jmv.25261
Thongprachum, A., Fujimoto, T., Takanashi, S., Saito, H., Okitsu, S., Shimizu, H., et al. (2018). Detection of nineteen enteric viruses in raw sewage in Japan. Infection, Genetics and Evolution. https://doi.org/10.1016/j.meegid.2018.05.006
Tsugawa, T., Akane, Y., Honjo, S., Kondo, K., & Kawasaki, Y. (2021). Rotavirus vaccination in Japan: Efficacy and safety of vaccines, changes in genotype, and surveillance efforts. Journal of Infection and Chemotherapy, 27(7), 940–948. https://doi.org/10.1016/j.jiac.2021.04.002
Ushijima, H., Hikita, T., Kobayashi, M., Pham, N. T. K., Onda-Shimizu, Y., Kawagishi, T., et al. (2021). The Detection of Rotavirus Antigenemia by Immunochromatographic Kits: a Case Series. Clinical Laboratory. https://doi.org/10.7754/Clin.Lab.2021.210125
Wang, J., Liu, J., Fang, F., Wu, J., Ji, T., Yang, Y., et al. (2022). Genomic surveillance of coxsackievirus A10 reveals genetic features and recent appearance of genogroup D in Shanghai, China, 2016–2020. Virologica Sinica, 37(2), 177–186. https://doi.org/10.1016/j.virs.2022.01.028
WHO. (2022). Wastewater surveillance of SARS-CoV-2: Questions and answers. Retrieved 28th Sept, 2022, from https://apps.who.int/iris/bitstream/handle/10665/353058/WHO-EURO-2022-5274-45038-64164-eng.pdf?sequence=4&isAllowed=y
Yi, L., Zhang, L., Feng, L., Luan, X., Zhao, Q., Xu, P., et al. (2022). Genomic analysis of a recombinant coxsackievirus A19 identified in Xinxiang, China, in 2019. Archives of Virology, 167(6), 1405–1420. https://doi.org/10.1007/s00705-022-05433-7
Zhang, J., Cao, J., & Ye, Q. (2022). Nonpharmaceutical interventions against the COVID-19 pandemic significantly decreased the spread of enterovirus in children. Journal of Medical Virology, 94(8), 3581–3588. https://doi.org/10.1002/jmv.27806
Zhou, F., Kong, F., Wang, B., McPhie, K., Gilbert, G. L., & Dwyer, D. E. (2011). Molecular characterization of enterovirus 71 and coxsackievirus A16 using the 5’ untranslated region and VP1 region. Journal of Medical Microbiology, 60(Pt 3), 349–358. https://doi.org/10.1099/jmm.0.025056-0
Zhou, H., Liu, L., Li, R., Qin, Y., Fang, Q., Balasubramaniam, V. R., et al. (2017). Detection and genetic characterization of canine astroviruses in pet dogs in Guangxi, China. Virology Journal, 14(1), 156. https://doi.org/10.1186/s12985-017-0823-4
Acknowledgements
The research was performed by an International Research Fellow of the Japan Society for the Promotion of Science (Long-term Invitational Fellowships for Research in Japan). This work was supported by Grants-in-Aid for Japan Agency for Medical Research and Development (AMED) [grant number JP22fk0108122 and JP22wm0225006 to S.S., T.K., and H.U; and the Nihon University Research Grant for 2022. We express utmost gratitude to the former Director, Prof. Dr. M. A. Malek, and present Director, Prof. Dr. Ishtiaque M. Syed, of the Centre for Advanced Research in Sciences (CARS), the University of Dhaka, Bangladesh for allowing this collaborative study between the University of Dhaka, Bangladesh, and Nihon University School of Medicine, Japan. We gratefully acknowledge all members of the Division of Microbiology, Department of Pathology and Microbiology, Nihon University School of Medicine, for their generous cooperation throughout this work.
Author information
Authors and Affiliations
Contributions
SAH performed the major portion of the study and wrote the manuscript. TOK performed the qPCR portion and analyzed the data. NTKP and YO helped in the study. SS, TAK, and HU received the grant and designed the study. SO, YY, NM, and HU helped with data analysis, manuscript editing, and critical comments. SH and HU supervised the whole study. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no financial or proprietary interests in any material discussed in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hoque, S.A., Kotaki, T., Pham, N.T.K. et al. Genotype Diversity of Enteric Viruses in Wastewater Amid the COVID-19 Pandemic. Food Environ Virol 15, 176–191 (2023). https://doi.org/10.1007/s12560-023-09553-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12560-023-09553-4