
Abstract
Mapping the distribution of genetic diversity of species over their geographic range is crucial from a conservation perspective. We investigated nesting populations of an iconic and endangered species, Chelonia mydas, the green sea turtle, in French Polynesia. Sequences of the mtDNA control region of 97 specimens were analyzed using Bayesian phylogeny reconstruction, Bayesian dating, and skyline plots. Samples from French Polynesia belonged to four of the nine recognized Indo-Pacific lineages, with two lineages (III and IV) recognized as widespread and two other lineages (V and VI) geographically restricted to the South Pacific. More than half of the specimens belonged to the geographically restricted lineages, and suggested the existence of two South Pacific refugia during glacial periods, a western one (represented by lineage V) and an eastern one (represented by lineage VI), herein revealed from French Polynesia. Expansions of populations were recovered in most of the lineages and were all dated after the Last Glacial Maximum, similarly to the Atlantic populations of C. mydas. Finally, as the likely existence of a glacial refugium near French Polynesia matches a previously published predicted refugium under future climatic conditions, this makes the area particularly promising for the conservation of green sea turtle populations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Understanding the effects of past climate changes on biodiversity is of the utmost importance in evolutionary and conservation biology (Pauls et al. 2013). A historical perspective on the contemporary distribution of genetic diversity is indeed necessary as modern conservation policies must include the conservation of evolutionary processes (Crandall et al. 2000; Moritz 2002) and the areas holding these processes, coined evolutionary hotspots (Vandergast et al. 2008; Hoareau et al. 2013). In this regard, past glacial refugia are particularly important as they usually hold great and/or unique genetic diversity (Hewitt 2004) and shelter therefore a species’ adaptive potential. However, in the northern hemisphere, past refugium areas were recently highlighted as the zones most likely to be at risk under future climate conditions (Pauls et al. 2013; Pfenninger et al. 2012; Razgour et al. 2013). It is thus critical to retrace the evolutionary history of species in a spatial context in order to identify endemic lineages and pinpoint past refugium areas. Tracking historical changes in distribution and abundances enables us to better predict future changes (Fordham et al. 2014).
However, even for iconic and endangered species, such as the green sea turtle, Chelonia mydas, our knowledge on genetic distribution and past demographic changes is far from complete. Exemplifying our incomplete knowledge of marine biodiversity, an endemic lineage was only recently recovered from the Ogasawara Islands in the northwestern Pacific (Hamabata et al. 2014). This lineage adds up to the eight other mitochondrial lineages reported from the Indo-Pacific region and the two from the Atlantic Ocean (Bowen et al. 1992; Dethmers et al. 2006; Bowen and Karl 2007; Dutton et al. 2014; Jensen et al. 2019). These Indo-Pacific lineages show contrasting distributions, some being widespread (e.g., lineage VIII (formerly clade I) in the Indian Ocean and West Pacific and lineage III (formerly clade V) in the Central and West Pacific) and some more geographically restricted (lineage V (formerly clade II) and lineage VI (formerly clade III) to the South Pacific and lineage VII (formerly clade IV) to the Ryukyu Archipelago and Ogasawara Islands). These contrasting patterns can be explained by the fact that C. mydas shows a strong female homing behavior (Bowen et al. 1992) but long-distance migration events have also been reported (Balazs et al. 1995; Luschi et al. 1996; Lohman et al. 1999; Scott et al. 2014).
Regarding past demographic changes, the Atlantic populations of Chelonia mydas were recently confirmed to have experienced expansions in the southern and the northern mitochondrial lineages likely following the Last Glacial Maximum (Naro-Maciel et al. 2014). It is particularly important to better understand the effects of past climatic changes as the influence of climate is strong on resources, breeding success, and sex-ratio for marine sea turtles. Indeed, the green sea turtle populations from the Great Barrier Reef have undergone feminization for the past two decades, stronger in the warmer northern sites (Jensen et al. 2018). Additionally, both Hamabata et al. (2014) and Naro-Maciel et al. (2014) point towards the existence of distinct glacial refugia and independent evolution of the lineages, which has important implications for conservation strategy. Reid et al. (2019) recently showed that most lineages of marine turtles underwent population expansions after the Last Glacial Maximum. Similarly, the latest review on phylogeographic patterns of green sea turtles suggests a strong influence of past climatic events, and the survival in multiple glacial refugia during the last glaciations (Jensen et al. 2019).
However, some areas of the species range, such as French Polynesia, have not been thoroughly investigated so far and authors acknowledge the fact that the region is still under-sampled and needs further investigation (Seminoff et al. 2015; Jensen et al. 2019). Dutton et al. (2014) showed that their nine specimens from Mopelia Island, French Polynesia, belonged to the rare lineage VI. Jensen et al. (2019) also reported on the southern part of the Central Pacific (American Samoa and French Polynesia) as an area of high nucleotide diversity, warranting further investigation. The populations of green sea turtles of French Polynesia, together with that of American Samoa, belong to a distinct population segment (DSP), the Central South Pacific (Seminoff et al. 2015). However, even with limited sampling, there is evidence of spatial structuring between Samoan and French Polynesian green turtle populations (Seminoff et el. 2015). Nevertheless, long-distance migrations between the breeding sites of French Polynesia and the foraging grounds around Fiji and Samoa have been reported on multiple occasions (Balazs et al. 1995; Touron et al. 2018).
Regarding population size, no overall review exists on the number of nesters per island in French Polynesia. Early estimations suggested a total population of 1000 females in French Polynesia (Groombridge & Luxmoore 1989). A recent report on Tetiaroa Island indicates 96 distinct females for the nesting season 2017–2018 and an estimation of 120–130 adult females for Tetiaroa and its islets (Touron et al. 2018). At Scilly Atoll, during the 1970s, 1980s, and 1990s, observations suggested that 300 to 400 females occurred there annually (Lebeau 1985; Balazs et al. 1995). Therefore, the population of French Polynesia is not as large as in other parts of the species range (e.g., Great Barrier Reef), somewhat spatially disconnected, and urgently needs further investigation.
Here, we surveyed the genetic diversity of green sea turtle nesting populations in French Polynesia in order to (i) determine what genetic lineages are present here and thus enabling a better understanding of the global distribution of genetic diversity in this iconic species and (ii) retrace past demographic changes, and verify if their timing matches past climatic fluctuations and the expansion of the Atlantic populations of Chelonia mydas.
Materials and methods
Sampling, DNA extraction, and mtDNA control region amplification
Tissue samples were collected from nesting populations of Chelonia mydas in French Polynesia (Moorea, Scilly, and Tetiaroa Islands), from both nesting females and hatchlings. Sampling was realized in accordance with relevant guidelines and regulations by Direction de l’Environnement de Polynesie Française, and all experimental protocols were approved by the Haut Commissariat de la République en Polynesie Française (export permit number FR1298700118-E). DNA was extracted from tissue using the QIAxtractor robot (Qiagen, Hilden, Germany). A portion of the mitochondrial control region (850 bp) was amplified for 176 specimens using the primers H950 and LCM15382 and cycling parameters as described in Abreu-Grobois et al. (2006). These specimens are part of a broader study using microsatellite markers to investigate the occurrence of multi-paternity in green sea turtle nests in French Polynesia (unpublished). The mtDNA control region dataset was compared with the microsatellite dataset and paternity results (320 specimens) to keep only one representative of maternal lineage per nest (i.e., removing siblings of the mitochondrial dataset to avoid overrepresentation of some haplotypes), resulting in 97 mtDNA control region sequences used in the subsequent analyses (Online Resources 1). To place the population of French Polynesia in the broader Indo-Pacific context, we analyzed these 97 sequences together with long sequences (i.e., approx. 800 bp) of Chelonia mydas from the Indo-Pacific and the Atlantic available in GenBank, mostly from Dutton et al. (2014), Hamabata et al. (2014), and Jensen et al. (2016) (see Fig. 1 for GenBank Accession numbers).

Maximum likelihood phylogenetic reconstruction of Chelonia mydas mtDNA control region haplotypes. Numbers above branches are “Time to the most recent common ancestor” (Tmrcas) in million years and their 95% highest posterior probabilities from the Bayesian reconstruction. Numbers below branches are bootstrap values for the neighbor joining reconstruction/bootstrap values for the maximum likelihood reconstruction/posterior probabilities of the Bayesian reconstruction. The nine haplotypes recovered from French Polynesia specimens are indicated in blue
Phylogeny and demography reconstructions
Sequences were aligned using Mafft online (Katoh et al. 2002). MrAic (Nylander 2004) was used to determine the best-fit model of nucleotide substitutions. Neighbor joining and maximum likelihood reconstructions were performed using Mega v.5.05 (Tamura et al. 2011), with a bootstrapping procedure. The software Beast v1.8 (Drummond et al. 2012) was used to perform Bayesian phylogenetic reconstructions. Bayes factors were used to choose between 3 clock models (strict, lognormal relaxed, exponential relaxed) and two tree models (coalescent constant size and exponential growth). Following Formia et al. (2006) and Naro-Maciel et al. (2014), a mean mutation rate of 1.75 × 10−8 substitutions/site/year was used to estimate the “Time to the most recent common ancestor” (Tmrca) of each lineage and of the species. A total of 107 generations was run, recorded every 100th generation. Tracer v1.5 (Rambaut et al. 2014) was used to ensure sufficient samples were recorded (i.e., ESS > 200) and to obtain parameter estimates. TreeAnnotator was used to summarize the information from 10,000 trees. Furthermore, the Bayesian Skyline Plot framework available in Beast was used to reconstruct past demographic changes of the different lineages separately. Similar to the previous step, 107 generations were performed, recorded every 100th generation, using the same mutation rate. Tracer v1.5 was used to compute the Bayesian Skyline Plots. The X-axis indicates the time in years while the Y-axis indicates the relative female effective population size (NeT, with T = generation time).
Nuclear data
Additionally, the 97 specimens were also analyzed at 12 microsatellite markers. Microsatellite markers, as well as amplification and cycling parameters, were from Dutton and Frey (2009) and Fitzsimmons et al. (1995). Principal coordinates analyses and estimation of population differentiation indices were performed in GenAlex v6.502 (Peakall and Smouse, 2012) to verify if grouping of samples occurred according to the mitochondrial lineages they belong to or according to their geographic origin.
Results
Lineages present in French Polynesia
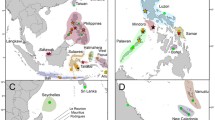
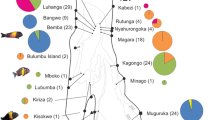
The sequences of 97 Chelonia mydas individuals collected from French Polynesia corresponded to nine haplotypes (Online Resources 1). They belong to four of the nine lineages known from the Indo-Pacific region (Fig. 1). Noticeably, 50 of the 97 sequences belong to lineage VI, while one sequence belongs to lineage V, 44 sequences belong to lineage III, and two sequences belong to lineage IV. So far, lineage VI was only recovered from seven individuals from the Great Barrier Reef, 20 from New Caledonia, nine from Mopelia (French Polynesia), four from American Samoa, one from the Marshalls, and one from Palau at breeding sites (Fig. 2), while another 38 specimens from foraging grounds of eastern Australia also belong to this lineage. While the sequences belonging to lineage IV, V, and lineage VI correspond to known haplotypes (CmP186.1, CmP65.1, and CmP56.1, see Online Resources 1), two of the six haplotypes of lineage III represent new haplotypes (see Online Resources 1; GenBank accession numbers: MH893824-MH893825).

Frequencies of observed lineages of Chelonia mydas in French Polynesia from this study (Scilly, Moorea, and Tetiaroa). Frequencies of lineages in breeding sites of other regions are compiled from Dethmers et al. (2006), Dutton et al. (2014), Hamabata et al. (2014), and Jensen et al. (2016). Numbers in pie charts indicate the number of specimens sequenced. Numbers along the map frame indicate longitude and latitude coordinates. Map generated with QGIS Lyon (version 2.12.3) a Free and Open Source Geographic Information System, available at http://www.qgis.org/fr/
Bayesian dating and Bayesian skyline reconstructions
The Tmrca of all lineages belonging to C. mydas was estimated at about 4 My (Fig. 1). Tmrcas of each lineage ranged between 0.54 and 0.71 My. The Bayesian Skyline Plot analyses revealed an expansion of populations for all Indo-Pacific lineages except lineage VI (Fig. 3). The expansion events were all estimated to have occurred after the LGM (about 10,000–15,000 years ago).

Bayesian skyline plots reconstructed from all Chelonia mydas mtDNA control region sequences belonging to the Indo-Pacific lineages LIII to LVIII. The X-axis indicates the time in years; the Y-axis indicates the female effective population size (NeT, with T = generation time). The vertical dotted lines indicate the lower 95% highest posterior density of the root height (i.e., time to the most recent common ancestor of each lineage)
Nuclear data
The 97 specimens from lineages III, IV, V, and VI analyzed at the 12 microsatellite loci form a homogeneous group on the PCoA with no clustering of specimens according to their lineage (see Online Resources 2: Fig. S1a). Similarly, when considering the three islands analyzed, Moorea, Scilly, and Tetiaroa, specimens cluster together on the PCoA and do not show genetic segregation (see Online Resources 2: Fig. S1b). The FST values were all low and all but one non-significant, suggesting important gene flow between islands and lineages (see Online Resources 2).
Discussion
Current geographic distribution of mitochondrial lineages
Combining our data with other data available for Chelonia mydas from the Indo-Pacific region (Figs. 1 and 2) confirmed three common and widespread lineages (III, IV, and VIII), and three rare and geographically restricted lineages (V, VI, VII). Lineage VIII, common, and occurring from the eastern coast of Africa to the western Pacific, was confirmed absent from French Polynesia. Lineage VII, so far endemic to the northwestern Pacific (Hamabata et al. 2014), was also absent from French Polynesia. Lineage III, widespread from the eastern coast of Africa to the south-central Pacific, was present in our dataset. Roughly half of our sequences belonged to this widespread lineage, with two new haplotypes. Two specimens belonged to lineage IV, a lineage present in the Southwest Pacific and the Southwest Indian Ocean, thus now expanding the range of this lineage to southern part of the Central Pacific. Specimens from lineage V were mainly derived from Australasian populations (Dethmers et al. 2006; Dutton et al. 2014). This lineage range is now extended to French Polynesia as one of the adult specimens analyzed here belongs to this lineage and suggests long-distance migration between the two regions. Long-distance migrations between west and central Pacific islands were already reported from tagged C. mydas individuals (Balazs et al. 1995; Touron et al. 2018). Lineage VI is rare and mostly restricted to the South Pacific, with most of the specimens sampled in French Polynesia and New Caledonia. Interestingly, more than half of our sequences belong to this rare lineage and French Polynesia could thus represent the center of the range for this lineage. Similarly to green sea turtles, the hawksbill turtle, Eretmochelys imbricata, shows five Indo-Pacific lineages with contrasting distribution patterns that demonstrate the complex evolutionary history of these species linked to paleoclimate changes in the region (Vargas et al. 2016). More recently, both Reid et al. (2019) and Jensen et al. (2019) emphasized the complex evolutionary history of sea turtles linked to paleoclimates and their survival in multiple refugia.
Past demographic changes
Signs of population expansions were detected in all lineages except lineage VI. These demographic events were all estimated between 10,000 and 15,000 years ago, after the Last Glacial Maximum (LGM, Fig. 3). This is in accordance with populations of C. mydas from the Atlantic where the expansions of the southern and northern lineages were also both dated after the LGM at about 20,000 years ago (Naro-Maciel et al. 2014). Similarly, many marine organisms showed an expansion of their populations after the LGM, when conditions became more suitable and habitats were newly available (Provan and Bennett 2008; Boissin et al. 2016). In particular, hawksbill and leatherback turtles also showed expansion of their populations dated after the LGM (Molfetti et al. 2013; Vargas et al. 2016). This is also in accordance with a recent review on population co-expansions of most sea turtle lineages, dated after the LGM (Reid et al. 2019). In contrast, lineage VI did not show a signature of expansion. It is not clear if this lack of signature comes from this lineage having a constant population size over time or if it is from the fewer number of specimens recorded from this rare lineage so far (Dethmers et al. 2006; Jensen et al. 2016). Small sample sizes are known to affect demographic reconstructions (Grant 2015). However, the shape of the network for this lineage is not star-like as the other lineages and this lineage might have a more complex evolutionary history (Jensen et al. 2019: Fig. 3). Overall, these signals of expansion of populations and the occurrence of distinct mitochondrial lineages highlight the sensitivity of green turtles to climate fluctuations (Reid et al. 2019; Jensen et al. 2019).
Past refugium areas, adaptive potential, and conservation units
Endemic or geographically restricted lineages are particularly interesting as they should point towards the areas of former refuges (Avise 2000). A northern refuge was recently suggested for the endemic lineage VII of Ogasawara Islands, Japan (Hamabata et al. 2014). In our study, French Polynesia was shown to shelter four lineages of green sea turtles. The presence of the geographically restricted lineages V and VI, mainly found in the South Pacific Ocean, points towards the putative existence of refugium areas. Found primarily in the South West Pacific, lineage V might have evolved from a western South Pacific glacial refugium, near New Caledonia and northeastern Australia where a glacial refugium area was already suggested for several species (Wörheide et al. 2002; Lukoschek et al. 2007; Tillett et al. 2012). Furthermore, with many specimens of our study belonging to lineage VI, a glacial refugium seems also likely in the south-central Pacific, possibly near French Polynesia. Even if this lineage is present in eastern Australia, it is minor over the thousand green sea turtles sampled (Dethmers et al. 2006; Jensen et al. 2016). However, private haplotypes belonging to this lineage and restricted so far to the south west Pacific exist (see Jensen et al. 2019). The structure of this lineage seems to be complex, and this lineage could have survived in several localities of the south and central west Pacific. Specimens of this lineage present in Australia and New Caledonia could represent a relict population. Alternatively, specimens from the Southwest Pacific could have colonized French Polynesia, where only two haplotypes have been recorded so far. Studies of an increasing number of specimens from this lineage might help understand better its complex evolutionary history. Nevertheless, Polynesia was already suggested as a likely glacial refuge during the LGM for a goby fish (Hoareau et al. 2012). Additionally, Jensen et al. (2019) recorded a high nucleotide diversity in the south-central Pacific (American Samoa and French Polynesia) and pinpointed the region as a likely past refugium area for green sea turtles. Overall, these results suggest that the two green sea turtle populations from French Polynesia and eastern Australia each have a unique evolutionary history, resulting from distinct glacial refugia. These evolutionary significant units match the regional management units defined for green sea turtles in the South Pacific region (Wallace et al. 2010, Seminoff et al. 2015; Jensen et al. 2019), and thus, the long-term and short-term conservation needs (Moritz 1994) coincide in this region for C. mydas. This distinctiveness due to an independent genetic history could provide promising adaptive potential under future climatic conditions, particularly given that the mitochondrial genome codes for important enzymes of metabolic pathways (such as cytochrome oxidases and ATPases). Duchene et al. (2012), studying complete mitogenomes, revealed differences between the Atlantic and Indo-Pacific lineages of C. mydas, whose effects on adaptive potential remain unknown so far. Finally, it is particularly interesting to note that French Polynesia is projected to serve as a refuge under future climatic conditions (van Hooidonk et al. 2013; Freeman 2015). Regarding sea turtles, a refuge under future warm conditions would likely lower the impact on habitat availability (availability of beaches to lay eggs) and will possibly limit feminization of populations linked to warming. Areas that align with past glacial refugia and those predicted to serve as refugia under future climatic conditions should be a priority for conservation, as they have the potential to maximize the preservation of genetic diversity and unique genetic lineages.
Conclusions
Four of the nine Indo-Pacific lineages were recovered from these French Polynesian specimens. This study extends the range of lineage V and confirms the absence of lineage VIII in the South-Central Pacific. As half of our sequences belong mainly to a rare lineage (VI), this could suggest that French Polynesia was a former glacial refugium for green sea turtles. Past climatic change effects can be seen on most of the lineages that show signs of population expansion dated after the Last Glacial Maximum, similar to the Atlantic populations of C. mydas. This study confirms the unique evolutionary trajectory of French Polynesian and eastern Australian populations of green sea turtles. Finally, French Polynesia seems a key conservation area for C. mydas; given that it was likely a past glacial refugium, it is predicted to be a refugium under future warm conditions and that four of the nine Indo-Pacific lineages of C. mydas occur there.
References
Abreu-Grobois FA, Horrocks J, Formia A, LeRoux R, Velez-Zuazo X, Dutton P, Soares L, Meylan P, D Brown (2006) New mtDNA Dloop primers which work for a variety of marine turtle species may increase the resolution capacity of mixed stock analysis. In: Frick M, Panagopoulou A, Rees AF, Williams K (eds) Book of abstracts of the 26th annual symposium on sea turtle biology and conservation, Crete, Greece, 3–8 April 2006, p 179
Avise JC (2000) Phylogeography – the history and formation of species. Harvard University Press, Cambridge
Balazs GH, Siu P, Landret JP (1995) Ecological aspects of green turtles nesting at Scilly Atoll in French Polynesia. Pages 7–10. In: Richardson JI, Richardson TH (eds) Proceedings of the 12th Annual Workshop on Sea Turtle Biology and Conservation. NOAA Tech. Memo. NMFS-SEFSC-361
Boissin E, Micu D, Janczyszyn-Le Goff M, Neglia V, Bat L, Todorova V, Panayotova M, Kruschel C, Macic V, Milchakova N, Keskin C, Anastasopoulou A, Nasto I, Zane L, Planes S (2016) Contemporary genetic structure and post-glacial demographic history of the black scorpionfish, Scorpaena porcus, in the Mediterranean and the Black Seas. Mol Ecol. https://doi.org/10.1111/mec.13616
Bowen BW, Karl SA (2007) Population genetics and phylogeography of sea turtles. Mol Ecol 16:4886–4907. https://doi.org/10.1111/j.1365-294X.2007.03542.x
Bowen BW, Meylan AB, Ross JP, Limpus CJ, Balazs GH, Avise JC (1992) Global population structure and natural history of the green turtle (Chelonia mydas) in terms of matriarchal phylogeny. Evolution 46:865–881. https://doi.org/10.1111/j.1558-5646.1992.tb00605.x
Crandall KA, Bininda-Emonds ORP, Mace GM, Wayne RK (2000) Considering evolutionary processes in conservation biology. Trends Ecol Evol 15:290–295. https://doi.org/10.1016/s0169-5347(00)01876-0
Dethmers KEM, Broderick D, Moritz C, Fitzsimmons NN, Limpus CJ, Lavery S, Whiting S, Guinea M, Prince RIT, Kennett R (2006) The genetic structure of Australasian green turtles (Chelonia mydas): exploring the geographical scale of genetic exchange. Mol Ecol 15:3931–3946. https://doi.org/10.1111/j.1365-294X.2006.03070.x
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. https://doi.org/10.1093/molbev/mss075
Duchene S, Frey A, Alfaro-Nunez A, Dutton PH, Gilbert MTP, Morin PA (2012) Marine turtle mitogenome phylogenetics and evolution. Mol Phylogenet Evol 65:241–250. https://doi.org/10.1016/j.ympev.2012.06.010
Dutton PH, Frey A (2009) Characterization of polymorphic microsatellite markers for the green turtle (Chelonia mydas). Mol Ecol Resour 9:354–356. https://doi.org/10.1111/j.1755-0998.2008.02443.x
Dutton PH, Jensen MP, Frutchey K, Frey A, LaCasella E, Balazs GH, Cruce J, Tagarino A, Farman R, Tatarata M (2014) Genetic stock structure of green turtle (Chelonia mydas) nesting populations across the Pacific Islands. Pac Sci 68:451–464. https://doi.org/10.2984/68.4.1
Fitzsimmons NN, Moritz C, Moore SS (1995) Conservation and dynamics of microsatellite loci over 300-million years of marine turtle evolution. Mol Biol Evol 12:432–440
Fordham DA, Brook BW, Moritz C, Nogues-Bravo D (2014) Better forecasts of range dynamics using genetic data. Trends Ecol Evol 29:436–443. https://doi.org/10.1016/j.tree.2014.05.007
Formia A, Godley BJ, Dontaine JF, Bruford MW (2006) Mitochondrial DNA diversity and phylogeography of endangered green turtle (Chelonia mydas) populations in Africa. Conserv Genet 7:353–369. https://doi.org/10.1007/s10592-005-9047-z
Freeman LA (2015) Robust performance of marginal Pacific coral reef habitats in future climate scenarios. PLoS One 10(16):e0128875. https://doi.org/10.1371/journal.pone.0128875
Grant WS (2015) Problems and cautions with sequence mismatch analysis and Bayesian skyline plots to infer historical demography. J Hered 106:333–346. https://doi.org/10.1093/jhered/esv020
Groombridge B, Luxmoore R (1989) The green turtle and hawksbill (Reptilia: Cheloniidae): world status, exploitation and trade, Flora. CITES, Lausanne
Hamabata T, Kamezaki N, Hikida T (2014) Genetic structure of green turtle (Chelonia mydas) peripheral populations nesting in the northwestern Pacific rookeries: evidence for northern refugia and postglacial colonization. Mar Biol 161:495–507. https://doi.org/10.1007/s00227-013-2352-z
Hewitt GM (2004) Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc Lond Ser B-Biol Sci 359:183–195. https://doi.org/10.1098/rstb.2003.1388
Hoareau TB, Boissin E, Berrebi P (2012) Evolutionary history of a widespread Indo-Pacific goby: the role of Pleistocene sea-level changes on demographic contraction/expansion dynamics. Mol Phylogenet Evol 62:566–572. https://doi.org/10.1016/j.ympev.2011.10.004
Hoareau TB, Boissin E, Paulay G, Bruggemann JH (2013) The Southwestern Indian Ocean as a potential marine evolutionary hotspot: perspectives from comparative phylogeography of reef brittle-stars. J Biogeogr 40:2167–2179. https://doi.org/10.1111/jbi.12155
Jensen MP, Bell I, Limpus CJ, Hamann M, Ambar S, Whap T, David C, FitzSimmons NN (2016) Spatial and temporal genetic variation among size classes of green turtles (Chelonia mydas) provides information on oceanic dispersal and population dynamics. Mar Ecol-Prog Ser 543:241–256. https://doi.org/10.3354/meps11521
Jensen MP, Allen CD, Eguchi T, Bell IP, LaCasella EL, Hilton WA, Hof CAM, Dutton PH (2018) Environmental warming and feminization of one of the largest sea turtle populations in the world. Curr Biol 28:154–159. https://doi.org/10.1016/j.cub.2017.11.057
Jensen MP, FitzSimmons NN, Bourjea J, Hamabata T, Reece J, Dutton PH (2019) The evolutionary history and global phylogeography of the green turtle (Chelonia mydas). J Biogeogr 46:860–870. https://doi.org/10.1111/jbi.13483
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. https://doi.org/10.1093/nar/gkf436
Lebeau A (1985) Essai d’évaluation des pontes de la tortue verte Chelonia mydas sur l’atoll de Scilly (iles-sous-le-vent, Polynésie française) au cours des saisons 1982-1983 et 1983-1984. Proceedings of the Fifth Intercontinental Coral Reef Congress, Tahiti. 5:487–493
Lohmann KJ, Hester JT, Lohmann CMF (1999) Long-distance navigation in sea turtles. Ethol Ecol Evol 11:1–23. https://doi.org/10.1080/08927014.1999.9522838
Lukoschek V, Waycott M, Marsh H (2007) Phylogeography of the olive sea snake, Aipysurus laevis (Hydrophiinae) indicates Pleistocene range expansion around northern Australia but low contemporary gene flow. Mol Ecol 16:3406–3422. https://doi.org/10.1111/j.1365-294X.2007.03392.x
Luschi P, Papi F, Liew HC, Chan EH, Bonadonna F (1996) Long-distance migration and homing after displacement in the green turtle (Chelonia mydas): a satellite tracking study. J Comp Physiol A-Sens Neural Behav Physiol 178:447–452
Molfetti E, Vilaca ST, Georges JY, Plot V, Delcroix E, Le Scao R, Lavergne A, Barrioz S, dos Santos FR, de Thoisy B (2013) Recent demographic history and present fine-scale structure in the Northwest Atlantic leatherback (Dermochelys coriacea) turtle population. PLoS One 8:11. https://doi.org/10.1371/journal.pone.0058061
Moritz C (1994) Defining evolutionarily significant units for conservation. Trends Ecol Evol 9:373–375. https://doi.org/10.1016/0169-5347(94)90057-4
Moritz C (2002) Strategies to protect biological diversity and the evolutionary processes that sustain it. Syst Biol 51:238–254. https://doi.org/10.1080/10635150252899752
Naro-Maciel E, Reid BN, Alter SE, Amato G, Bjorndal KA, Bolten AB, Martin M, Nairn CJ, Shamblin B, Pineda-Catalan O (2014) From refugia to rookeries: phylogeography of Atlantic green turtles. J Exp Mar Biol Ecol 461:306–316. https://doi.org/10.1016/j.jembe.2014.08.020
Nylander J (2004) MrAIC.pl. Program distributed by the author, Evolutionary Biology Centre, Uppsala University, Sweden
Pauls SU, Nowak C, Balint M, Pfenninger M (2013) The impact of global climate change on genetic diversity within populations and species. Mol Ecol 22:925–946. https://doi.org/10.1111/mec.12152
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Pfenninger M, Balint M, Pauls SU (2012) Methodological framework for projecting the potential loss of intraspecific genetic diversity due to global climate change. Bmc Evol Biol 12: 13 doi 224https://doi.org/10.1186/1471-2148-12-224
Provan J, Bennett KD (2008) Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol 23:564–571. https://doi.org/10.1016/j.tree.2008.06.010
Rambaut A, Suchard MA, Xie D, Drummond AJ (2014) Tracer v1.6, Available from http://beast.bio.ed.ac.uk/Tracer
Razgour O, Juste J, Ibanez C, Kiefer A, Rebelo H, Puechmaille SJ, Arlettaz R, Burke T, Dawson DA, Beaumont M, Jones G (2013) The shaping of genetic variation in edge-of-range populations under past and future climate change. Ecol Lett 16:1258–1266. https://doi.org/10.1111/ele.12158
Reid BN, Naro-Maciel E, Torres Hahn A, FitzSimmons NN, Gehara M (2019) Geography best explains global patterns of genetic diversity and post-glacial co-expansion in marine turtles. Mol Ecol. https://doi.org/10.1111/mec.15165
Scott R, Marsh R, Hays GC (2014) Ontogeny of long-distance migration. Ecology 95:2840–2850. https://doi.org/10.1890/13-2164.1
Seminoff JA, Allen CD, Balazs GH, Dutton PH, Eguchi T, Haas HL, Hargrove SA, Jensen MP, Klemm DL, Lauritsen AM, MacPherson SL, Opay P, Possardt EE, Pultz SL, Seney EE, Van Houtan KS, Waples RS (2015) Status review of the green turtle (Chelonia mydas) under the endangered species act. NOAA technical memorandum, NOAA-TM-NMFS-SWFSC-539, 571 pp
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Tillett BJ, Meekan MG, Broderick D, Field IC, Cliff G, Ovenden JR (2012) Pleistocene isolation, secondary introgression and restricted contemporary gene flow in the pig-eye shark, Carcharhinus amboinensis across northern Australia. Conserv Genet 13:99–115. https://doi.org/10.1007/s10592-011-0268-z
Touron M, Genet Q, Gaspar C (2018) Final report on the green sea turtle (Chelonia mydas) egg-laying season of 2017–2018 on the atoll of Tetiaroa, French Polynesia. Te Mana O Te Moana. 54 pp
van Hooidonk R, Maynard JA, Planes S (2013) Temporary refugia for coral reefs in a warming world. Nat Clim Chang 3:508–511
Vandergast AG, Bohonak AJ, Hathaway SA, Boys J, Fisher RN (2008) Are hotspots of evolutionary potential adequately protected in southern California? Biol Conserv 141:1648–1664. https://doi.org/10.1016/j.biocon.2008.04.009
Vargas SM, Jensen MP, Ho SYW, Mobaraki A, Broderick D, Mortimer JA, Whiting SD, Miller J, Prince RIT, Bell IP, Hoenner X, Limpus CJ, Santos FR, FitzSimmons NN (2016) Phylogeography, genetic diversity, and management units of hawksbill turtles in the Indo-Pacific. J Hered 107:199–213. https://doi.org/10.1093/jhered/esv091
Wallace BP, DiMatteo AD, Hurley BJ, Finkbeiner EM, Bolten AB, Chaloupka MY, Hutchinson BJ, Abreu-Grobois FA, Amorocho D, Bjorndal KA, Bourjea J, Bowen BW, Duenas RB, Casale P, Choudhury BC, Costa A, Dutton PH, Fallabrino A, Girard A, Girondot M, Godfrey MH, Hamann M, Lopez-Mendilaharsu M, Marcovaldi MA, Mortimer JA, Musick JA, Nel R, Pilcher NJ, Seminoff JA, Troeng S, Witherington B, Mast RB (2010) Regional management units for marine turtles: a novel framework for prioritizing conservation and research across multiple scales. PLoS One 5:11. https://doi.org/10.1371/journal.pone.0015465
Wörheide G, Hooper JNA, Degnan BM (2002) Phylogeography of western Pacific Leucetta ‘chagosensis’ (Porifera: Calcarea) from ribosomal DNA sequences: implications for population history and conservation of the Great Barrier Reef World Heritage Area (Australia). Mol Ecol 11:1753–1768. https://doi.org/10.1046/j.1365-294X.2002.01570.x
Acknowledgments
We are grateful to three anonymous reviewers for their comments that greatly helped to improve the manuscript. We are grateful to G. Iwankow for help with preparation of Fig. 2 and to J. Almany for English corrections. EB was supported by a European Marie Curie fellowship MC-CIG-618480.
Funding
This study was funded by Marie Curie Fellowship (grant number MC-CIG-618480).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed by the authors.
Sampling and field studies
All necessary permits for sampling and observational field studies have been obtained by the authors from the competent authorities and are mentioned in the acknowledgments, if applicable.
Data availability
The datasets generated during and/or analyzed during the current study are available in the GenBank repository (MH893824-MH893825) and NOAA SWFSC CmP (haplotype number CmP21.1 and CmP249.1 ).
Additional information
Communicated by B. W. Hoeksema
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Boissin, E., Neglia, V., Boulet Colomb D’hauteserre, F. et al. Evolutionary history of green turtle populations, Chelonia mydas, from French Polynesia highlights the putative existence of a glacial refugium. Mar. Biodivers. 49, 2725–2733 (2019). https://doi.org/10.1007/s12526-019-01001-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12526-019-01001-6