
Abstract
The shift in belowground biodiversity driven by wetland degradation is a hotspot research in global change ecology. However, little is known about the association of microbial diversity with alteration in soil environment along a degradation gradient. This study aimed to identify the specific response of bacterial taxa to change in soil variables along wetland degradation gradient, using Illumina high-throughput sequencing technology. Four stages, including primary wetland (for reference), swamp meadow (transitional stage), meadow (degraded stage), and reclaimed wetland (severely degraded stage), were employed to identify the abundance change of different bacterial taxa along degradation gradient in the Napahai plateau. The relative abundance of dominant taxa, i.e., Acidobacteria (8.5–25.6%) and δ-Proteobacteria (7.2–21.7%), varied with wetland degradation. The primary and transitional stages distributed higher δ-Proteobacteria abundance (19–21.7%), while degraded stages had higher Acidobacteria (22.7–25.6%). We observed a conspicuous decrease (1.5–5.3 times) in soil water, organic matter, and total and available nitrogen, but a distinct increase (1.1–2.8 times) in carbon to nitrogen ratio, and total phosphorus compared with primary wetland. The δ-proteobacteria abundance was mainly determined by concentrations of soil water, organic matter, and total and available nitrogen, whereas Acidobacteria abundance was closely associated with carbon to nitrogen ratio and total phosphorus. Therefore, our results indicate that alterations in edaphic variables (e.g., soil water, carbon, nitrogen, and phosphorus) can serve as crucial predictors for shift in taxa abundance of bacterial community along degradation gradient.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
At present, predicting the influence of wetland degradation on global biodiversity has become a hotspot of global change research, as wetlands are extremely sensitive to human interference and global climate change (Mamo and Bekele 2020; Aíza et al. 2021). Wetlands often suffer from serious degradation due to climate changes (e.g., increasing temperature, decreasing precipitation) and human interference (e.g., anthropogenic drainage, peat exploitation, and land reclamation) (Shen 2019; Cui et al. 2021). These changes have resulted in a series of degradations in water, soil, and vegetation of wetlands (Mamo and Bekele 2020). For example, the artificial drainage for grain and livestock production lowers wetland water table, resulting in gradual disappearance of hydrophytes and the invasion of dominant mesophytes (Hu et al. 2015). Ultimately, wetland degradation is subjected to strong recessions in native marsh soils. This leads to loss of organic matter, humus, and peat horizon from wetland soils, which may reduce microbial diversity in soils (Steinmuller and Chambers, 2019). In particular, wetland degradation can induce the change in aerobic and anoxic soil conditions, affecting microbial composition and diversity (Mandal et al. 2020). Thus, it is crucial to identify how the wetland degradation can shape the change in microbial community.
The response of microbial community to wetland degradation is important but unclear in predicting the alteration in nutrient cycling of ecosystems. Soil bacteria account for 70–90% of the total soil microorganisms, which play key roles in regulating biogeochemical cycles of wetland ecosystems (Chapman et al. 2017). At present, the severe degradation of world’s wetlands had resulted in crucial effects on soil bacterial communities (Lamsal et al. 2015). Furthermore, bacterial communities can respond to alterations in wetland soil environments more quickly than plants (Andersen et al. 2013; He et al. 2021). However, little is known about the influencing extent of the increase or decrease of soil variations on taxonomic composition of bacterial community during wetland degradation (Gu et al. 2018). Therefore, the responses of bacterial groups to wetland degradation need to be better determined for predicting their effects on soil carbon and nitrogen cycling.
It is pivotal to determine the linkage of soil properties (or biogeochemical cycling) with bacterial community during wetland degradation. The edaphic variables can differ in effects on bacterial community, as bacterial taxa have different demands for soil water and nutrients (Bai et al. 2013; Panico et al. 2020). For example, soil bacterial taxa such as copiotrophs or oligotrophs have distinct difference of nutritional requirements (Koch 2001;Romero-Trigueros et al. 2021). The change in soil variables can thus drive the shift in bacteria composition and diversity during wetland degradation (Gu et al. 2018). Wetland degradation can also induce the alteration in abundance of specific bacterial taxa. For example, Bacteroidetes were widely distributed in primary swamp wetlands, while ɑ-Proteobacteria were more dominant in degraded wetlands (Tang et al. 2012). The shift in bacterial taxa may be critical for predicting the changes in ecosystem composition and functioning. However, there is limited knowledge about the response of specific bacterial taxa to wetland degradation. Probably, the reduction of soil nutrients due to wetland degradation results in a selective effect on specific bacterial taxa (Cederlunda et al. 2014; Stone et al. 2021). Therefore, the change in specific bacterial taxa would be valuable indicators for the changes in soil nutrient environments during wetland degradation.
Napahai plateau wetlands are hot spots for conservation of global biodiversity, as they are internationally famous for their rich species and diverse habitats. However, Napahai wetlands have suffered from a series of degradations due to global change and human activities such as drainage and farming since 1960s (Guo et al. 2013a, b; Luan et al. 2014). These thus formed four typically degraded stages, including primary wetland (PW, reference site), swamp meadow (SM, transitional stage), meadow (M, degraded stage), and reclaimed wetland (RW, severely degraded stage) (Lu 2018). For this study, four stages (i.e., PW, SM, M and RW) along a degradation gradient were employed to determine the response of bacterial communities to wetland degradation, using Illumina high-throughput sequencing technology. The specific queries of this study were (1) to explore the shift in soil bacterial communities along degradation gradient, (2) to identify the specific taxa governed by different soil variations, and (3) to link the possible association of specific bacterial taxa with soil variables. We hypothesized that (1) soil bacterial communities vary with wetland degradation, and (2) soil variations show different effects on specific bacterial taxa during wetland degradation. Our research can provide basic data for predicting the interaction of bacterial community and soil variables during wetland degradation.
Materials and methods
Site trait
We conducted this study in the Napahai wetlands (27°49′ ~ 55′ N, 99°37′ ~ 43′ E), which located in the northwestern transverse margin of Qinghai-Tibet Plateau, Shangrila, Yunnan, southwest of China. The wetlands have a typical western monsoon climate, characterized by high cold, long frost period, low precipitation, and distinct dry–wet seasons (Xiao et al. 2008). Mean annual precipitation is about 620 mm, with 87% precipitation occurred in the wet seasons (from May to October). The wetlands have specially closed and semi-closed terrain, favor a high biodiversity, and thus become hot spots for biodiversity conservation in the world (Guo et al. 2013a, b). However, a series of degraded wetland stages were formed due to global change and human interference (e.g., draining and reclamation). Four stages (i.e., PW, SM, M, and RW) along a degradation gradient were applied to explore the response of bacterial community to wetland degradation (Table 1; Figs. 1 and 2; Lu 2018).

Sketch map of sampling sites in the Napahai plateau wetlands

Ground landscape in dry and wet seasons along a degradation gradient in the Napahai wetlands
Soil sampling and analysis
We randomly sampled three sites (50 × 60 m quadrats, at least 200 m apart) for each of four degraded stages. Three plots (10 × 10 m, 20 m apart) were also randomly sampled for each site. Soils were sampled in each replicated site at 20 cm soil layer in January 2016. Soil cores were taken from five points (20 × 20 cm, 3-m intervals), and then thoroughly mixed into one composite sample for each plot. Soil cores (5 cm diameter) were then sampled to determine soil variables. One portion of these samples was utilized to analyze soil water content (SW) gravimetrically, while another portion was used for analysis of other soil variables. A glass electrode was used for determining soil pH in a 1:2.5 soil/water solution (w/v). The procedure of external heating dichromate oxidation was applied to determine soil organic matter (SOM). Kjeldahl digestion method was used for the analysis of total nitrogen (TN), and available nitrogen (AN) was analyzed by alkali diffusion method. The colorimetry of H2SO4–HClO4 elimination–Mo-Sb was applied to determine total phosphorus (TP), and total potassium (TK) was determined by NaOH melting-flame photometry. Available phosphorus (AP) was measured by Mo–Sb colorimetric method. Available potassium (AK) was analyzed by a flame photometry method. All selected soil characteristics were determined by the method described by Lu 2004).
DNA extraction and high-throughput sequencing
DNAs in soil samples were extracted using a PowerSoil DNA Isolation kit (MoBio Laboratories, catalog no. 12888–50, USA), following the manufacturer’s instructions. The extracted DNAs were checked by 1% agarose gel electrophoresis, and then stored below − 20 ℃ until use.
The primers 341F: CCCTACACGACGCTCTTCCGATCTG (barcode) CCTACGGGNGGCWGCAG and 805R: GACTGGAGTTCCTTGGCACCCGAGAATTCCAGACTACHVGGGTATCTAATCC were applied to amplify V3-V4 regions of the 16S rRNA gene. The primers contained 10-bp multiplex identifiers (MIDs) and the Roche 454-pyrosequencing adaptor Lib-L (Zhao et al. 2016). The conditions of first PCR amplification were 95 ℃ for 5 min with 27 cycles of three steps (i.e., 95 ℃ for 30 s, 55 ℃ for 30 s, and 72 ℃ for 30 s), with a final step at 72 ℃ for 10 min. TruSeq DNA HT Sample Prep Kit (Catalog No. FC-121–2001, Illumina) was applied to purify the PCR products, and a Quanti Fluor TM-ST system (Promega Corporation, Madison, WI, USA) was used for quantifying them. An Illumina Sequencer Miseq platform (San Diego, CA, USA) was applied for high-throughput sequencing in Sangon Biotech (Shanghai, China) Co., Ltd. The obtained sequences were filtered by quality and length control (Phred score threshold 25, length range from 200 to 600 bp) (Embarcadero-Jiménez et al. 2016).
Grouping and statistical analysis
The sequences were clustered into operational taxonomic units (OTUs) at 97% similarity threshold using the Mothur program (http://mothur.org, version 1.30.1). Microbial alpha diversity (i.e., Chao, ACE, and Shannon and Simpson index) was determined by Mothur for each sample (Zhao et al. 2016). Principal component analysis (PCA) was utilized to identify the relationship between spatial soil heterogeneity and soil bacterial communities across wetland degradation. The classifier of Ribosomal Database Project (RDP) was applied to determine taxonomic sequence classifications at an 80% confidence threshold, according to recommended sequences in the length range (Zhao et al. 2016; Embarcadero-Jiménez et al. 2016). A one-way ANOVA (analysis of variance) was used to determine the differences of soil variables and bacterial community among the four degradation stages. Redundancy analysis (RDA) was conducted to address the links of relative abundance of taxonomic groups with soil variations. The correlation between soil variables and the relative abundance of bacteria phyla was illustrated using Adobe Illustrator. Correlation matrix analyses were applied to show the relationships between bacterial diversity and measured soil variables. The association of OTUs richness and diversity to soil variations was examined by correlation matrix analyses. The data were confirmed for variance normality and homogeneity before analysis. Multiple comparisons were conducted through a Duncan test. Means were compared by least significant difference test (LSD) (P < 0.05). The whole process was conducted on computation with Canoco software (version 4.5, Centre for Biometry, Wageningen, The Netherlands).
Result
Variation in soil variables along degradation gradient
There was a distinct separation of soil variables in four degradation stages, indicating that wetland degradation resulted in a clear spatial heterogeneity for the development of bacterial community (R > 0.9, P < 0.001) (Fig. 3). Soil variables increased or decreased along wetland degradation in the Napahai plateau (Table 2; P < 0.01). The concentrations of SW, SOM, TN, and AN generally decreased along degradation gradient, whereas there was an increase in values of C/N and TP (Table 2). The highest concentrations of TK, AP, and AK were observed in reclaimed wetland, while the lowest values were in meadow wetland (Table 2). Soil pH also varied significantly among the four degradation stages, with the lowest values observed in reclaimed wetland (5.92 ± 0.35; n = 9, P < 0.05). Therefore, soil properties can differ in response to wetland degradation.

Principal component analysis (PCA) showing effect of wetland degradation on spatial soil heterogeneity (e.g., SW, pH, and C, N, P, K nutrients) in the Napahai plateau
Alteration of soil bacterial taxa along degradation gradient
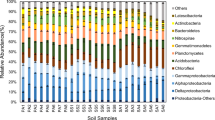
Soil bacterial communities had similar assemblages, with 12 major phylum present at four stages of degradation succession in the Napahai plateau wetlands (Fig. 4: P < 0.05). The composition of bacterial communities was dominated by average abundances of Acidobacteria (17.3%) and δ-Proteobacteria (14.6%). The common taxa were α-Proteobacteria (8.6%), Firmicutes (7.8%), β-Proteobacteria (7.3%), γ-Proteobacteria (6.1%), Actinobacteria (6.0%), Chloroflexi (6.0%), Bacteroidetes (5.4%), Verrucomicrobia (5.2%), Planctomycetes (4.1%), and Gemmatimonadetes (2.3%) (Fig. 4). These bacterial groups together accounted for 90.8% (on average) of the taxa obtained from soils in the four stages (Fig. 4).

Effect of wetland degradation on taxonomic composition of bacterial communities in the Napahai plateau. PW, SM, M, and RW were primary wetland, swamp meadow, meadow, and reclaimed wetland, respectively. PW1, PW2, and PW3: three replicated primary wetlands; SM1, SM2, and SM3: three replicated swamp meadows; M1, M2, and M3: three replicated meadows; RW1, RW2, and RW3: three replications of reclaimed wetlands
We observed that the relative abundances of bacterial taxa varied markedly along degradation succession (Fig. 4). In primary wetland, the average relative abundance of Acidobacteria was the lowest (8.5%), while that of δ-Proteobacteria was higher in initial (19.0% in primary wetlands) and transitional (21.7% in swamp meadows) stages than in the degraded stages (i.e., meadows and reclaimed wetlands). The abundances (8.2–9.6%) of β-Proteobacteria, γ-Proteobacteria, Chloroflexi, and Bacteroidetes were the highest in primary wetlands (P < 0.01), whereas ɑ-Proteobacteria (12.8%) and Gemmatimonadetes (3.6%) were the most abundant in reclaimed wetlands. The abundances of δ-Proteobacteria (21.7%) and Firmicutes (14.3%) were highest in swamp meadow, but those (7.5–25.6%) of Acidobacteria, Actinobacteria, Planctomycetes, and Verrucomicrobia were the highest in meadows.
Soil bacterial community was dominated by δ-Proteobacteria in primary wetland, while δ-Proteobacteria and Firmicutes were the dominate groups in swamp meadow (Fig. 4). The soil bacterial community was dominated by Acidobacteria, Verrucomicrobia, and δ-Proteobacteria in meadow, whereas Acidobacteria and a-Proteobacteria were the main groups in reclaimed wetland. The OTU richness and indices of Shannon, ACE, and Chao of bacterial communities were lower, but Simpson index was higher in primary wetlands than in transitional (swamp meadows) and degraded wetlands (i.e., meadows and reclaimed wetlands) (Table 2; P < 0.01). In conclusion, wetland degradation can exert a significant effect on relative abundance, composition, and diversity of soil bacterial communities.
Response of bacterial taxa to change in soil variables
Wetland degradation resulted in a significant alteration in soil variables (Table 2), which exerted a positive or negative effect on bacterial community composition (Fig. 5). Soil C/N and TP had positive association with the abundances of ɑ-Proteobacteria, Acidobacteria, Actinobacteria, Verrucomicrobia, Planctomycetes, and Gemmatimonadetes (r = 0.58–0.90; P < 0.05 and 0.01), while AP and AK were only positively related with those of Actinobacteria and Verrucomicrobia (r = 0.55–0.76; P < 0.05 and 0.01: Figs. 5 and 6). The abundances of Bacteroidetes and γ-Proteobacteria had positive responses to changes in soil pH and TK (r = 0.50–0.87; P < 0.05 and 0.01). The abundances of δ-Proteobacteria, β-Proteobacteria, Chloroflexi, and Bacteroidetes were positively correlated with the changes in SW, SOM, TN, and AN along the wetland degradation (r = 0.51–0.93; P < 0.05 and 0.01: Figs. 5 and 6). In primary wetland, soil pH and TK had main effects on Bacteroidetes abundances, while γ-Proteobacteria and Chloroflexi abundances were obviously affected by SOM, TN, and AN (Fig. 5). The abundances of Acidobacteria, Actinobacteria, and Verrucomicrobia were affected by C/N and TP in meadow and reclaimed wetlands (Fig. 5).

Redundancy analysis (RDA) showing effect of wetland degradation on bacterial community ordination based upon the relative abundance of bacterial taxonomic groups and measured soil properties. SW soil water, SOM soil organic matter, TN total nitrogen, C/N ration of C to N, AN available nitrogen, TP total phosphorus, AP available phosphorus, TK total potassium, AK available potassium. PW, SM, M, and RW were primary wetland, swamp meadow, meadow, and reclaimed wetland, respectively. PW1, PW2, and PW3: three replicated primary wetlands; SM1, SM2, and SM3: three replicated swamp meadows; M1, M2, and M3: three replicated meadows; RW1, RW2, and RW3: three replications of reclaimed wetlands

Adobe illustrator for correlation between soil properties and the relative abundance of soil bacteria phyla in the Napahai plateau wetlands
Soil variables had close associations with OTUs richness and diversity of soil bacterial communities during wetland degradation (Fig. 7). We observed a positive prediction for the changes in OTU richness of bacterial communities by TP (r = 0.67, P < 0.05), while a negative prediction by the concentrations of SW, SOM, TN, and AN (r from − 0.93 to − 0.98, P < 0.01: Fig. 7). The diversity (i.e., Shannon, ACE and Chao indices) of bacterial community was negatively related with SW, SOM, TN, and AN (r from − 0.86 to − 0.99, P < 0.01), but positively related with TP (r = 0.64–0.78, P < 0.05: Fig. 7). Simpson index was positively related with SW, SOM, TN, and AN (r = 0.95–0.99, P < 0.01), but negatively related with TP (r = − 0.72, P < 0.05). In particular, soil AP only had a positive association with Shannon diversity (Fig. 7; r = 0.64, P < 0.05).

Correlation matrix analysis showing association of bacterial diversity with measured soil properties during wetland degradation in the Napahai plateau. SW soil water, SOM soil organic matter, TN total nitrogen, C/N ration of C to N AN available nitrogen, TP total phosphorus, AP available phosphorus, TK total potassium, AK available potassium
Discussion
Effect of wetland degradation on soil variables
Understanding the process of wetland degradation is crucial for land management, as wetland diversity and functioning have been severely affected by global change and anthropogenic activities (Cui et al. 2018). The wetland degradation often results in soil degradation from protogenic swamp to degraded meadow, and even reclaimed wetland (Guo et al. 2013a, b). This deterioration can lead to a loss of soil humus and peat horizon, and a decrease in some soil nutrients, which affects bacterial diversity and ecosystem functions (Luan et al. 2014; Steinmuller and Chambers 2019). For this study, the pronounced decrease in soil organic matter and total nitrogen may be attributed to the fact that nutrient mineralization was promoted by increased soil permeability due to decreased soil and surface water content during primary wetland degraded into reclaimed land (Fang et al. 2020). In contrast, the conspicuous increase in concentrations of phosphorus and potassium may be owing to increased nutrient release through the mineralization of soil organic matter, as richer soil oxygen in degraded wetland can result in higher microbial activity compared with primary wetland (Bostic et al. 2010). The degradation of soil fertility can shape bacterial composition and diversity, as well as bacterial acclimatization and evolution (He et al. 2021; Yang et al. 2016). Therefore, information about alterations of soil variables along wetland degradation may be crucial for predicting the effect of wetland degradation on bacterial community.
Effect of wetland degradation on bacterial abundance composition
The wetland degradation often results in alterations of soil biogeochemical cycling (or soil properties), which can shape bacterial abundance composition. It may be attributed to the specific effects of soil variables on metabolic path and growth of soil bacteria (Buckley et al. 2006; He et al. 2021). For this study, different metabolic types of soil bacterial taxa had differed responses to the alternations in soil carbon and nitrogen availability during wetland degradation. The abundances of copiotrophic δ-Proteobacteria were higher in primary and transitional wetlands where there were higher concentration of carbon and nitrogen in soils. The Proteobacteria (e.g., Deltaproteobacteria and Betaproteobacteria) are often stimulated in nutrient-rich soil environment (Zhang and Xu 2008; Röskea et al. 2012). In contrast, oligotrophic Acidobacteria were more abundant in degraded soils where there were lower carbon and nitrogen concentrations, as they include many oligotrophs that prefer for less organic matter (Nemergut et al. 2010; Ling et al. 2021). Therefore, the change in soil nutrients can be important for predicting the shift in bacterial community along wetland degradation.
The alteration in water table during wetland degradation directly affects oxic-anoxic interfaces and indirectly changes the status of soil nutrients, which ultimately governs bacterial composition and abundance (Jaatinen et al. 2007; Tian et al. 2012). The primary wetland usually favors anoxic, neutrophilic, and copiotrophic bacteria, while the degraded wetland facilitates the development of oxic, eosinophilic, and oligotrophic bacteria (He et al. 2021; Zhang et al. 2020). This was why soil pH and total potassium had main contribution to the change in Bacteroidetes abundances, and soil organic matter and total and available nitrogen facilitated the development of γ-Proteobacteria and Chloroflexi in primary and swamp wetlands. In contrast, there was a greater contribution of carbon/nitrogen ratio to the abundances of Acidobacteria, Actinobacteria, and Verrucomicrobia in meadow and reclaimed wetlands. Furthermore, soil variables differed in effects on bacterial groups that had different behaviors of nutrient adsorption and immobilization during wetland degradation (Feng et al. 2020). The increase in concentrations of soil water, soil organic matter, and total and available nitrogen obviously facilitated the development of δ-Proteobacteria and β-Proteobacteria, through affecting the fixation pathways of autotrophic carbon and nitrogen (Jesser et al. 2013). In particular, β-Proteobacteria abundance can be stirred by soil phosphorus through increase of polyphosphate accumulations and ribosomal RNA synthesis (Liu et al. 2021). Therefore, wetland degradation differed in modifications on soil variables, which can exert specific effects on the abundance composition of soil bacterial communities.
Effect of wetland degradation on bacterial diversity
Wetland degradation had a crucial regulation on OTU richness and diversity of bacterial community. During the wetland degradation, the abundance of dominated taxa (e.g., Acidobacteria and ɑ-Proteobacteria) was decreased, which unexpectedly resulted in an increase in bacterial diversity. Some studies found that bacterial diversity decreased along wetland degradation, which may be closely linked with increased soil pH (Tang et al. 2012). In contrast, our results observed that the decreased bacterial diversity was closely related with the increase in concentrations of total and available phosphorus during wetland degradation. This is similar with some other studies that reported the association of total and available phosphorus with bacterial OTUs and diversity (Huang et al. 2016; Zhou et al. 2020). A number of mechanisms can explain this association. Soil microbes need a lot of phosphorus to meet their metabolisms and growths (Turner et al. 2013). Some studies documented that soil phosphorus directly accelerated the abundance of specific bacterial taxa through stirring the growth of their cells (Marks et al. 2019), which may affect bacterial diversity. Therefore, our results indicate that total and available phosphorus are good predictors for shift in bacterial diversity along wetland degradation. Our findings can be crucial in determining how the wetland degradation can affect soil bacterial community. Furthermore, the shift in specific bacterial taxa may be valuable in predicting the effect of wetland degradation on biogeochemical cycling of ecosystem.
Conclusions
Our results showed that wetland degradation changed bacterial community from copiotrophs to oligotrophs. The shift in abundance composition and diversity of soil bacterial communities was governed by specific soil variables. Wetland degradation severely decreased the concentrations (1.5–5.3 times) of soil water, soil organic matter, and total and available nitrogen, disfavoring the development of copiotrophic δ-Proteobacteria and Bacteroidetes. In contrast, wetland degradation strongly increased (1.1–2.8 times) the ratio of carbon to nitrogen, which increased the abundances of oligotrophic Acidobacteria and ɑ-Proteobacteria. Furthermore, the increases in total and available phosphorus were closely linked with increased OTU richness and diversity of bacterial communities along wetland degradation. Therefore, soil properties exert a crucial effect on nutrient metabolism and development of specific bacterial taxa, which can serve as crucial predictors of the shift in bacterial community composition and diversity.
References
Aíza LG, Ruiz BK, Low-Pfeng AM, Vallejos Escalona LML, García-Meneses PM (2021) Perceptions and sustainable actions under land degradation and climate change: the case of a remnant wetland in Mexico City. Environ Dev Sustain 23:4984–5003
Andersen R, Chapman SJ, Artz RRE (2013) Microbial communities in natural and disturbed peatlands: a review. Soil Biol Biochem 57:979–994
Bai XY, Wang SJ, Xiong KN (2013) Assessing spatial–temporal evolution processes of karst rocky desertification land: Indications for restoration strategies. Land Degrad Dev 24:47–56
Bostic EM, White JR, Corstanje R, Reddy KR (2010) Redistribution of wetland soil phosphorus ten years after the conclusion of nutrient loading. Soil Sci Soc Am J 74:1808–1815
Buckley DH, Huangyutitham V, Nelson TA, Rumberger A, Thies JE (2006) Diversity of planctomycetes in soil in relation to soil history and environmental heterogeneity. Appl Environ Microb 72:4522–4531
Cederlunda H, Wessén E, Enwall K, Jones CM, Juhanson J, Pell M, Philippot L, Hallin S (2014) Soil carbon quality and nitrogen fertilization structure bacterial communities with predictable responses of major bacterial phyla. Appl Soil Ecol 84:62–68
Chapman EJ, Hinsby Cadillo-Quiroz L, Daniel C, Merritt R, Turetsky MP, Waldrop, (2017) Soil microbial community composition is correlated to soil carbon processing along a boreal wetland formation gradient. Eur J Soil Biol 82:17–26
Cui LL, Li GS, Ouyang NL, Mu FY, Yan F, Zhang YT, Huang XY (2018) Analyzing coastal wetland degradation and its key restoration technologies in the coastal area of Jiangsu, China. Wetlands 38:525–537
Cui Q, Ammar ME, Iravani M, Kariyeva J, Faramarzi M (2021) Regional wetland water storage changes: The influence of future climate on geographically isolated wetlands. Ecol Indic 120:106941
Embarcadero-Jiménez S, Flor N, Rivera-Orduña Wang ET (2016) Bacterial communities estimated by pyrosequencing in the soils of chinampa, a traditional sustainable agro-ecosystem in Mexico. J Soil Sediment 16:1001–1011
Fang J, Deng Y, Che R, Han C, Zhong W (2020) Bacterial community composition in soils covered by different vegetation types in the Yancheng tidal marsh. Envir Sci Pollut R 27:21517–21532
Feng W, Li Y, Lin Z, Luo Y, Wang S, R, (2020) The influence on biosorption potentials of metal-resistant bacteria enterobacter sp. EG16 and bacillus subtilis DBM by typical red soil minerals. J Soil Sediment 20:3217–3229
Gu YF, Bai Y, Xiang QJ, Yu XM, Zhao K, Zhang XP, Li CN, Liu SQ, Chen Q (2018) Degradation shaped bacterial and archaeal communities with predictable taxa and their association patterns in Zoige wetland at Tibet plateau. Sci Rep 8:3884
Guo XH, Xiao DR, Tian K, Yu HZ (2013a) Biomass production and litter decomposition of lakeshore plants in Napahai Wetland, Northwestern Yunnan Plateau, China. Acta Ecol Sin 33:1425–1432
Guo XJ, Du W, Wang X, Yang ZF (2013b) Degradation and structure change of humic acids corresponding to water decline in Zoige peatland, Qinghai-Tibet Plateau. Sci Total Environ 445–446:231–236
He S, Hu W, Jin X, Han J (2021) Soil bacterial community composition and diversity respond to soil environment in the Ebinur Lake Wetland. Arch Microbiol 203:1175–1182
Ho J, Boughton EH, Jenkins DG, Sonnier G, Bohlen PJ, Chambers LG (2018) Ranching practices interactively affect soil nutrients in subtropical wetlands. Agr Ecosyst Environ 254:130–137
Hu YX, Huang JL, Du Y, Han PP, Wang JL, Huang W (2015) Monitoring wetland vegetation pattern response to water-level change resulting from the three Gorges Project in the two largest freshwater lakes of China. Ecol Eng 74:274–285
Huang JS, Hu B, Qi KB, Chen WJ, Pang XY, Bao WK, Tian GL (2016) Effects of phosphorus addition on soil microbial biomass and community composition in a subalpine spruce plantation. Eur J Soil Sci 72:35–41
Jaatinen K, Fritze H, Laine J, Laiho R (2007) Effects of short- and long-term water-level drawdown on the populations and activity of aerobic decomposers in a boreal peatland. Global Change Biol 13:491–510
Jesser KJ, Fullerton H, Hilton TS, Kimber J, Hager K, Moyer CL (2013) qPCR analysis of carbon, nitrogen, and arsenic cycling in Zetaproteobacteria-dominated microbial mats. AGU Fall Meeting Abstracts.
Koch AL (2001) Oligotrophs versus copiotrophs. Bioessays 23:657–661
Lamsal P, Pant K, Kumar L, Atreya K (2015) Sustainable livelihoods through conservation of wetland resources: a case of economic benefits from Ghodaghodi Lake, western Nepal. Ecol Soc 20:10
Ling L, Fu Y, Jeewani PH, Tang C, Pan S, Reid BJ, Gunina A, Li Y, Li Y, Cai Y, Kuzyakov Y, Li Y, Su W, Singhi BP, Luo Y, Xu J (2021) Organic matter chemistryand bacterial community structure regulate decomposition processes in post-fire forest soils. Soil Biol Biochem 160:108311
Liu Z, Shang H, Han F, Zhang M, Li Q, Zhou W (2021) Improvement of nitrogen and phosphorus availability by pseudoalteromonas sp. during salt-washing in saline-alkali soil. App Soil Ecol 168(6):104117
Lu RK (2004) Analysis Method of Soil Agricultural Chemistry. China Agricultural Science and Technology Press. Beijing
Lu M (2018) Effects of wetlands degradation on structure and diversity of soil microbial community in plateau wetlands. Beijing Forestry University, Beijing (Doctoral dissertation).
Luan JW, Cui LJ, Xiang CH, Wu JH, Song HT, Ma QF (2014) Soil carbon stocks and quality across intact and degraded alpine wetlands in Zoige, East Qinghai-Tibet Plateau. Wetl Ecol Manag 22:427–438
Mamo GS, Bekele T (2020) Review on wetland ecosystem destruction. Int J Sci Res Civ Eng 2(2):2456–6667
Mandal SD, Laskar F, Panda A.K, Mishra R (2020). Microbial diversity and functional potential in wetland ecosystems. In book: Recent Advancements in Microbial Diversity (pp.289–314).
Marks EAN, Montero O, Rad C (2019) The biostimulating effects of viable microalgal cells applied to a calcareous soil: increases in bacterial biomass, phosphorus scavenging, and precipitation of carbonates. Sci Total Environ 692(20):784–790
Nemergut DR, Cleveland CC, Wieder WR, Washenberger CL, Townsend AR (2010) Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest. Soil Biol Biochem 42:2153–2160
Panico SC, Ceccherini MT, Memoli V, Maisto G, Marco AD (2020) Effects of different vegetation types on burnt soil properties and microbial communities. Int J Wildland Fire 29(7):628–636
Romero-Trigueros C, Díaz-López M, Vivaldi GA, Camposeo S, Bastida F (2021) Plant and soil microbial community responses to different water management strategies in an almond crop. Sci Total Environ 778(1):146148
Röskea K, Sachse R, Scheerer C, Röske I (2012) Microbial diversity and composition of the sediment in the drinking water reservoir Saidenbach (Saxonia, Germany). Syst Appl Microbiol 35:35–44
Shen G (2019) Original articles remote sensing and evaluation of the wetland ecological degradation process of the Zoige Plateau Wetland in China. Ecol Indic 104:48–59
Shen CC, Ge Y, Yang T, Chu HY (2017) Verrucomicrobial elevational distribution was strongly influenced by soil pH and carbon/nitrogen ratio. J Soil Sediment 17:2449–2456
Steinmuller HE, Chambers LG (2019) Characterization of coastal wetland soil organic matter: implications for wetland submergence. Sci Total Environ 677:648–659
Stone BW, Li J, Koch BJ, Blazewicz SJ, Hungate BA (2021) Nutrients cause consolidation of soil carbon flux to small proportion of bacterial community. Nat Commun 12:3381
Tang J, Ding X, Wang LM, Xu QR, Yang ZR, Zhao J, Sun Q, Feng S, Zhang J (2012) Effects of wetland degradation on bacterial community in the Zoige Wetland of Qinghai-Tibetan plateau (China). World J Microb Biot 28:649–657
Tian JQ, Zhu YB, Kang XM, Dong XZ, Li W, Chen H, Wang YF (2012) Effects of drought on the archaeal community in soil of the Zoige wetlands of the Qinghai-Tibetan plateau. Eur J Soil Sci 52:84–90
Turner BL, Lambers H, Condron LM, Cramer MD, Leake JR, Richardson AE, Smith SE (2013) Soil microbial biomass and the fate of phosphorus during long-term ecosystem development. Plant Soil 367(1–2):225–234
Wagner M, Loy A (2002) Bacterial community composition and function in sewage treatment systems. Curr Opin Biotech 13:218–227
Xiao DR, Tian K, Yuan H, Yang YM, Li NY, Xu SG (2008) Distribution patterns and changes of aquatic plant communities in Napahai Wetland in Northwestern Yunnan Plateau, China. Front Biol China 3:338–343
Yang J, Ma L, Jiang H, Wu G, Dong H (2016) Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci Rep 6:25078
Zhang L, Xu ZH (2008) Assessing bacterial diversity in soil. J Soil Sediment 8:379–388
Zhang X, Li W, Zhong Z, Zhang Q, Yang G, Wang X, Han X, Ren C (2020) Response of soil microbial community to C:N: P stoichiometry along a caragana korshinskii restoration gradient on the loess plateau, china. Forests 11(8):823
Zhao XY, Yang JX, Bai SW, Ma F, Wang L (2016) Microbial population dynamics in response to bioaugmentation in a constructed wetland system under 10℃. Bioresource Technol 205:166–173
Zhou CF, Heal KV, Tigabu M, Xia L, Ma X (2020) Biochar addition to forest plantation soil enhances phosphorus availability and soil bacterial community diversity. Forest Ecol Manag 455:117635
Funding
Our study was supported by National Nature Science Foundation of China (42067011), Fundamental Research of Yunnan Province (202001AT070113), Yunnan Province Talent Reserve Project for training young and middle-aged academics and technology leaders; Fund Project for Doctor to start science research in Southwest Forestry University (111901).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Haroun Chenchouni
Rights and permissions
About this article

Cite this article
Lu, M., Li, C., Ren, Y. et al. Bacterial taxa have different responses to alterations in soil variables along a degradation gradient in the Napahai wetlands. Arab J Geosci 15, 607 (2022). https://doi.org/10.1007/s12517-021-09273-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12517-021-09273-8