
Abstract
We examine the structure and phylogeography of the pig-eye shark (Carcharhinus amboinensis) common in shallow coastal environments in northern Australia using two types of genetic markers, two mitochondrial (control region and NADH hydrogenase 4) and two nuclear (microsatellite and Rag 1) DNA. Two populations were defined within northern Australia on the basis of mitochondrial DNA evidence, but this result was not supported by nuclear microsatellite or Rag 1 markers. One possibility for this structure might be sex-specific behaviours such as female philopatry, although we argue it is doubtful that sufficient time has elapsed for any potential signatures from this behaviour to be expressed in nuclear markers. It is more likely that the observed pattern represents ancient populations repeatedly isolated and connected during episodic sea level changes during the Pleistocene epoch, until current day with restricted contemporary gene flow maintaining population genetic structure. Our results show the need for an understanding of both the history and ecology of a species in order to interpret patterns in genetic structure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genetic diversity is fundamental to species persistence and hence effective species management. An understanding of the origin and maintenance of patterns of genetic diversity requires knowledge of both the ecology (mating systems etc.) and history of a species across geological time. The combination of this information offers managers the ability to predict how genetic diversity may alter in the future, a capacity that is particularly valuable in a world facing the possibility of large-scale changes in climate and structure of ecosystems.
The coastal habitats of the Indo-Australian archipelago harbour a very diverse range of elasmobranchs that includes many endemics (White et al. 2006; Last and Stevens 2009). This diversity is thought to reflect the geological history of the region with large changes in sea levels during Pleistocene ice ages significantly altering inshore habitats, (Williams et al. 2009) repeatedly fracturing then reconnecting populations. This process of geological change has been particularly severe for species inhabiting the shallow coastal shelves of northern Australia. For almost 90% of the last 150,000 years, the Torres Strait has been closed due to the formation of a land-bridge between Cape York and Papua New Guinea; isolating eastern populations from north-west Australia (Voris 2000). The Gulf of Carpentaria was periodically isolated as one or more freshwater lakes and swamps. Furthermore, sea surface temperatures were up to 4°C cooler due to changes in the intensity and dynamics of the Indonesian Throughflow Current; the major current system in the region (Kuhnt et al. 2004; Williams et al. 2009).
In addition to events occurring in geological time, ecological processes also serve to structure populations. Complex patterns of habitat use are a good example of such phenomena, with differential patterns of male migration and female site fidelity or philopatry restricting gene flow in species such as white (Carcharodon carcharias, Pardini et al. 2001), blacktip (Carcharhinus limbatus, Keeney et al. 2005), bull (C. leucas, Tillett et al. In review) and lemon sharks (Negaprion brevirostris, Feildheim et al. 2002). Other behaviours such as the distance travelled to find a mate (isolation by distance) can also restrict gene-flow.
Carcharhinus amboinensis is a large (2.8 m total length within Australia) apex predator common in shallow coastal waters of northern Australia, and along tropical and sub-tropical coasts throughout much of the Indian Ocean (Last and Stevens 2009). Little demographic data exists for this species and much of our information about its distribution and abundance may be confounded by problems with accurate species identification due to morphological similarities between closely related species (Last and Stevens 2009). The habitat that it occupies predisposes C. amboinensis to the likelihood of high fishing pressure by growing human populations along coasts abutting most of its range (Field et al. 2009; Northern Territory Government 2009). However, we lack sufficient data to determine the degree of threat to the species, leading to the International Union for the Conservation of Nature (IUCN) to classify the pig-eye shark as ‘Data Deficient’ (IUCN 2010). The coastal waters of northern Australia remains one of the few strongholds for the species and offers the opportunity to examine regional patterns in the genetics largely unconfounded by the effects of selective harvest and declining populations.
This study describes the genetic structure of populations and intra-specific phylogeny of C. amboinensis across northern Australia. Patterns in mtDNA are compared with geological history, geographic distance and nuclear DNA markers in order to determine the relative effects of historical and ecological processes on species genetic diversity. We test firstly whether the regular formation of land bridges through the Torres Strait during glacial maxima has resulted in greater mtDNA genetic similarity between populations in Western Australia and the Northern Territory, than with populations from Queensland. Secondly, if this separation facilitated reproductive isolation generating current day sympatric cryptic species evidenced by genetic differences in nuclear Recombination Activation Gene (RAG 1) between any identified clades. Thirdly, whether genetic structure is influenced by geographic distance (Isolation by Distance hypothesis) where individuals from the same area are more genetically similar than those from distant habitats. And lastly, if patterns of population genetic structure differ between mtDNA and microsatellite markers commonly associated with sex-specific behaviours such as female philopatry or male-mediated dispersal. Other Indian Ocean populations (South Africa and Arabian Sea) were also included as outgroups to examine regional patterns of genetic diversity.
Materials and methods
Sample collection and preservation
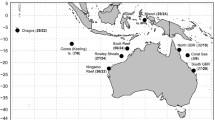
Tissue samples of C. amboinensis were obtained within Australia from both commercial fisheries collected by on-board scientific observers and fishery independent surveys. Samples were collected near Broome, Western Australia (WA); the north-eastern side of the Joseph Bonaparte Gulf to the Gulf of Carpentaria (GoC), Northern Territory (NT); Townsville, north Queensland (nth QLD) and from Moreton Bay (MB), near Brisbane also in Queensland. Samples from the Arabian Sea were collected from fish markets in Oman, Qatar and the United Arab Emirates. Samples from South Africa were collected in the shark control program of the KwaZulu-Natal sharks board (Cliff and Dudley 1991; Fig. 1). Refer to SOM for size distribution. Each sample consisted of approximately 5 g of white muscle tissue preserved in either 95% ethanol solution or 10% DMSO (dimethylsulphoxide in saturated 5 M NaCl solution).

Pig-eye shark (Carcharhinus amboinensis; total n = 324) capture locations. * Indicates capture locations. Circles indicate Australian population groupings: 1 = Western Australia (n = 41), 2 = Northern Territory (n = 205), 3 = Queensland (n = 54). Inset shows other Indian Ocean populations sampled; 4 = South Africa (n = 16); 5 = Arabian Sea (n = 8); PNG = Papua New Guniea (no samples)
In the field identifications of the species were based on morphological attributes. Due to the physical similarities among many Carcharhinus sp. and the presence of numerous congenerics in the region, mtDNA ND4 and control region sequences were compared with known reference collections including museum voucher specimens where possible. Sequences from this study were compared against the sandbar (C. plumbeus) (Nardo), the whitecheek (C. dussumieri) (Müller and Henle), the bignose (C. altimus) (Springer), the common blacktip (C. limbatus) (Müller and Henle), the Australian blacktip (C. tilstoni) (Whitley), the graceful (C. amblyrhychoides) (Whitley), the bull (C. leucas) (Valenciennes), the spinner (C. brevipinna) (Müller and Henle) and the spot-tail sharks (C. sorrah) (Müller and Henle).
Genomic DNA extraction
Total Genomic DNA was extracted from 50 mg of preserved tissue using the Chelex method (Walsh et al. 1991; Estoup et al. 1996). Tissue was placed in a small vial containing a 200 μl solution of 10% Chelex 100 in TE buffer (5 mM TrisCL pH 8.0 with 0.5 mM EDTA). The enzyme proteinase K (100 ng) was then added (5 μl) to the vial, and heated at 55°C for 3 h on a shaking platform to facilitate tissue digestion. The mixture was subsequently boiled for 8 min and then centrifuged at 13000×g for 5 min to precipitate the Chelex resin and bind polyvalent metal ions from the denatured DNA in solution. The supernatant containing the extracted DNA was then transferred to a fresh vial for manipulation and storage (Walsh et al. 1991; Estoup et al. 1996).
Amplification and sequencing—mitochondrial DNA (mtDNA)
Mitochondrial control region and NADH dehydrogenase subunit 4 (ND4) genes were selected as these markers are solely maternally inherited (evidencing female movement patterns) and do not undergo recombination (genetic signatures are not mixed during reproduction). Genes were amplified from 324 individual C. amboinensis using the polymerase chain reaction (PCR) methods and sequenced. The 5′ end of the ND4 gene was amplified and sequenced using the forward primer, ND4 (CACCTATGACTACCAAAAGCTCATGTAGAAGC) (Arevalo et al. 1994) and the reverse primer, H12293-LEU (TTGCACCAAGAGTTTTTGGTTCCTAAGACC) (Inoue et al. 2001). Refer to Table 1 for PCR reaction conditions. PCR products were purified using commercial QIAquick PCR purification kits (Qiagen, Doncaster, Vic, Australia) and viewed on a 1.5% agarose TAE (containing Tris base, acetic acid and EDTA) gel stained with ethidium bromide. PCR products were cycle sequenced using ABI Big Dye Terminator v3.1®. Fragment separation was carried out by capillary electrophoresis (Applied Biosystems 3130xl) under conditions recommended by the manufacturer producing 823 base pairs of sequence.
The 5′end of the control region was amplified using the forward primer GWF (CTGCCCTTGGCTCCCAAAGC) (Pardini et al. 2001) and a reverse primer that was designed from preliminary C. amboinensis sequence, CL2 (GGAAAAATATACGTCGGCCCTCG). The primer was designed using Primer3 v 0.4.0 (Rozen and Skaletsky 2000). Refer to Table 1 for PCR reaction conditions. PCR product purification and cycle sequencing followed the same protocol used for the ND4 gene, although C. amboinensis control regions were sequenced with the designed internal reverse primer CAR1 (TTTCCAAACCCGGGGTGAGT). Primers were designed following above methods. A fragment of 831 base pairs was produced.
Amplification and sequencing—nuclear DNA (Recombination activating gene 1)
Nuclear Recombination Activating Genes (RAG 1) were amplified to examine whether present day genetic-mixing occurs between clades identified in mtDNA intra-specific phylogenies. Thirty Carcharhinus amboinensis (ten from each clade identified in mtDNA) were amplified using the polymerase chain reaction (PCR) methods and sequenced. The 5′ end of the RAG1 gene was amplified and sequenced using the designed forward primer, RAG1F (CCCTCTATAGATGCCTTGCATTG) and the designed reverse primer, RAG1R (CCAAYTCATARCTTTTGGACTGC). Primers were designed following the fore-mentioned methods (Rozen and Skaletsky 2000). Refer to Table 1 for PCR reaction conditions. PCR purification and sequencing was performed as described above. A fragment of 565 base pairs was produced.
Amplification and genotyping—microsatellites
Unlike RAG1 gene, microsatellites are non-coding sections of DNA and as such conform to different modes of evolution, providing additional information on genetic exchange. Pig eye sharks (n = 222) were screened for 14 and genotyped for five microsatellite loci developed for species (C. tilstoni, C. limbatus, C. plumbeus) other than C. amboinensis (Ovenden et al. 2006; Portnoy et al. 2007). Loci were selected based on their successful cross-species amplification (Ovenden et al. 2006) and the highest number of polymorphic alleles between distant phylogenetic clades. Due to the overall lack of nuclear variation between both clades and population structure identified in mtDNA, the remaining individuals were not assayed. Amplification was achieved using polymerase chain reaction methods. Reaction mixtures (total volume of 6 μl) contained 1.18 μl of milli-Q water; 3 μl of 2× QIAGEN Multiplex PCR Master Mix® (QIAGEN, Doncaster, Vic, Australia) containing a pre-optimised mix of Taq DNA polymerase, dNTPs and providing a final concentration of 6 mM MgCl2; 0.02 μl of 10 μM forward primer with an M13 extension (Schuelke 2000); 0.2 μl of 10 μM reverse primer; 0.1 μl of fluro-labelled M13 primer; 1 μl of DNA template (12–40 ng) and 0.6 μl of 5× Q solution® (QIAGEN, Doncaster, VIC, Australia). The DNA template and reaction mix were initially denatured at 95°C for 15 min and then underwent 37 cycles of a denature period at 94°C for 30 s, an annealing period with loci specific temperatures of 50°C for loci CLi-110, CS-10, and CLi-12 and 52°C for loci CS-02 and CS-08 for 45 s and an extension time of 72°C for 1 min 30 s. The thermocycling was completed with a final extension time of 72°C for 45 min. Loci were individually amplified but subsequently combined for fragment separation according to label colour and fragment size. Microsatellite fragment separation and scoring was performed using capillary electrophoresis (ABI3130xl). The size of microsatellite amplicons (in base pairs) was calculated to two decimal place and amplicons were allocated to a “bin” that represented the mean allele size.
Restricted gene-flow due to Pleistocene sea-level changes
Mitochondrial DNA sequences were aligned and edited using the software Geneious™ v4.65 (Drummond et al. 2009). No premature stop codons were identified in the protein coding ND4 gene. Identical sequences were condensed into unique haplotypes and the polymorphisms defined by eye and then confirmed using Arlequin v3.11 (Excoffier et al. 2005) and MEGA 4.0 (Kumar et al. 2008) software.
The best fit model of nucleotide substitution and its associated gamma shape for both mtDNA genes were determined by performing hierarchical likelihood ratio test and by calculating approximate Akaike Information Criteria using MrModelTest v2.2 (Possada and Crandall 1998) implemented in Paup 4.0b10 (Swofford 2000).
Populations were defined as the Northern Territory, Western Australia and north Queensland (Fig. 1). As sample sizes from the Gulf of Carpentaria and Moreton Bay were significantly lower than the above three populations, these were pooled with other Australian locations. Nine samples from the Gulf of Carpentaria were grouped with the Northern Territory in accordance with the a priori hypothesis of geographic isolation due to land bridge formation in the Torres Strait. Eleven individuals from Moreton Bay did not differ genetically from any of the other locations and therefore grouped with Queensland due to geographic proximity.
Haplotype diversity (h), (likelihood of randomly choosing two different haplotypes from the one population), nucleotide diversity (π), (likelihood that two homologous base positions from two different haplotypes from the same population are different) and the number of polymorphic sites were estimated for each Australian population (Tajima 1983; Nei 1987).
Gene-flow among these populations was examined using F-statistics through a series of pairwise comparisons using Arlequin v.311 (Excoffier et al. 2005). Patterns of population genetic structure were quantified by PHIST measures for each gene region both separately and concatenated (supported by the total linkage of the genes due to their common origin within the mitochondria). This index incorporates the molecular evolution of haplotypes (Tamura and Nei model of nucleotide evolution gamma corrected) and ranges from 0 (identical allele frequencies) to 1 (no shared alleles). Analysis of Molecular Variance (AMOVA) was then used to assess the hierarchical contribution of molecular variance both within (PHISC) and among populations within groups (PHIST); and among groups (PHICT) to the overall measure of molecular variance.
Connectivity between populations was further confirmed through measuring evolutionary distance (Reynold’s D and Slatkin’s D) and the absolute number of migrants (M-values), again for both genes individually and concatenated. Estimates of evolutionary distance between populations measures the time in either generation time (Reynold’s D) or coalescence time (Slatkin’s D) required to generate the observed population pairwise genetic difference, assuming that the variation between populations’ increases linearly with time (Reynolds et al. 1983; Slatkin 1995). Similarly, the number of migrants exchanged was estimated as the exchange of migrants required to generate the observed population pairwise differences under the assumption that populations were of equal size and the mutation rate was negligible compared to the migration rate (Slatkin 1991).
Demographic consequences in C. amboinensis populations such as bottlenecks or expansions due to Pleistocene sea-level changes were investigated using Fu’s Fs and Tajima’s D statistics. Tajima’s D compared estimates of the mutational parameter (θ) based on the number of polymorphic sites and the mean number of pairwise differences (Tajima 1983, 1996). Significant differences in these estimates confirm higher or lower frequency of haplotypes than expected if mutations were evolving randomly. Fu’s F S calculates the probability of observing k or less alleles in a neutral (randomly evolving) population, based on the observed average number of pairwise differences (Fu 1997). A negative value of either statistic is evidence of an excess of low frequency haplotypes as expected from a recent population expansion or secondary contact between previously allopatric populations, while a positive value is evidence for a deficiency of low frequency haplotypes expected from a recent population bottleneck (Ramos-Onsins and Rozas 2002).
Evidence for either population bottlenecks in microsatellite DNA as shown by heterozygosity excess or population expansion indicated by heterozygosity deficiency were analysed using the Wilcoxon test in the program Bottleneck assuming Infinite Alleles Model (I.A.M) and 10,000 iterations (Cornuet and Luikart 1997).
Phylogenetic support for historic geographic isolation was investigated by reconstructing intra-specific phylogenies among unique mtDNA haplotypes and mapping their distribution across northern Australia. Both character-based (Neighbour-Joining and Maximum Parsimony) and model-based (Maximum Likelihood and Bayesian Inference) methods were used. All analyses were done on each gene region individually and then with the two gene regions concatenated. Mutations were unweighted and indels were treated as a fifth state. Indian Ocean and South African populations were included as outgroups. Maximum Likelihood and Maximum Parsimony analysis were performed using the software Paup 4.0b10 (Swofford 2000) and Bayesian Inference was performed using the software MrBayes v3.1 (Huelsenbeck and Ronquist 2001). Concatenated sequences were partitioned for Bayesian Inferences accommodating different models of evolution for the each gene region. Priors for Maximum Likelihood and Bayesian Inference were determined by performing hierarchical likelihood ratio test and by calculating Akaike Information Criteria using the software MrModelTest v2.2 (Possada and Crandall 1998). Heuristic tree searches were performed with 1000 random addition replications and the statistical support for nodes was determined via 1000 non parametric bootstrap replicates. A majority-rule consensus tree was also constructed based on the 1000 bootstrap replicates. Bayesian Inference was run using the Metropolis-coupled Markov Chain Monte Carlo (MCMC) algorithm from randomly generated starting trees for three million generations, sampling trees every 1000 generations. Two simultaneous runs were performed with three heated chains and one cold chain each with a temperature parameter of 0.2. The standard deviation of split frequencies was used as a convergence diagnostics to determine that when posterior probability distribution had reached stationarity. The burnin was set to discard the initial 25% of samples following guidelines outlined in the manual. Only Bayesian trees are presented as other phylogenetic reconstructions produced similar topologies. In addition to conventional phylogenetic reconstructions, statistical parsimony networks (TCS) were also generated (Clement et al. 2000). Unlike traditional methods, parsimony networks assume that the ancestral haplotype is present in the current sample, incorporates homoplasy and is not limited to bifurcation at branch nodes. Gaps were again treated as a fifth state and the connection limit was set to 95%. Divergence time (million years) between clades was estimated by determining the percent sequence divergence and then assuming similar mutation rate as defined for lemon, Negraprion brevirostris (Schultz et al. 2008) and scalloped hammerhead, Sphyrna lewini sharks converting these values to divergence per million years (Duncan et al. 2006).
Phylogeographic patterns were simplified by graphically representing the frequency of each clade within Western Australia, the Northern Territory, Gulf of Carpentaria and north Queensland.
Current day sympatric cryptic species
Rag 1 sequences were aligned and edited using the software Geneious™ v4.65 (Drummond et al. 2009). Identical sequences were condensed into unique haplotypes and the polymorphisms defined by eye and then confirmed using Arlequin v3.11 (Excoffier et al. 2005) and MEGA 4.0 (Kumar et al. 2008) software. Phylogenetic structure identified with mitochondrial DNA was tested by pairwise comparisons and Analysis of Molecular Variance (AMOVA).
Isolation by distance
The ‘Isolation by Distance’ hypothesis was also tested to determine if populations distributed continuously along the north Australia coastline were structured by geographic distance. This hypothesis assumes that individuals are not embarking on long-distance travel, and for this reason genetic distance (PHIST) should increase in a linear fashion with geographic distance (km). Genetic distances between capture locations (n = 12) were calculated based on the Tamura and Nei model of nucleotide gamma corrected evolution using Arlequin v.311 (Excoffier et al. 2005) and were correlated using a Mantel test with geographical distances (by sea). Slope and intercept estimates were subsequently assessed using reduced major regression analysis using the ‘Isolation by Distance Web Service’ (Jensen et al. 2005). Only concatenated sequences were compared as they were most variable.
Population genetic structure—nuclear marker (Microsatellites)
Prior to population structure analysis of microsatellite DNA, the null hypothesis of Hardy–Weinberg equilibrium was tested using Arlequin v3.11 (Excoffier et al. 2005) and GenAlex v 6.1 (Peakall and Smouse 2005). In addition, the software, Microchecker v. 2.2.3 (van Oosterhout et al. 2004) was implemented to identify possible causes for any deviations from Hardy–Weinberg equilibrium. Microsatellite genetic diversity was characterised by the number of alleles per locus (Na), expected (HE) and unbiased (UHE) heterozygosity, observed heterozygosity (HO) and fixation index (F) using Arlequin v3.11 (Excoffier et al. 2005) and GenAlex v 6.1 (Peakall and Smouse, 2005). The probability of rejecting the null hypothesis of genotypic disequilibrium between pairs of loci across populations was estimated by Arlequin v3.11 (Excoffier et al. 2005). Population structure identified with mitochondrial DNA was tested by pairwise comparisons and Analysis of Molecular Variance (AMOVA).
Results
Restricted gene-flow due to Pleistocene sea-level changes
MtDNA supported genetic similarity between Western Australia and the Northern Territory and the difference of both of these locations from Queensland. The model of nucleotide evolution was GTR + I and HKY + I + G (gamma = 0.9871) for ND4 and control region respectively. Both mtDNA genes were highly diverse. Fourteen unique ND4 and 29 control region haplotypes were defined (refer to online supplementary data, SOM for individual gene region summary tables and GenBank accession numbers). Subsequent population statistics refer to concatenated gene regions. Nucleotide diversity (π, as %) was high (0.50 ± 0.31 to 0.89 ± 0.45) compared with other inshore carcharhinids (0.0067 ± 0.0095 to 0.535 ± 0.351; control region C. sorrah) (Ovenden et al. 2009). Haplotype diversity (h) was slightly higher in Western Australia and the Northern Territory (~0.85) than the Gulf of Carpentaria, north Queensland or Moreton Bay (~0.6–0.75) (Table 2).
Three main concatenated mtDNA haplotypes (CN02, CN04 and CN06) were present in all locations but in differing frequencies (Table 2). Haplotype CN06 was overall most abundant decreasing in frequency from Moreton Bay (67%) to Western Australia (22%). Haplotype CN02 displayed a complementary pattern and was most abundant in Western Australia accounting for 20% of haplotypes, and decreased in frequency eastwards so that it accounted for only 4% of haplotypes in north Queensland. Haplotype CN04 was similarly abundant in Western Australia, the Northern Territory and north Queensland (5–10%) but represented 30% of haplotypes within the Gulf of Carpentaria. The occurrence of less frequent haplotypes differed between locations but overall, samples from the Northern Territory and Western Australia had more shared haplotype frequencies than those from Queensland.
AMOVA confirmed that sharks from Queensland waters were genetically distinct from those from Western Australia and Northern Territory populations (PHIST = 0.029 P < 0.025; PHIST = 0.025 P < 0.035; PHIST = 0.027 P < 0.027 ND4, control region and concatenated sequences respectively; refer to Table 3 for population pairwise PHIST values). Connectivity among populations was further supported by the highest evolutionary distance (Slatkins D and Reynolds D) and fewest exchanges of migrants (M-values) between Western Australia and Queensland and a complementary pattern between Western Australia and the Northern Territory (Table 4).
There was no support in any population for bottlenecks or expansion between previously allopatric populations (indicated by insignificant Tajima’s D and Fu’s F S ; Table 5). Additionally there was no evidence in microsatellite DNA for influences of Pleistocene sea-level changes on population structure. One-tailed Wilcoxon tests did not support recent bottlenecks or expansions (P < 0.84, P < 0.43; P < 0.9, P < 0.15; P < 0.84, P < 0.43 heterozygosity excess and deficiency for Western Australia, Northern Territory and Queensland populations respectively).
Phylogeographic analysis of mtDNA identified three distinct clades of C. amboinensis across northern Australia. Clade one was the most abundant, while clades two and three were a half and a third as abundant respectively (Fig. 2). The basal clade (one) was most abundant in the east Australian and western Indian Ocean populations (South Africa and Arabian Sea) and decreased in frequency moving west along the Australia coastline. The second clade was equally abundant across Australia, perhaps more frequent in the Gulf of Carpentaria and not present in both western Indian Ocean populations, while the third clade was most abundant in Western Australia and decreased in frequency eastwards. It was also not present in western Indian Ocean populations (Fig. 3). Sequence divergence between clade one and two was 0.24% and correlated to an isolation period of 300,000 to 360,000 years during the Pleistocene era. Sequence divergence between clade one and three was 1.2% correlating to an isolation period of 1.6 to 2 million years, again during the Pleistocene (Fig. 2).

Inferred phylogeny of concatenated mitochondrial NADH dehydrogenase subunit 4 (ND4; 823 bases) and control region (831 bases) reconstructed using 95% statistical parsimony network. Haplotype numbers from Table 1 are given next to each pie chart. The size of the pie chart represents the frequency of the haplotype. Inset Bayesian Inference; rooted with Carcharhinus amblyrhyncoides and C. leucas, nodal support given as Bayesian posterior probabilities (n = 324)

Frequency of each clade identified in 95% statistical parsimony network by location for Carcharhinus amboinensis for each gene region individually, NADH dehydrogenase subunit 4 (ND4; 823 bases) and control region (831 bases); and concatenated. Total n = 324. Dashed circles represent population structure tested
Current day sympatric cryptic species
All 30 C. amboinensis RAG 1 sequences were the same haplotype.
Isolation by distance
A Mantel test and reduced major regression analysis for a positive relationship between genetic similarity and geographic distance based on concatenated gene regions between capture locations was supported (r = 0.2446; P < 0.03; Fig. 4).

Reduced major regression analysis between pairwise geographic distances by sea (km) and pairwise genetic distances (mtDNA PHIST) between un-pooled captured locations (total n = 12). Regression y = 1.35x − 5.487; R 2 = 0.059; P < 0.03
Population genetic structure—nuclear marker (microsatellites)
Analysis of microsatellite DNA did not support the population structures identified in mtDNA (Global FST = −0.00016, P < 0.46). The CS02 microsatellite locus deviated from Hardy–Weinberg expectations for all Australian populations and was omitted for population structure and phylogenetic analysis. All other loci remained within these expectations and did not show evidence of linkage disequilibrium. Sample sizes of genotypes assayed with the remaining microsatellite loci for C. amboinensis were 66 ± 11.351 (mean ± SE) over all three populations and four microsatellite loci. The un-biased heterozygosity was 0.683 ± 0.08. The mean number of alleles (±SE) was 3 ± 0.000 for Cli110, 15.667 ± 3.283 for Cli12, 24.667 ± 0.882 for CS08 and 14.667 ± 3.180 for CS10 (Table 6).
Discussion
We provide the first report of the genetic structure of populations of the pig-eye shark, Carcharhinus amboinensis across its known distribution. Despite large total lengths obtained by C.amboinensis and subsequent potential for mobility, genetic diversity (each mitochondrial gene region individually and concatenated) was partitioned across northern Australia so that populations from Western Australia and the Northern Territory grouped together, separate from Queensland. This Pacific/Indian Ocean barrier is common in coastal north Australian species (Chenoweth et al. 1998; Lukoschek et al. 2007) and is argued to be a consequence of the land bridge between Cape York and Papua New Guinea that formed during the Pleistocene, which formed a barrier to movement and gene flow of marine animals between east and west coasts of Australia. The unexpected genetic similarity between Gulf of Carpentaria and Queensland populations suggests that gene-flow across the Torres Strait has occurred supporting secondary introgression, but due to the low sample size in the Gulf of Carpentaria we were unable to conclusively quantify the rate of exchange.
The occurrence of a third clade in phylogenetic reconstructions suggests that present day genetics are not solely shaped by this land bridge which would result in only two groups, but rather multiple isolating events such as those that occurred during the Pleistocene epoch. Once these barriers to dispersal were removed gene flow occurred, accounting for the spread of each clade (albeit in different proportions) among all populations.
The occurrence of clade one in both western Indian Ocean populations suggests that this is ancestral and that the other two uniquely Australian clades have resulted from recent isolating mechanisms. The east–west cline in mtDNA haplotype frequencies across northern Australia in clades one and three suggests earlier isolation of populations with divergence occurring 1.6–2 million years ago during the Pleistocene, followed by more recent movement, most likely since the last opening of the Torres Strait approximately 6000 years ago (Voris 2000). Interestingly, despite also evolving independently (divergence time of 300,000–360,000 years ago during the Pleistocene era), the second clade does not show an east–west cline in frequencies, possibly due to different evolutionary constraints. If the higher abundance of this clade within the Gulf of Carpentaria is not simply due to sampling error, then this pattern is suggestive of regional isolation that has subsequently dispersed evenly east and west as sea-levels rose. Conversely, if this elevated abundance does not represent true frequencies and this clade is evenly distributed across Australia, this may indicate admixing with Indonesian populations during lower sea-levels that forced previously allopatric populations together. Further research incorporating a greater number of samples from within the Gulf of Carpentaria and from Indonesia would tease out the origin of clade two, and increasing the number of sampling locations across the Indian Ocean would resolve broad-scale colonisation and dispersal events.
Attributing which of many scenarios in this topographically and hydrologically complex region has resulted in the current population structure is challenged by the lack of phylogeographic patterns (indicated by the occurrence of all three clades in each Australian location) and episodic nature of vicariance events that occurred during the Pleistocene. What is clear is that the identification of these three clades support multiple incidences of independent evolution during the Pleistocene and subsequent introgression and movement across Australia. This movement has mixed and increased population sizes sufficiently to prevent Tajima’s D and Fu’s F S from detecting changes in allele frequencies due to sudden reduction or admixing between populations expected during the Pleistocene to generate the phylogenies, and also prevents further analysis to define likely refugia. Phylogenetic discontinuties in the absence of current spatial separation as we identified in C. amboinensis are rare, but have been recorded by Avise et al. (1987) and attributed to hybrid swarming arising from secondary contact between allopatrically evolved populations.
The lack of variation among both nuclear markers (Rag 1 and microsatellites) confirms present day patterns of unrestricted genetic mixing between mitochondrial clades verifying the absence of a cryptic species of C. amboinensis in north Australia. The lack of population structure in these markers may indicate sex-specific behaviours such as female philopatry maintaining mtDNA structure generated through the Pleistocene, although we cannot eliminate the possibility that not enough time has elapsed (given the generation time of 13 years and likely effective population size greater than 100,000 individuals) for this pattern to occur in nuclear markers, reinforcing the idea that genetic structures have an ancient origin (Frankham et al. 2002). Furthermore, preliminary evidence suggests that pig-eye sharks may not utilise distinct nurseries such as those occupied by juvenile bull or lemon sharks which are known to be philopatric (Feldheim et al. 2004; Chapman et al. 2009; Tillett et al. 2011; Tillett et al., in review), and there is no record of adults undergoing large oceanic migrations indicative of dispersal phases which have been recorded for morphologically similar bull sharks (Carlson et al. 2010). Future studies with increased power in microsatellite loci are needed to provide robust information on male movement patterns, particularly male-mediated dispersal. ‘Isolation by Distance’ (only explaining 6% of variance among populations) effects indicate contemporary restricted movement of individuals across north Australia and may explain the susceptibility of C. amboinensis to isolating mechanisms during geological time.
In conclusion, results suggests that coastal changes during the Pleistocene epoch, such as the repeated formation of land bridges in the Torres Strait, separated east and west Australian populations of Carcharhinus amboinensis, although this isolation was not sufficient to generate current day sympatric cryptic species. Phylogenetic re-constructions indicate that this has occurred multiple times through geological history. In addition to geological events, ecological processes such as habitat use and distance travelled to find a mate have also influenced population genetic structure. Discriminating between these causes is vital for successfully attributing how species genetic diversity is maintained, but is challenged by not only by the complex ecologies of sharks (different ecological function of age cohorts and the potential for high mobility), but also complex geology. We caution hastily simplifying factors driving current population genetic structure providing an example where geological history is a major contributor. Shark biodiversity in the Indo-Australian Archipelago requires conservation as regional pressures increase and this hinges on understanding the origin and maintenance of genetic diversity. Phylogenetic reconstructions reiterate the susceptibility of C. amboinensis to changes in shallow coastal environments, such as those that occurred during the Pleistocene, and predicted to occur under a changing world climate.
References
Arevalo E, Davis SK, Sites JW (1994) Mitochondrial-DNA sequence divergence and phylogenetic-relationships among 8 chromosome races of the Sceloporus grammicus complex (Phrynosomatidae) in central Mexico. Syst Biol 43:387–418
Avise JC, Arnold J, Ball RM, Bermingham E, Lamb T, Neigel JE, Reeb CA, Saunders NC (1987) Intraspecific phylogeography—the mitochondrial DNA bridge bewteen population-genetics and systematics. Annu Rev Ecol Syst 18:489–522
Carlson JK, Ribera MM, Conrath CL, Heupel MR, Burgess GH (2010) Habitat use and movement patterns of bull sharks, Carcharhinus leucas determined using po-up satellite archival tags. J Fish Biol 77:661–675
Chapman DD, Babcock EA, Gruber SH, Dibattista JD, Franks BR, Kessel SA, Guttridge T, Pikitch EK, Feldheim KA (2009) Long-term natal site-fidelity by immature lemon sharks (Negaprion brevirostris) at a subtropical island. Mol Ecol 18:3500–3507
Chenoweth SF, Hughes JM, Keenan CP, Lavery S (1998) When oceans meet: a teleost shows secondary intergradation at an Indian-Pacific interface. Proceedings of the Royal Society of London Series B-Biological Sciences, vol 265, pp 415–420
Clement M, Possada D, Crandall K (2000) TCS: a computer program to estimate genealogies. Mol Ecol 9:1657–1660
Cliff G, Dudley J (1991) Sharks caught in the protective nets off Natal, South Africa. 5. The Java Shark Carcharhinus amboinensis (Müller and Henle). S Afr J Mar Sci 11:443–453
Cornuet JM, Luikart G (1997) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequence data. Genetics 144:2001–2014
Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M, Moir R, Stones-Harves S, Thrierer T, Wilson A (2009) Geneious v4.65
Duncan KM, Martin AP, Bowen BW, De Couet HG (2006) Global phylogeography of the scalloped hammerhead shark (Sphyrna lewini). Mol Ecol 15:2239–2251
Estoup A, Largiader CR, Perrot E, Chourrout D (1996) Rapid one-tube DNA extraction for reliable PCR detection of fish polymorphic markers and transgenes. Mol Mar Biol Biotechnol 5:295–298
Excoffier L, Laval G, Schneider S (2005) Arlequin ver 3.0. An integrated software package for population genetics data analysis. Evol Bioinformat Online 1:47–50
Feildheim KA, Gruber SH, Ashley MV (2002) The breeding biology of lemon shark at a tropical nursery lagoon. In: Proceedings of the royal society biolgical sciences, London, pp 1655–1661
Feldheim KA, Gruber SH, Ashley MV (2004) Reconstruction of parental microsatellite genotypes reveals female polyandry and philopatry in the lemon shark, Negaprion brevirostris. Evolution 58:2332–2342
Field IC, Meekan MG, Buckworth RC, Bradshaw CJA (2009) Protein mining the world’s oceans: Australasia as an example of illegal expansion-and-displacement fishing. Fish Fish 10:323–328
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to the conservation genetics. Cambridge University Press, Cambridge
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Inoue JG, Miya M, Tsukamoto K, Nishida M (2001) A mitogenomic perspective on the basal teleostean phylogeny: resolving higher-level relationships with longer DNA sequences. Mol Phylogenet Evol 20:275–285
IUCN (2010) Red list of threatened species. Version 2010.3
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:1–6
Keeney DB, Heupel MR, Hueter RE, Heist EJ (2005) Microsatellite and mitochondrial DNA analyses of the genetic structure of blacktip shark (Carcharhinus limbatus) nurseries in the northwestern Atlantic, Gulf of Mexico, and Caribbean Sea. Mol Ecol 14:1911–1923
Kuhnt W, Holbourn A, Hall R, Zuvela M, Kase R (2004) Neogene history of the Indonesian throughflow. In: Clift P, Kuhnt W, Wang P, Hayes D (eds.) Continent–ocean interactions within East Asian Marginal Seas, pp 299–320
Kumar S, Dudley J, Nei M, Tamura K (2008) MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Bioinformatics 9:299–306
Last PR, Stevens JD (2009) Sharks and rays of Australia. CSIRO, Victoria
Lukoschek V, Waycott M, Marsh H (2007) Phylogeography of the olive sea snake, Aipysurus laevis (Hydrophiinae) indicates Pleistocene range expansion around northern Australia but low contemporary gene flow. Mol Ecol 16:3406–3422
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Northern Territory Government (2009) Fishery status reports 2008. Department of Resources, Darwin
Ovenden JR, Street R, Broderick D (2006) New microsatellite loci for Carcharhinid sharks (Carcharhinus tilstoni and C-sorrah) and their cross-amplification in other shark species. Mol Ecol Notes 6:415–418
Ovenden JR, Kashiwagi T, Broderick D, Giles J, Salini J (2009) The extent of population genetic subdivision differs among four co-distributed shark species in the Indo-Australian archipelago. BMC Evol Biol 9:40
Pardini AT, Jones CS, Noble LR, Kreiser B, Malcolm H, Bruce BD, Stevens JD, Cliff G, Scholl MC, Francis M, Duffy CAJ, Martin AP (2001) Sex-biased dispersal of great white sharks. Nature 412:139
Peakall R, Smouse PE (2005) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Portnoy DS, Piercy AN, Musick JA, Burgess GH, Graves JE (2007) Genetic polyandry and sexual conflict in the sandbar shark, Carcharhinus plumbeus, in the western North Atlantic and Gulf of Mexico. Mol Ecol 16:187–197
Possada D, Crandall KA (1998) Modeltest: testing the model of DNA substitutions. Bioinformatics 14:817–818
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100
Reynolds J, Weir BS, Cockerham CC (1983) Estimation of the co-ancestry coefficient—basis for a short-term genetic distance. Genetics 105:767–779
Rozen S, Skaletsky H (2000) Primer 3 on the WWW for general users and for biologist programmers. Methods Mol Methodol 132:365–386
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Schultz JK, Feldheim KA, Gruber SH, Ashley MV, McGovern TM, Bowen BW (2008) Global phylogeography and seascape genetics of the lemon sharks (genus Negaprion). Mol Ecol 17:5336–5348
Slatkin M (1991) Inbreeding coefficients and coalescence times. Genet Res 58:167–175
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequences. Genetics 139:1463–1563
Swofford DL (2000) PAUP* phylogenetic analysis using parsimon (*and other methods). Sinauer Associates, Sunderland, MA
Tajima F (1983) Evolutionary relstionship of DNA sequences in finite populations. Genetics 123:585–595
Tajima F (1996) The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 143:1457–1465
Tillett BJ, Parry DL, Munksgaard NC, Meekan MJ, Field IC, Bradshaw CJA, Thorburn D (2011) Decoding fingerprints—elemental composition of vertebrae map ontogenetic habitat partitioning between two morphologically similar apex predators in northern Australia. Mar Ecol Prog Ser 434:133–142
Tillett BJ, Meekan MJ, Field I, Thorburn D, Ovenden J (in review) Evidence for reproductive philopatry in the bull shark, Carcharhinus leucas in northern Australia. J Fish Biol
van Oosterhout C, Hutchinson WF, Wills PM, Siple P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Voris HK (2000) Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J Biogeogr 27:1153–1167
Walsh PS, Metzger DA, Higuchi R (1991) Chelex-100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
White WT, Last PR, Stevens JD, Yearsley GK, Fahmi, Dharmadi (2006) Economically important sharks and rays of Indonesia. CSIRO Publishing, Canberra
Williams M, Cook E, van der Kaars S, Barrows T, Shulmeister J, Kershaw P (2009) Glacial and deglacial climatic patterns in Australia and surrounding regions from 35000 to 10000 years ago reconstructed from terrestrial and near-shore proxy data. Quat Sci Rev 28:2398–2419
Acknowledgments
Funded by Tropical Rivers and Coastal Knowledge Research Hub, Charles Darwin University, Darwin and the Molecular Fisheries Laboratory, Department of Employment, Economic Development and Innovation, Queensland Government. Sampling was undertaken with the kind support of fishermen; Wildlife Resources Inc; Kakadu National Park; Fishing and Fisheries Research, Centre James Cook University; Department of Fisheries—Western Australia, Fish for the Future, RSK Environment LTD/University of Bangor and the Department of Resources—Fisheries, Northern Territory. We also thank R. Street and J. Morgan for their technical expertise and Bioscience North Australia for their support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Tillett, B.J., Meekan, M.G., Broderick, D. et al. Pleistocene isolation, secondary introgression and restricted contemporary gene flow in the pig-eye shark, Carcharhinus amboinensis across northern Australia. Conserv Genet 13, 99–115 (2012). https://doi.org/10.1007/s10592-011-0268-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-011-0268-z