
Abstract
Mangroves are tropical wetlands that are among the most productive ecosystems on Earth which cover up to 75% of the coastlines. Humic substances are the ever-present natural organic compounds, being a major component of organic carbon in the global carbon cycle. The present study focuses on the organic carbon dynamics as well as spectroscopic characterization (UV, FT- IR, and NMR) of humic substances—humic acids (HAs) and fulvic acids (FAs)—isolated from three tropical mangrove sediments representing different environmental settings belonging to Kerala, in the southwest coast of India during a premonsoon sampling conducted in April 2018. The study revealed a higher concentration of HAs than FAs in all the stations which was also complemented by a higher concentration of tannin and lignin as well as TOC. The E4/E6 ratios of FAs were also higher than that of HAs confirming its low molecular weight and less polymerized nature. The FTIR spectrum of both HAs and FAs showed peaks corresponding to the existence of carboxyl, phenol, carbonyl, and amide group. The proton NMR spectrum showed the presence of aliphatic regions slightly more controlled with long chains and/or alicyclic moieties rather than methyl groups which are also supported by the higher hydrogen content in HAs than FAs, and these peaks were absent in FAs. It also showed the presence of more or less common aromatic core involved in unsaturated structures as well as other aromatic groups like phenols and ionized carboxylic groups slightly higher in FAs than in HAs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mangroves or tropical tidal wetlands are among the most productive coastal habitats and dynamic zones of fishery potential areas (Kaladharan et al. 2003) and cover up to 75% of the coastlines (Pernetta 1993; Ranjan et al. 2010). The average productivity of mangroves is 2500 mg C day−1 (Kristensen et al. 2008) making them one of the most productive ecosystems on the earth (Rani et al. 2021; Bouillon et al. 2008; Jennerjahn and Ittekkot 2002). Due to high productivity and turnover rates, mangroves play a crucial role in the biogeochemical cycling of P, C, N, and other nutrients in coastal environments (Kristensen et al. 2008; Singh et al. 2005; Ranjan et al. 2010). Although mangroves cover only 0.1% of the continents’ surface, they can account for more than 10% of the dissolved organic matter transported to the oceans (Jennerjahn and Ittekkot 2002; Dittamar et al. 2006). Organic matter of both autochthonous and allochthonous origin derived from coastal wetlands, mangrove forests, riverine input, freshwater, and marine phytoplankton accumulates in these ecosystems (Ranjan et al. 2010). Mangroves act not only as a carbon source (carbon out-welling) but also as a carbon sink for the burial of mangrove assimilated carbon in sediments (Enog 1993; Zhang et al. 2011; Rani et al. 2021). The average sediment carbon density of mangroves swamps is 0.05 ±0.004 g/cm3 (Chmura et al. 2003; Ferreira et al. 2009; Zhang et al. 2011) contributing to a large pool of carbon which plays a major role in the global carbon biogeochemical cycle in the coastal system (Zhang et al. 2011). The studies dealing with soil organic matters in mangrove sediments are limited (Sierra et al. 2004; Ferreira et al. 2009; Ranjan et al. 2010; Zhang et al. 2011). It was considered by high liquid content and low solid content. The large number of holes that were occupied air, water, gas (Matsui et al. 2015) and is going to limit the biogeochemical processes involved in sedimentary organic matter in mangrove areas (Zhang et al. 2011). The elevated organic carbon production of mangrove wetlands influenced the accumulation of this aliphatic rich humic-composition. As the chief constituent of soil organic matter, humus is the central exporter of fractional carbon and nutrient pools, and the degree of humification is one of the significant features disturbing the build-up of soil organic matter in forest biomes (Kwiatkowska-Malina 2018; Yang et al. 2019).
Humic substances (HS) are the most well-known and ever-present natural non-living organic materials in aquatic and terrestrial environments, and characterize a major proportion of the organic carbon in the global carbon cycle (Allard et al. 1991; Giovanela et al. 2004; Yang et al. 2019). HS constitutes a major fraction of the soil organic matter (up to 80%) and the largest fraction (up to 60% of the dissolved organic carbon) of natural organic matter in aquatic systems (Stevenson 1994; Giovanela et al. 2004; Fernandes et al. 2010; Sarlaki et al. 2020). The properties and structure of HS may vary significantly, depending on the characteristics of the water or soil, of the compounds of origin, maturation of the HS, and specific conditions of extraction (Fong and Mohamed 2007; Garcia et al. 2016; Muscolo et al. 2013; Rigobello et al. 2017). Humic substances originate from the decomposition and further subsequent polymerization of organic deposits of plant and animal origin leading to heterogeneous supramolecular species with large molecular weights (Reddy et al. 2014; Carlose et al. 2011; Martin et al. 2014) being polydispersive molecules with different functional groups, the elemental configuration and properties of which differ rendering to the source of organic matter and the surroundings in which it is designed (Rashid 1985; Fernandes et al. 2010). Conversely, the richness of carboxylic and phenolic substituents is associated with terrestrial contributions, demonstrating that lignins and, perhaps, tannins play a significant role in the construction of humus in continental settings (Stevenson 1994; Fernandes et al. 2010). Generally, the degradation of humic substances give rise to their decolorization, and the constitutions of humic substances with a lighter color change gradually (Ikeya et al. 2015;Yangi and shindo 2016;Yang et al. 2019). Based on their solubility, HS are classified into three main fractions, humic acids (HA), which are soluble in alkali, but insoluble in acid; fulvic acids (FA), soluble in alkali and acid; and humins, insoluble in both (Rigobello et al. 2017). These fractions vary significantly in molecular size and functional group content, the main functional groups being carboxylic and phenolic along with minor amounts of other groups like amino acids (Cezikova et al. 2001; Anuradha et al. 2011), and HAs are generally larger than FA (Samios et al. 2007; Fong and Mohamed 2007; Rigobello et al. 2017).
No single analytical tool, however, can provide definitive structural or functional information about the natural organic matter because of its heterogeneous, ill-defined nature (Chen et al. 2002). Spectroscopic techniques such as nuclear magnetic resonance (NMR), Fourier-transform infrared (FTIR), electron paramagnetic resonance (EPR), ultraviolet-visible (UV/Vis), and fluorescence have been applied for both quantitative and qualitative characterization of NOM (Stevenson and Goh 1971; Schnitzer and LeVesque 1979; Schnitzer and Preston 1986; Leenheer et al. 1987; Sensi et al. 1989; Sensi et al. 1991; Gu et al. 1994; Chen et al. 2002; Anuradha et al. 2011; Prashob Peter 2014; Rigobello et al. 2017; Jennees et al. 2019). The objective of this study is the isolation and characterization of humic substances such as humic acid and fulvic acid from three different mangrove ecosystems belonging to the state of Kerala situated along the southwest coast of India. This study makes use of sp\ectroscopic techniques as well as the elemental composition of isolated humic materials from these diverse mangrove environments situated in a tropical region like Kerala to get an idea about the structural characteristics of isolated materials concerning environmental settings.
Materials and methods
Study area and sampling
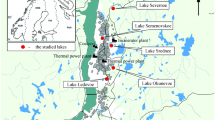
Mangroves in India account for about 5% of the world’s mangrove vegetation which expands over an area of about 4500 km2 beside the coastal States/UTs of the country (Mini et al. 2012). Mangroves are highly localized in Kerala, but the species diversity and their associates are comparatively rich and are confined to estuaries, lagoons, backwaters, and creeks. In Kerala, mangrove forests that once occupied about 700 km2 have now dwindled to 17 km2; the vegetation has diminished in its extent drastically and has acquired a “threatened” status in Kerala (Basha 1991, 1992). In Kerala, the maximum extent of mangroves is reported from Kannur district, belonging to Northern Kerala (1100 ha.), followed by Ernakulam (600 ha.), and the minimum extent is reported in three districts, Malappuram (26 ha.), Thiruvanthapuram (belonging to southern Kerala as well as the state capital (28 ha.), and Thrissur (30 ha.) (Vidyasagaran and Madhusoodann 2014). Figure 1 represents the location map of the study area.

Map showing of sampling area
In the present study, mangrove sediments were collected from three locations by geographically dividing it as the northern, central, and southern part of Kerala and also based on the mangrove area covered by the respective stations, northern side maximum, followed by central and the southern side with minimum mangrove cover. Station 1 (Dhrmadam) is located in Kannur district belonging to Northern Kerala between 11° 47′ 24.27795″ N latitude and 75° 27′ 51.43521″ E longitude. Dharmadam is on the estuary of the Anjarakkandy River, which surrounds the island on three sides (the Laccadives Sea is on the fourth). Station 2 is Mangalavanam located in Ernakulum district between 9° 59′ 133″ N latitude and 76° 16′ 26″ E longitude, and in this study, it is taken as a representative of the central region of Kerala. This mangrove is an ecologically sensitive area situated at the center of the city of Kochi and covers about 2.74 hectares. This acts as a nesting ground for a large variety of migratory birds and also supporting a variety of mangroves and is often regarded as the “green lung of Kochi” since it plays a pivotal role in controlling the air pollution of Kochi city.
Station 3 is Asramam Adventure Park situated in Kollam district belonging to southern Kerala, near the backwaters of Ashtamudi Lake (between 8° 53′ 7560″ N Latitude and 76° 35′ 1151″ E longitude). In addition to being a major spawning ground for several edible marine species, the Asramam mangroves are also home to otters as well as several migratory birds.
In this study, mangrove sediment samples were collected during the pre-monsoon season (April 2018) from the three geographically distinct regions of Kerala. The surface sediment samples were collected from each station using a pre-cleaned plastic scoop. Core samples were collected from these sites using PVC pipes with 75-mm diameter and 1.5-m length. The core samples were divided into two parts such as the surface sediments and the core sediments representing 1-m depth on each sampling site. The samples were carried to the laboratory in an icebox and kept in a deep freezer at −200 °C until analysis. The samples were freeze-dried for further analysis.
Analytical methods
Extraction of humic substances
The humic substances were isolated using the procedure IHSS 2010. In this method, 200 g freeze-dried sediment was washed with 1 l of 0.1 M HCl to discard weakly bound molecules like hydroxides, carbonates, and sulfates. The sediment/HCl mixture was continuously stirred and allowed to settle. The supernatant was then separated. One liter of 0.1 M NaOH was added to the sediment mixture. For the next 24 h, it was kept on a rotatory shaker. The dark-colored extract was separated from sediment by centrifugation at 10,000 rpm for 30 min. 6 M HCl was then added to it and the pH was adjusted between 1 and 2. The precipitate thus obtained was maintained at −4 °C for 16 h. The precipitate was then separated again by centrifugation for 10 min at 10,000 rpm. The obtained precipitate of humic acid (HA) was freeze-dried for further analysis while the supernatant was used for the analysis of fulvic acids (FAs).
The above acid-soluble supernatant containing FA solution was allowed to pass through a XAD-8 resin previously cleaned and then desalted by passing Milli-Q water and ensured it was chloride-free by testing with 0.1 M AgNO3. To desorb the absorbed FA, 0.1 M NaOH was passed through the column and then the eluate was passed through a previously cleaned cation-exchange resin (Collins et al. 1971). The FA solution was freeze-dried for further studies.
Characterization of isolated HAs and FAs
The characterization of isolated HAs and FAs by the above methods was done by using UV-visible, FTIR, 1H NMR spectroscopic methods, CHNS elemental analysis, and TOC.
The total organic carbon (TOC) in the samples was analyzed by direct analysis done with the aid of the PRIMACSMCS TOC analyzer of SKALAR. The UV-visible spectroscopic analysis was done with the Thermo Fischer Model No 117. For this, 2 mg humic substance was dissolved in 10 ml, 0.05 N sodium bicarbonate of pH 8, and observed the optical densities at 465 and 665 nm and hence the E4/E6 ratio was determined (Chen et al. 1977). The E2/E3 ratio (optical densities at 250 and 365 nm (Chen et al. 1977)) was calculated by dissolving 2 mg humic substance in 10 ml of 0.05 N sodium hydroxide followed by determining optical densities at 270 and 407 nm.
C, H, N, and S compositions were determined by direct analysis on an ash-free basis with the aid of the CHNS Elemental analyzer of Elementar Vario ELIII. The oxygen level was calculated using the formula 100 − (sum of the percentages of CHNS).
FTIR spectra of HAs and FAs are documented using Avatar 370 (FTIR spectrometer) with a high resolution of 4 cm−1 obtained from the mid-infrared region (4000–400 cm−1). The samples were prepared to eliminate excess moisture; aliquots of humic substance (HAs and FAs) were placed over P2O5 at 45 °C overnight. Four hundred milligrams of spectroscopic grade KBr was mixed with 30 mg dried humic substance samples (HA and FA), and then the combination was squeezed to form pellets. The spectra were picked up with high resolution.
1H NMR spectroscopic characterization was done using TMS, and the signals were determined with Bruker Advance 400 MHz NMR spectrometer fitted with QNP 1H probe.
The tannin and lignin concentrations were estimated by using the standard procedure adopted by the sodium tungstate phosphomolybdic acid method (APHA 1995). The textural characteristics of the sediments were analyzed by using a particle analyzer, LISST-PORTABLE | XRD (SEQUOIA).
Results and discussions
Quantification of humic substances
In this study, the quantities of humic acids and fulvic acids isolated were higher in the surface compared to the bottom sediments (Table 1). The surface samples of station 1, taken as a representative of central Kerala mangroves, showed (12.53 g) higher concentration of HAs compared to the other stations as well as depth. This was almost double than that of isolated from the surface samples stations 1 (5.62 g) and 3 (5.32 g). Station 3, representing southern Kerala, showed slightly lower quantities of HAs for both surface (5.32 g) as well as bottom (1.02 g) sediments. The amount of fulvic acids isolated from all the three stations was much lower than that of the HAs. Though not much different, the quantity of isolated FAs followed the order station 2 (central) > station 1 (northern) > station 3 (southern).
Station 2 was characterized by the higher amount of HAs compared with other stations maybe because of the rich organic content provided by the migratory birds and plant foliage that are plenty in this bird sanctuary. This mangrove forest is currently a site that is being highly protected for preservation. The large quantities of humic substances are isolated from areas characterized by high decomposition from plant foliage, microbial action, and soil condition, for example, pH and temperature (Swift et al. 1979). This station was noticed by a higher percentage of TOC, i.e., 4.9399% for surface and 3.1768% for bottom sediments (Table 2) compared to the other two stations which may be due to extraordinary primary production , the elevated contribution of autochthonous sedimentary organic matter, and also high sedimentation rate (Marchand 2017; Rani et al. 2021).
Station 2 was also noted for its higher percentage of silt (more than 50% for the surface as well as the bottom) and a significantly higher proportion of tannin and lignin compared to the other stations (25.03 mg/g for surface and 18.69 mg/g for bottom) (Table 2). In station 3, the quantity of humic substances isolated was less and it is also supported by low TOC (3.03% in surface and 0.002% in the bottom) as well as predominantly sandy sediment texture ( > 80% was sandy for the surface as well as the bottom). This is due to the improvement of mud in the mangroves area over a long period in dissimilar stages under tidal effects and deposition of wind instinctive material. Maybe at the previous stage, there was high involvement of sand deposits (from sea and wind) and then the coarser material transformed to finer material through physical weathering or the settling of fine particles at the surface through sedimentation in suspension (Alsumaiti and Shahid 2018; Suthhof et al. 2000). The association of organic matter with fine-grained sediment is well established (Nair et al. 1993; Sarala Devi et al. 1995; Hedges and Keli 1995).
The surface samples of all three stations are noted for the higher TOC values (Table 2) which may be due to the decomposition of dead organisms and mangrove detritus, domestic sewage, and anthropogenic inputs. The high levels of organic carbon in mangrove sediments are due to decomposition of dead organisms, decomposition of mangrove detritus, and anthropogenic inputs, particularly oil spills, sewage, etc. which are transported by tides and strand on low energy ecosystems such as mangrove forests (Rini 2002). The TOC content demonstrated a sharp decrease from top to bottom which recommended forceful mineralization (Bouillon et al. 2004; Manju et al. 2020). In station 3, tidal flushing might have removed the organic matter from the surface sediment and sandy nature allows the decomposition to takes place more rapidly allowing the microbes and oxygen to penetrate the sediment, hence resulting in a comparatively low percentage of TOC (Table 2).
Tannin and lignin, familiar aromatic polycyclic phenolic compounds biosynthesized by complex plants (Hernes and Hedges 2000), have been introduced into an aquatic environment through terrestrial runoff (Kalesh et al. 2001). The tannin content of mangrove leaves is higher and more polymerized than that found for 40 other dicotyledonous plant species (Hernes et al. 2001). Lignins have been implicated as major source material for terrestrial humic substances (Flaig et al. 1975; Kalesh et al. 2001). Mangrove species exhibit a typical vascular-plant lignin signature, with great variations between leaves and wood, the latter being richer in lignin oxidation product (Kristensen et al. 2008). They act as a major fraction of refractory organic matter, and its quantitative resolution offers valuable information on the input of terrestrially derived organic detritus into the sediments (Manju et al. 2016). In this study, the tannin and lignin concentrations (mg/g) varied from 8.99 to 25.03 which showed a higher concentration in the surface than the bottom sediments (Table 2). The majority of the species of mangrove leaves contained a greater amount of tannin and lignin during pre-monsoon (Katherisan and Veera 1990). Since these leaves are the main source, its concentration was maximum in the system during this season (Geetha 2002). The maximum amount of tannin and lignin was noted during pre-monsoon in selected mangrove ecosystems of Kerala, especially from Mangalavanam (station 2 in this study) (Geetha et al. 2008). Rhizophora mucronata is profusely distributed in Kannur and Ernakulam districts whereas it was not seen in Kottayam and Trivandrum districts (Vidyasagaran and Madhusoodann 2014). Mangroves, especially those belonging to the Rhizophoraceae family, contain a large amount of tannins (Bask et al. 1999; Hernes and Hedges 2004; Maie et al. 2008).
Elemental analysis
Tables 3 and 4 display the elemental composition (CHNS %) as well as the elemental ratios of humic acids and fulvic acids respectively of the isolated samples under study.
The content of C, H, O, N, and S provides essential information on the origin of samples. The elemental ratios like H/C, O/C, and C/N indicate the structure and molecular shapes of isolated humic substances. Atomic ratios (H/C and N/C) have been preferentially employed to establish the organic matter source, degree of condensation, diagenetic transformation, and environmental conditions under which the humic substances are generated (Stuemer et al. 1978; Rice and Mac Carthy 1991; Meyers and Ishiwatari 1993). Marine sediments are normally less aerated than superficial sediments and have an aliphatic and nitrogen-rich source of organic matter, and produce HS with high H/C and N/C ratios (Rashid 1985; Rice and Mac Carthy 1991). Due to the large inputs of terrestrial plants, terrestrial samples are rich in lignin moieties presenting, consequently, low H/C and N/C ratios (Stuemer et al. 1978).
While observing the elemental composition of isolated HAs, it is seen that in most of the stations, the percentage of carbon was higher than that of oxygen. In the bottom soils of stations 1 and 3, the percentage of oxygen was higher than that of carbon. Next to carbon and oxygen, the percentage of hydrogen was higher in all the samples of HAs. The percentage of sulfur was higher than that of nitrogen especially in the samples of station 3 (both surface and bottom) and also in the bottom sediments of stations 1 and 2 (Table 3). While discussing the percentagewise composition of elements in fulvic acids, it is seen that the highest percentage was for oxygen, followed by carbon, hydrogen, sulfur, and nitrogen (Table 4). The values obtained are similar to those attained by previous researchers (Rice and Mac Carthy 1991; Giovanela et al. 2004; Anuradha et al. 2011; Prashob Peter 2014).
When the sample is aliphatic than aromatic, it is generally observed that the hydrogen content will be higher (Traina et al. 1990). In the samples collected, the humic acids showed higher hydrogen content, indicating the aliphaticity of the isolated humic acids (Bravard and Richi 1991; Rigobello et al. 2017). An increase in hydrogen content indicates a greater number of aliphatic carbons (CH 2) than the aromatic carbons (C=C) (Traina et al. 1990). The carbon content was also observed to be higher in humic acids compared to fulvic acids. On the other hand, the oxygen content was greater in fulvic acid when compared to humic acid (Tables 3 and 4). HAs contained more carbon than nitrogen; the formation of FA is accompanied by the loss of carbon and nitrogen and the gain of oxygen (Schnitzer 1999). The percentage of nitrogen was higher in HAs than FAs (1.01–4.6% in HAs and 0.65–1.92% in FAs) which can be attributed to the fact that unlike FA they are not completely separated from the more hydrophilic and possibly nitrogenous compounds for example proteins and amino acids, by the adsorption step onto XAD-8 resins (Giovanela et al. 2004).
While discussing the elemental ratios, the H/C ratio of the fulvic acid was found to be lower (0.14–0.49%) than that of humic acid (0.14–1.14%), indicating that humic acids had more aromatic fractions and contained a large fraction of unsaturated structures (zhang et al. 2011; Rigobello et al. 2017; Abakumov et al. 2018). Prashob Peter (2014) reported an H/C ratio of 1.1 in Kochi mangrove sediments. Low H/C values support lignin mineralization rather than humification, whereas, increased O/H values support accelerated humification (Anuradha et al. 2011). In this study, the reported range of O/C was 0.5369–3.39 in HAs and 1.15–7.80 in FAs (Tables 3 and 4), and Prashob Peter (2014) reported O/C ratio of 0.566 in HAs of Kochi mangroves that is almost similar to our values reported from station 2 which is a Kochi mangrove (Mangalavanam). The O/C ratios are low in humic acids when compared to that of fulvic acids, which indicates the higher humification of the complex (Bravard and Richi 1991; Giovanela et al. 2004; Sierra et al. 2004). The O/C atomic ratio is related to the carbohydrate and carboxylic acid content (both aromatic and aliphatic) and degree of oxidation of HS. The lower values of this ratio indicate a higher degree of humification due to the reduction in the carbohydrate content in the O-bearing structures (polysaccharides) ( Prashob Peter 2014). The N/C and H/C ratio in samples from all stations in HAs and FAs was found to be low, indicating the geographical location as “terrestrial” due to high lignin mineralization (Stuemer et al. 1978; Giovanela et al. 2004; Rigobello et al. 2017).
The high C/N ratios have a large contribution to humification resulting from the decomposition of vascular plants (Rocha et al. 2007). Humification is prominent in static mangrove sediments where organic molecules are plenty in the form of leaf litter and organic wastes like bird excreta such as in the case of station 2 in this study. Mangalavanam is a tiny forest and a shelter of several exotic and more varieties of migratory birds of different extremes (Anuradha et al. 2011). The C/N ratios of HAs and FAs reported are comparatively higher (10.03–39.65 for HAs and 15.87–42.17 in FAs). In stations 1 and 2, C/N ratios are higher for the bottom samples than the surface in both HAs and FAs (Tables 3 and 4). Low C/N ratio indicates that N is preferentially mineralized relative to carbon (Resmi 2004). Organic compounds rich in nitrogen (low C:N ratio) favor net mineralization whereas those poorer in N (high C:N) favor net immobilization (Resmi 2004). Nonvascular aquatic plants contained a low C/N ratio, normally among 2.0 and 10, while vascular land plants, which enclose cellulose, lignin, and tannins, have ratios of 20 and upper (Giovanela et al. 2010). The C/N ratios in this study varied from 10.03 to 39.65 in HAs (Table 3) and 15.87 to 42.17 (Table 4) in FAs. These values are comparatively high, which designates that terrestrial vascular plants are the main sponsors of these samples.
The contribution of lignin moieties to the HAs can be inferred from the N/C ratio (Sainto and Hayano 1981). HAs with lower N/C ratios are generally rich in lignin moieties (Stuemer et al. 1978; Sierra et al. 2004). The N/C ratios reported in this study are in the range 0.0252–0.0996 for HAs (Table 3) and 0.04–0.189 for FAs (Table 4), and Prashob Peter (2014) reported N/C ratio of HAs as 0.074 for Kochi mangrove sediments and our study reported N/C ratio of 0.0851 in station 2, which is a mangrove from Kochi (Mangalavanam). A higher N/C ratio in terrestrial humic acids suggests the presence of vanillyl and syringyl acids, aldehydes, and ketones, along with cinnamic compounds (p- coumaric and ferulic acids) (Ertel and Hedges 1984; Lallier et al. 2008). The most likely occurrence of varying degrees of vascular lignin moieties and non-vascular lignin moieties is confirmed by higher C/N ratios (Prashob Peter 2014).
UV-visible spectroscopic methods
The ratio of the absorbance of dilute aqueous humic substance solutions at 465 and 665 nm is extensively used for the determination of the characterization and to evaluate the degree of humification and investigate the aliphatic or aromatic nature of the humic substance (Prashob Peter 2014). This ratio, which is discussed as the E4/E6 ratio, is originated to be independent of the concentration of humic materials, but it varies for humic materials, extracted from diverse soil types (Kononova 1966; Schintzer and Khan 1972). E4/E6 ratio (condensation degrees) designed from the investigational data is used to clarify the rigidity of the condensed aromatic rings in HS (i.e., degree of aromaticity), particle size, molecular weight, and acidity (Chen et al. 1977; Uyguner and Bekbolet 2004). Many of these complexes are held to be joint structural subunits in humic matter and E2/E3 as absorbance ratios measured at 250 and 365 nm resemble the correlation of molecular size and aromaticity (Peuravuori and Pihlaja 1997).
The E4/E6 ratios and E2/E3 ratios of isolated HAs and FAs are displayed in Table 5. The E4/E6 ratio, i.e., a decrease in absorptivity with an increase in wavelength, characterizes the changing content of aliphatic and aromatic fragments existing in the HAs. The E4/E6 ratio of HAs was in the range varying between 3.54 and 11.5 and that of FAs was in the range varying between 5.97 and 18.61. These values lie in the upper side of the known range (3–5.9) (Kononova 1966; Chen et al. 1977) and recommended the occurrence of inflexible aromatic core in the isolated humic substances, and are more in FAs than HAs. The E4/E6 ratios of fulvic acid are usually higher than the HA, to associate the fact that the previous ones are with low molecular weights and are less polymerized. Comparatively extensive ratios of FAs than those of HAs reproduce a low degree of aromatization and the presence of a relatively large proportion of aliphatic structures in FAs (Stevenson 1994). Fulvic acid had a higher E4/E6 ratio in comparison to humic acid. It could be due to the higher degree of aromaticity in carbon atoms of humic acid (Eshwar et al. 2017) supported by Tahiri et al. 2016), Srilatha et al. (2013) and Satisha and Devarajan (2011). The low H/C values (Table 3 and Table 4) indicate greater aliphaticity and lesser content of aromatic structures in humic substances (Rigobello et al. 2017, Amir et al. 2004), and therefore, its decrease infers an increase in the degree of humification. Stevenson (1994) displayed that absorbance at wavelength 465 nm is the same as light absorption of components related to the first phases of the humification process (young humic substances). Light absorption at 665 nm is related to healthy humified components. Small E4/E6 ratio values (< 4) show high (HS) quality. In contrast, a high E4/E6 ratio of pure humic acid shows that the molecule is smaller in size and has low molecular weight and C content, rich in O content, COOH group and total acidity and assumes the occurrence of comparatively large proportions of aliphatic structures (Stevenson 1994; Prashob Peter 2014). This is also supported by the fact that HAs in this study presented lower O/C values than the FAs. But, for all samples, the HA fraction offered values greater than 0.5, suggesting the presence of a larger amount of oxygenated functional groups such as carboxylic and carbohydrates (Tan 2014).
The surface sample gives a higher E4/E6 ratio than the core samples (Table 5) in both HAs and FAs. This is because the surface samples were of young origin, whereas the bottom regions are more ancient due to higher microbial activity and decomposition (Stevenson 1994; Zeck et al. 1997). While comparing among the stations, E4/E6 ratio in station 3 was slightly less than that of stations 1 and 2. Since E4/E6 is an index of the maturity of the HAs, the low E4/E6 ratio indicates a lesser degree of humification and maturity (Yavmetdinov et al. 2003). The reduced free acidic groups in the isolated compounds can also lead to increased ratios (Prashob Peter 2014). A high E465/E665 ratio (or a rapid decrease in absorptivity with an increase in wavelength) may result primarily from the absorption by ketonic C=O functional, and on the other hand, a low E465/E665 ratio may be largely attributed to the absorption by aromatic C=C functional groups (Prashob Peter 2014). Additionally, a high degree of condensation of the aromatic rings and the large molecular weight of SHA are believed to contribute to its relatively high absorption in the visible range (Schintzer and Khan 1972). Typically E4/E6 is larger for non-humified material by the presence of proteins and carbohydrates, which increase the absorptivity at the UV region of the spectrum (Faustino et al. 2009). These results are in line with those of Haddad et al. (2015), Petrus et al. (2009), and Banik and Sanyal (2006). Higher E2/E3 ratios are normally connected with lower molecular weight and a lesser degree of aromaticity (Vergnoux et al. 2011). The E2/E3 ratios varied from 0.53 to 1.96 in HAs and 0.36 to 0.66 in FAs, besides, to associate the fact that FAs have more aromatic nature than HAs which is also confirmed by E4/E6 ratios. To determine the aromaticity of the molecule, an equation proposed by Peuravuori and Pihlaja (1997) was used, in which aromaticity = 52.5 − 6.78 E2/E3.
FTIR spectroscopic studies
Fourier-transform infrared spectroscopy has been commonly used for the description of hydroxyls, aromatic and aliphatic carboxyls and carbonyls, aliphatic C-H and amides that are typical to HAs (Stevenson 1994; Amir et al. 2004; Sierra et al. 2004), which can offer considerable evidence about the structural as well as functional group properties of these compounds. Irrespective of their origin, all FAs and HAs isolated in this study offered nearly similar spectra maybe because of the presence of polar chemical functionalities absorbing infrared radiation such as phenolic and alcoholic hydroxyls, aromatic and aliphatic carboxyls and carbonyls, aliphatic C-H and amides, in all the HAs and FAs, irrespective of their origin (Giovanela et al. 2004). Such explanation is in line with the conceptions attributed to humus mixtures, which around that it consists of associations of molecules, with a “universal average formula unit,” held together via supramolecular interactions (Schintzer and Khan 1972; Piccolo et al. 1996; Sein Jr et al. 1999; Cozzolino et al. 2001; Giovanela et al. 2004).
The FTIR spectral characteristics of the isolated humic acids and fulvic acids are given in Fig. 2 (for HAs) and Fig. 3 (for FAs). A broad, high-intensity band at 3400 cm−1 displays the occurrence of H-bonded –OH groups of alcohols and phenols as well as hydrogen-bonded N - H groups (Amir et al. 2004; Sierra et al. 2004; Stevenson 1994). These peaks are evident in the FTIR spectrum of isolated humic acids and fulvic acids of this study. In the 1600–1720 cm−1 region, two peaks were detected: a fewer intensive absorption band around 1720 cm−1 which has been recognized to carbonyl groups in aldehydes, ketones, and carbonic acids, and a sharp high-intensity absorption band about 1635 cm−1 which has numerous assignments with aromatic –C=C– vibrations, –C=O stretching of amide groups (amide I band), strongly H-bonded –C=O of quinones, and/or H-bonded and conjugated carbonyl –C=O (Stevenson 1994; Smid et al. 2015). The absorbance at these wavelengths recommends the existence of extremely conjugated delocalized π electron through condensed double bond character (–C=O) of quinones (Silverstein et al. 1991). The peak around 1635 cm−1 is existing in all the samples in both isolated HAs and FAs.

FTIR spectrum of HAs. a S1. b S2. c S3

FTIR spectrum of FAs. a S1. b S2. c S3
Absorption bands of nitrogen-containing functional groups in HAs were perceived in the FTIR absorption region between 1570 and 1510 cm−1 (Orlov 1985). The occurrence of protein-like fractions was characterized by two peaks, one at 1660–1630 cm−1 (C=O stretching of amide groups) and additional at 1560–1510 cm−1 (N - H deformation and C ≡N stretching of amide groups) (Zhang et al. 2011). Peaks nearby 1590 cm−1 are realized in the isolated FAs only in station 1 (surface and bottom), station 2 (bottom), and station 3 (surface). In the 1400–1500 cm−1 range, peaks characterize OH deformation and CO stretching of phenolic OH, C-H deformation of CH 3 and CH2 groups and C-O – O- (Sierra et al. 2004; Stevenson 1994). These peaks are similarly joint in both isolated FAs and HAs in this study, but in station 1 bottom, station 3 (surface and bottom) for HAs and station 2 (surface and bottom) for FAs. The peak at 1400–1390 cm−1 (COO -1 antisymmetric stretching) and the peak around 1220 cm−1 (C- O stretching and OH deformation of COOH) designated the occurrence of a huge quantity of carboxylic acids in both the FA and HA (Zhang et al. 2011). Peaks conforming to this were further shared in FAs in this study than the HAs. This was also reinforced by the higher percentage of oxygen in FAs than HAs (Tables 3 and 4).
Peaks in the range 1100–1225 cm−1 are distinguished from the second overtone C-H stretching, NH3+ of lysine rocking, and occurrence of cellulose (Sierra et al. 2004; Adani et al. 2006). In the isolated FAs, sharp peak at 1120 cm−1 was perceived in all the samples from the three stations. In the isolated HAs, sharp peaks in this range were realized only in station 2 (surface along with bottom) and station 3 (surface in addition to bottom). Numerous earlier research work on humic substances has attained comparable results (Jennees et al. 2019, Prashob Peter 2014, Moreda–Pineiro et al. 2004, and Giovanela et al. 2004). The FTIR study clearly showed the existence of hydroxyl, methyl, methylene, carbonyl, carboxyl, phenol, alcohol, and amide groups in the humic substances isolated from sediment samples collected from different environmental settings.
1H NMR spectroscopic studies
The 1H NMR spectra of isolated humic acids and fulvic acids are given in Fig. 4 (for HAs) and Fig. 5 (For FAs). Resonances in the range 0.65–1.75 ppm arise from protons on terminal methyl groups of methylene chains and methyl groups of extremely branched alkyl groups and alicyclic compounds (Francioso et al. 2001; Graham et al. 2002). In this study, the 1H NMR spectrum of isolated HAs exhibited resonance peaks in this range for the surface as well as bottom sediment samples. In the surface sample of station 1, resonance peaks are detected at 0.842, 1.147, 1.227, and 1.470 whereas in the bottom sediments two resonance peaks were observed at 0.732 and 0.838. In station 2, in surface peaks are observed at 0.827, 1.149, 1.229, and 1.480. In this station, the peaks were obtained at 0.832, 0.870, 1.155, 1.224, and 1.646 in the bottom sediment sample. In station 3, surface sediment showed peaks at 0.791, 0.823, 1.144, and 1.222, whereas, in bottom sediment sample peaks were displayed at 0.842, 1.147, 1.227, and 1.470. Peaks in the range 1.10–1.40 are suggestive of methyl groups of highly branched aliphatic structures, and those in the region 1.4–1.65 designate the occurrence of methylene protons of alicyclic compounds. Amount of peaks further than 1.40 in station 1 (surface), station 2 (surface), station 2 (bottom), and station 3 (bottom) displays the occurrence of aliphatic regions slightly more controlled with long chains and/or alicyclic moieties rather than methyl groups (Hatcher et al. 1981; Graham et al. 2002). This is also supported by the increased hydrogen content in the isolated HAs than FAs which shows the occurrence of more number of aliphatic carbons (CH2) than aromatic carbons (C = C) (Traina et al. 1990; Prashob Peter 2014).

1H NMR spectrum of HA. a S1. b S2. c S3

1H NMR spectrum of FA. a S1. b S2. c S3
Aliphatic protons adjacent to carbon atoms together with highly electronegative atoms (O or N), unsaturated groups or aromatic groups, are usually seen in the range 2.00–3.00 (Sainto and Hayano 1981; Francioso et al. 2001; Graham et al. 2002; Qi et al. 2004). Peaks corresponding to this are observed at 2.175 and 2.5 (station 1 surface), at 2.5 (station 1 bottom), at 2.18 and 2.5 (station 2 surface), at 2.5 (station 2 bottom), at 2.5 (station 3 surface), and at 2.175 and 2.5 (station 3 bottom). The occurrence of sharp resonance peaks detected in this region may be due to the protons near to benzylic carbons (Hatcher et al. 1980; Majid and Ripmeester 1990). Poorly established proton resonance in this range could also be due to the decreased presence of aliphatic protons nearby unsaturated or aromatic groups (Graham et al. 2002; Qi et al. 2004). In this study, all the samples reported a sharp peak at 2.5 ppm.
The resonance in the range 3.10–4.75 ppm is mostly due to the protons on the carbons bonded with amines, alcoholic hydroxyls (polysaccharides), proteins, and polyether or methoxyl linkages of HAs (Francioso et al. 2001; Adani et al. 2006). These peaks are noticed at 3.6 (station 1 surface, station 2 surface as well as the bottom), at 3.5 (station 1 bottom), and 3.4 (station 3 bottom). Peaks in the spectral range 3.3–3.8 exhibit the influence of proton maximum which takes place from –CHOH and -CH2 OH functional groups, which point out the major contribution of polysaccharides and/or lignin and lignin like moieties (Yasuda et al. 1999; Kingery et al. 2000; Francioso et al. 2001). A resonance peak was observed at 4.5 ppm in station 3 surface sample. The number of peaks in this region was relatively less for the isolated HAs. The occurrence of sugar like moieties in HAs decreased because of acidification process (pH < 2) carried out during the isolation of HAs from sediments that damaged a major amount of polysaccharides (Schnitzer and Preston 1983; Gonzales-Villa et al. 1999; Montoneri et al. 2003; Adani et al. 2006).
Resonances in the range 6.40–8.45 ppm originate from aromatic protons phenols and quinones (Hatcher et al. 1980; Wilson 1981; Wersaw 1985; Jokica et al. 2004). Sterically hindered aromatic protons and nitrogen heteroaromatics display peaks in this region. In this study, the bottom sediments of stations 1 and 2 did not show peaks in this region which concludes the absence of aromatic structures in the isolated HAs from these stations. In the surface sample of station1, four peaks were detected at 7.085, 7.212, 7.236, and 7.341. In this sample, two more peaks were detected at 9.153 and 9.183, and another peak was noticed at 9.399. In station 2 surface, peaks were displayed as follows: one at 6.669, two at 7.105 and 7.136, another two at 7.236 and 7.261, and two more at 7.362 and 7.387. Here one more peak was observed at 8.244. In station 3 surface, peaks were observed as follows, two at 7.059 and 7.084, two at 7.184 and 7.210, and two more at 7.315 and 7.339. In this sample, one peak was seen at 9.130, two more at 9.316 and 9.332, and one more at 9.417. In station 3, bottom sediment sample peaks were observed at 7.085, two at 7.212 and 7.236, one at 7.341 and two more at 9.153 and 9.183 and finally at 9.339. These results correspondingly recognized the occurrence of deshielded protons and sterically hindered aromatic protons (Grasso et al. 1990; Qi et al. 2004). This also concludes that all are having more or less common aromatic core closed to unsaturated carbons ( >CH = CR2) and also the presence of aromatic groups such as phenols, reduced quinines through ionized carboxylic groups which contributed to electron shuttling and redox properties related with HAs (Bradley et al. 1998; Hernandez and Newman 2001).
Peaks demonstrating terminal methyl groups of alkyl chains, methyl groups of highly branched aliphatic structures, and methylene protons of alicyclic compounds are absent in the 1H NMR spectrum of isolated FA samples which fully ruled out the probability of the presence of aliphatic side chains in FAs. Peaks in the range 2.00–2.30 display protons attached to α- methyl and methylene groups on aromatic rings. Single resonance peak is detected in this range in all the stations at 2.3 ppm (station 1 surface), 2.1 ppm (station 1 bottom), 2.2 ppm (station 2 surface and bottom), 2.1 ppm (station 3 surface) and 2.2. ppm (station 3 bottom). Peaks in the range 3.10–3.25 show the presence of aromatic amines. Very few peaks are observed corresponding to this as observed at 3.04 ppm (station 1 surface), 3.03 ppm (station 1 bottom), 3.028 ppm (station 3 surface), and 3.112 ppm (station 3 bottom).
Peaks in the range 3.65–4.00 showed the existence of protons attached to α- carbons to oxygen groups like methyl aryl ether and carbohydrate HCHO. The appearance of peaks is due to protons adjacent to ester. The main dissimilarity between the (Abakumov et al. 2018) H NMR spectra of HAs and FAs is that the occurrences of aliphatic and alicyclic groups are completely absent in FAs and they show the presence of aromatic rings with α- methyl and methylene groups, aromatic amines, oxygen groups like methyl aryl ether and carbohydrate HCHO (FAs contain more oxygen-containing functional groups than HAs). Peaks in the range 3.65–4.00 are seen in all the samples. This is detected at 3.844 ppm (station 1 surface), 3.852 ppm (station 1 bottom), 3.610 (station 2 surface), 3.843 (station 2 bottom), 3.848 (station 3 surface), and 3.871 (station 3 bottom). Peaks in the range 4.00–4.75 are also seen in the 1H NMR spectra of isolated FAs. In the surface sediment of station 1, peaks were observed at 4.079 ppm and 4.6 ppm, while in the bottom sediment sample peaks were displayed at 4.109 and 4.6. Also in station 2, the surface sediment showed peaks at 4.09 ppm and 4.7 ppm, while peaks were displayed at 4.078 ppm and 4.6 ppm in the bottom sediment sample of this station. In station 3, the surface sample showed peaks at 4.104 ppm and 4.6 ppm, but in the bottom sediment, resonance peak was seen at 4.6 ppm.
As said previously, resonances in the range 6.40–8.45 ppm arise from aromatic protons with phenols and quinones, sterically hindered aromatic protons and also of nitrogen heteroaromatics (Hatcher et al. 1980; Wilson 1981; Wersaw 1985; Jokica et al. 2004; Prashob Peter 2014). These peaks were generally in the case of isolated FAs in this study. They are observed at 8.244, 9.617, and 10.667 ppm in station 1 surface sample and at 8.309, 9.626, and 10.618 ppm in the bottom sediment of the same station. In station 2 also more or less similar peaks were observed at 8.281, 9.626, and 10.637 ppm in surface and 8.293, 9.608, and 10.647 ppm in the bottom sediment sample. In station 3, the surface sample exhibited peaks at 7.152 and 8.120 ppm, while in the bottom sediment it was seen at 10.628 ppm. As seen in the case of HAs, at this point also peaks are recognized at values > 9 ppm which displays the occurrence of deshielded protons and sterically hindered aromatic protons (Grasso et al. 1990; Qi et al. 2004). This confirms the probability of having more or less common aromatic core involved to unsaturated carbons (>CH = CR2) and aromatic groups such as phenols, decreased quinines through ionized carboxylic groups which may be slightly higher in FA than in HA (Bradley et al. 1998; Hernandez and Newman 2001; Prashob Peter 2014). The presence of signals in the low field region 7.1–10.7 ppm indicates the presence of more plentiful OH groups with the phenolic OH groups( Schnitzer and Skinner 1974), and that in the region 9.3–13.2 ppm labeled the existence of carboxylic OH groups (Chamberlain 1974 ).
Conclusion
The study revealed differences in the amount of isolated humic substances—humic and fulvic acids—with respect to differences in environmental settings. The existences of humic substances were found to be positively correlated with the organic carbon content of the sediments, tannin and lignin content, and fine texture of the sediment (silt), thus attesting the fact that environmental settings control the humification status of mangrove sediments. The results obtained from elemental analysis, UV-visible spectroscopic studies, FTIR, and NMR of the samples and their respective HS showed that almost all of the signals indicated the presence of the functional group properties and the higher degree of humification in the samples isolated from the study area.
Sediment samples collected from different mangrove wetlands contained higher level of carbon, nitrogen, hydrogen, and sulfur and also higher amount of humic substances which indicate higher rates of decomposition and humification happening in these environments. The presence of higher hydrogen content indicated the aliphaticity of the isolated humic substances from the Kerala mangroves. The N/C and H/C ratio in HAs and FAs in all the samples from the selected stations was found to be low, indicating the geographical location as “terrestrial” due to high lignin mineralization. The most likely occurrence of varying degrees of vascular lignin moieties and non-vascular lignin moieties is confirmed by higher C/N ratios from the Kerala mangroves. The spectral characteristics also supported the elemental ratios of the isolated humic substances. The E4/E6 ratios of fulvic acids were found to be higher than those of humic acids supporting the fact that the fulvic acids are with low molecular weights and are less polymerized. The surface samples from Kerala mangroves showed a higher E4/E6 ratio than the bottom sediment samples in both HAs and FAs which indicates that the surface samples were of young origin, whereas the bottom regions are more ancient due to higher microbial activity and decomposition. These ratios also help us to differentiate the study area based on humification degree and maturity; the low E4/E6 ratio indicates a lesser degree of humification and maturity which was observed towards the mangroves collected from the southern part of Kerala. The presence of hydroxyl, methyl, methylene, carbonyl, carboxyl, phenol, and amide in the humic substances was confirmed with FTIR spectrum of the isolated humic substances. 1H NMR spectral characteristics were also complementary to the FTIR results. The occurrence of active functional groups, diverse elements, and favorable structural properties observed in this study indicate the richness of isolated humic materials which enhances microbial activity, soil productivity, and the potential to form the varied complexes in the study area. The structural characterization helps us to understand the mechanism of interaction of these materials with contaminant metals and other toxic chemicals in the environment, which definitely has the scope of a spatial comparison with respect to mangroves from different environmental settings in this study.
References
Abakumov EV, Rodina OA, Eskov AK (2018) Humification and humic acid composition of suspended soil oligotrophous environments in South Vietnam. Hind Appl Environ Soil Sci 2018:1–8. https://doi.org/10.1155/2018/1026237
Adani F, Genevini P, Tambone F, Montoneri E (2006) Compost effect of soil humic acid :ANMR study. Chemosphere 65(8):414–1418. https://doi.org/10.1016/j.chemosphere.2006.03.070
Allard B, Boren H, Grimvall A (1991) Humic substances in the aquatic and terrestrial environment. Springer-Verlag, Berlin
Alsumaiti TS, Shahid SA (2018) Comprehensive analysis of mangrove soil in Eastern Lagoon National Park of Abu Dhabi Emirate. Inter J Buss Appl Soc Sci 4(5):39–56 https://ssrn.com/abstract=3187689
Amir S, Hafidi M, Merlina G, Hamdi H, Revel JC (2004) Elemental analysis, FTIR and 13C-NMR of humic acids from sewage sludge composting. Agronomie 24(1):13–18. https://doi.org/10.1051/agro:2003054
Anuradha V, Nair SM, Kumar NC (2011) Humic acids from the sediments of three ecologically different esturine systams-a comparison. Int J Environ Sci 2(1):174–184. https://doi.org/10.6088/ijes.00202010018
APHA (1995) Standard methods for the examination of water and wastewater 5:47–48
Banik GC, Sanyal SKA (2006) Study on chromium humic complexation :part 1.Characterization of humic substances. J Ind Soci Soil Sci 54(2):163–169
Basha SC (1991) Distribution of mangroves in Kerala. Indian Forester 117(6):439–448
Basha SC (1992) Mangroves of Kerala: a fast disappearing asset. Indian Forester 118(3):175–190
Bask UC, Das AB, Das P (1999) Organic constituents in leaves of 9 mangrove specious of Orissa Coast,India. Pak J Bot 31:55–62
Bouillon S, Moens T, Dehairs F (2004) Carbon sources supporting benthic minerilization in mangrove and adjacent seagrass sediments (Gazy Bay,Kenya). Biogeo SCi Discu 1(1):311–333 hal-00297748
Bouillon S, Connolly RM, Lee SY (2008) Organic matter exchange and cycling in mangrove ecosysytems : recent insights from stable isotops studies. J SeaRes 59:44–58. https://doi.org/10.1016/j.seares2007.05.001
Bradley PM, Chapelle FH, Lovely DR (1998) Humic acids as electron acceptors for anaerobic microbial oxidation of vinyl chloride and dichloroethene. Appl Environ Microbiol 64(8):3102–3105. https://doi.org/10.1128/AEM.64.8.3102-3105.1998
Bravard S, Richi D (1991) Characterization of fulvic and humic acids from an Oxisol Spodosol toposequence of Amazonia,Brazil. Geoderma 48:151–162. https://doi.org/10.1016/0016-7061(91)90013-J
Carlose L, Pedersen BW, Ogilby PR, Martire DO (2011) The role of humic acid aggregation on the kinetics of photosensitized singlet oxygen production and decay. Photochem Photobiol Sci 10:1080–1086. https://doi.org/10.1039/C1PP00003A
Cezikova J, Kozler J, Mardronova L, Novak J, Janos P (2001) Humic acids from coal of northbohemia coal field III. Metal binding properties of humic acids measurments in a coloumn arrangement. Hum Reac FunPoly 47(2):119–123. https://doi.org/10.1016/S1381-5148(00)00077-8
Chamberlain NF (1974) The practice of NMR spectroscopy .Plenum
Chen Y, Senci N, Schnitzer M (1977) Information provided on humic substance by E4/E6 ratios. Soil Sci Soc Am J 41:352–358. https://doi.org/10.2136/sssaj1977.03615995004100020037
Chen J, Baohua G, Eugene J, Boeuf L, Pan H, Dai S (2002) Spectroscopic characterization of the structural and functional properties of natural organic matter fractions. Chemosphere 48:59–68. https://doi.org/10.1016/S0045-6535(02)00041-3
Chmura GL, Anisfeld SC, Cahoon DR, Lynch JC (2003) Globel carbon sequestration in tidal saline wetland soil. Globel Biogeochemical Cycle 17:1–12. https://doi.org/10.1029/2002GB001917
Collins CH, Collins KE, Ackerhalt RE (1971) Cation exchange separation of 51 Cr –labelled species in aqueous Cr(VI)-Cr(III) solution. J Radioanal Chem 8:263–274. https://doi.org/10.1007/BF0251819167
Cozzolino A, Conte P, Piccolo A (2001) Conformational changes of humic substances induced by some hydroxyl keto and sulphonic acids. Soil Biol Biochem 33:563–571. https://doi.org/10.1016/S0038-0717(00)00196-6
Dittamar T, Hertkorn N, Kattner G, Lara RJ (2006) Mangroves ,a major source of dissolved organic carbon to the oceans. Glob Biogeo Chem Cycles 20(1):1–7. https://doi.org/10.1029/2005GB002570
Enog OJ (1993) Mangroves - a carbon source and sink. Chemosphere 27:1097–1107. https://doi.org/10.1016/0045-6535(93)90070-L
Ertel JR, Hedges JI (1984) The lignin component of humic substances : distribution along soil sedimentary humic ,fulvic and base insoluble fractions. Geochim Cosmochim Acta 48(10):2065–2074. https://doi.org/10.1016/0016-7037(84)90387-9
Eshwar M, Srilatha K, Rekha B, Harish Kumar Sharma S (2017) Characterization of humic substances by functional groups and spectroscopic methods. Int J Microbiol App Sci 6(10):1768–1774. https://doi.org/10.20546/ijcmas.2017.610.213
Faustino E, Vieyara M, Valeria I, Palazzi Maria I, de Pinto S, Cloudio D, Borsarelli (2009) Combined UV-Vis absorbance and flourescence properties of extracted humic substances- like forcharacterization of composting evolution of domestic solid waste. Geoderma 151:61–67. https://doi.org/10.1016/j.geoderma.2009.03.006
Fernandes AN, Giovanela M, Esteves VI (2010) Elemental and spectral properties of peat and soil samples and their respective humic substances. J.Mol.Str 971:33–38. https://doi.org/10.1016/j.molstru.2010.02.069
Ferreira FP, Vidal-Torrado P, Buurman P, Macias F, Otero XL, Boluda R (2009) Pyrolysis-gas chromatography /mass spectrometry of soil organic matter extracted from a Brazilian mangrove and Spanish salt marshes. Soil Sci Soc Am J 73:841–851. https://doi.org/10.2136/sssaj2008.0028
Flaig WHP, Beutelspacher, Reitz E (1975) Chemical composition and physical properties of humic substance. In: Gieseking JE (ed) Soil Components 11. Springer-Verlang, New York, pp 1–219
Fong SS, Mohamed M (2007) Chemical characterization of humic substaces occurring in the peats of Sarawak, Malaysia. Org Geochem 38:967–976. https://doi.org/10.1016/j.orggeochem.2006.12.010
Francioso O, Sanchez-Cortes S, Tugnoli V (2001) Spectroscopic study of (DRIFT,SERS and1 HNMR) of peat leonardite and lignite humic substances. J Mol Struct 565:481–485. https://doi.org/10.1016/S0022-2860(00)00905-4
Garcia AC, Souza LGA, Pereira MG, Castro RM, Garcia-Mina JM, Zonta E (2016) Structure-property –function relationship in humic substances to explane the biological activity in plants. Sci Rep 6:1–10. https://doi.org/10.1038/srep20798
Geetha R (2002) Modelling of Geochemical Processes in Mangrove Eccosystem.PhD Thesis,Cohin University of Science and Techonology
Geetha R, Chandramohankumar N, Mathews L (2008) Geochemical reactivity of surficial and core sediment of tropical mangove ecosystem. Int J Environ Res 2(4):329–342
Giovanela M, Parlanti E, Soriano-Sierra J, Soldi MS, Sierra MMD (2004) Elemental composition , FT-IR spectra and thermal behaviour of sedimentary fulvic and humic acids from aquatic and terrestrial environment. Geochem J 38:255–264. https://doi.org/10.2343/geochemj.38.255
Giovanela M, Crespo JS, Antunes M, Admatti DS, Fernandes AN, Barrison A, da Silva CWP, Guegan R, Montelica-Heino M, Sierra MMD (2010) Chemical and spectroscopic characterization of humic acid extracted from the bottom sediments of a Brazilian subtropical microbasin. JMolStr 981:111–119. https://doi.org/10.1016/j.molstruc.2010.07.038
Gonzales-Villa FJ, Almendros G, Madrid F (1999) Molecular alterations of organic fractions from urban waste in the course of composting and their further tranceformation in amended soil. Sci Total Environ 236(1):215–229. https://doi.org/10.1016/S0048-9697(99)00284-3
Graham B, Mayol-Bracero OL, Guyon P (2002) Water soluble organic compounds in biomass burning aerosols Amazonia 1.Characterization by NMR and GC-MS.J. GeophysRes-Atmos 107(D20):8047–8062. https://doi.org/10.1029/2001JD000336
Grasso D, Chin Y, Weber W (1990) Structural and behavioural characteristics of a commercial humic acid and natural dissolved aquatic organic matter. Chemosphere 21(10):1181–1197. https://doi.org/10.1016/0045-6535(90)90139-K
Gu B, Schmitt J, Chen Z, McCarthy JF (1994) Adsorption and desorption of natural organic matter on iron oxide:mechanisms and models. Environ Sci Technol 28:38–46. https://doi.org/10.1021/es00050a007
Haddad G, Ali FE, Mouneimne AH (2015) Humic matter of compost: determination of humic spectroscopic ratio(E4/E6). Curr Sci Inter 4(1):56–72
Hatcher PG, Rowan R, Mattingly MA (1980) 1H and 13 CNMR of Marine humic acid. Orga Geo chem 2(2):77–85. https://doi.org/10.1016/0146-6380(80)90023-6
Hatcher PG, Schnitzer M, Dennis LW, Maciel GE (1981) Aromaticity of humic substances in soil. Soil Sci Soci Ame J 45(6):1089–1094. https://doi.org/10.2136/sssaj1981.03615995004500060016x
Hedges JI, Keli RG (1995) Sedimentary organic preservation: an assessment and speculative synthesis. Mar Chem 49:81–115. https://doi.org/10.1016/0304-4203(95)00008-F
Hernandez ME, Newman DK (2001) Extracellular electron transfer. Cellular and Molecular Life ScienceCMLS 58(11):1562–1571. https://doi.org/10.1007/PL00000796
Hernes PJ, Hedges JI (2000) Determination of condensed tannin monomers in environmental samples by capillary gas chromatography of acid depolymerization extract. Annal Chem 72(20):5115–5124. https://doi.org/10.1021/ac991301y
Hernes PJ, Hedges JI (2004) Tanninsignature of barks,neeles ,leaves.cones and wood at molecular level. Geochim CosmochimActa 68:1293–1307
Hernes PJ, Benner R, Cowie GL, Goni MA, Bergamschi BA, Hedges JI (2001) Tannin diagenesis in mangrove leaves from tropical estuary: Anovel molecular approach. Geochim Cosmochim Acta 65:3109–3122. https://doi.org/10.1016/S0016-7037(01)00641-X
IHSS (2010) Natural organic matter research .Isolation of IHSS sample, available at http://www.humic substances .org/isolation.html
Ikeya K, Sleighter RL, Hatcher PG, Watanabe A (2015) Characterization of the chemical composition of soil humic acids using Fourier tranceform ion cyclotron resonance mass spectrometry. Geochim Cosmochim Acta 153:169–182. https://doi.org/10.1016/j.gca.2015.01.002
Jennees M, Shaji A, Gopinath A, Krishnan KP, Sanil VL, Anoop PP (2019) A characteristic study of humic acids isolated from Arctic Fjord sediment. Adv Polym Sci 30(1):24–31. https://doi.org/10.13679/j.advps.2019.1.00024
Jennerjahn TC, Ittekkot V (2002) Relevance of mangroves for the production and deposition of organic matter along tropical continental margins. Naturwissenscaffen 89:23–30. https://doi.org/10.1007/s00114-001-0283-x
Jokica A, Wangb MC, Liua C, Frenkelc AI, Hung PM (2004) Integration of poly phenol and Maillard reactions in to a unified abiotic pathway for Humification in nature : the role of d-MnO2. Org Geochem 35:747–762. https://doi.org/10.1016/j.orggeochem.2004.01.021
Kaladharan P, George JP, Nandakumar A, Khambadkar LR (2003) Humic acids in mangrove sediments of Kerala.J.mar.biol. Ass. India. 45(2):232–234
Kalesh NS, Sujatha CH, Muraleedharan Nair S (2001) Dissolved folin phenol active substances (tannin and lignin) the seawater along the West Coast of India. J Ocenography 57:29–36. https://doi.org/10.1023/A:1011118502643
Katherisan K, Veera RA (1990) Seasonal changes in Tannin content of mangrove leaves .Ind.Forester. p391
Kingery WL, Simpson AJ, MHB H, Locke MA, Hicks RP (2000) The application of multidimensional NMR to the study of soil Humic substances. Soil Sci 165(6):483–496. https://doi.org/10.1097/00010694-200006000-00004
Kononova MM (1966) Soil organic matter, second edn. Pergamon press, Oxford NewYork
Kristensen E, Bouillon S, Dittmar T, Marchand C (2008) Organic matter dynamics in mangrove ecosystems-a review. Aquat Bot 8:201–219. https://doi.org/10.1016/j.aquabot.2007.12.005
Kwiatkowska-Malina J (2018) Qualitative and quantitative soil organic matter estimation for sustainable soil management. J Soils Sediments 18:2801–2812. https://doi.org/10.1007/s11368-017-1891-1
Lallier VE, Marchand C, Disnara JR, Lottiera N (2008) Origin and diagenesis of lignin and carbohydrates in mangrove sediments of Guadeloupe (French West Indies):Evidence for two-step evolution of organic deposits. Chem Geol 255(3):338–398. https://doi.org/10.1016/j.chemgeo.2008.07.009
Leenheer JA, Wilson MA, Malcolm RL (1987) Presence of potential significance of aromatic –ketone groups in aquatic humic substances. Org Geochem 11:273–280. https://doi.org/10.1016/0146-6380(87)90038-6
Maie N, Pisani O, Jaffe R (2008) Mangrove tannins in aquatic ecosystems. Their fate and possible influence on dissolved organic carbon and nitrogen cycling. Limnol Oceanogr 53(1):160–171. https://doi.org/10.4319/lo.2008.53.1.0160
Majid A, Ripmeester JA (1990) Isolation and charecterization of humic acids from Alberta oil sands and related materials. Fuel 69(12):1527–1536. https://doi.org/10.1016/0016-2361(90)90202-2
Manju MN, Resmi P, Ratheesh Kumar CS, Gireeshkumar TR, Manju Mary Joseph CN (2016) Biochemical and stable carbon isotope records of mangrove derived organic matter in the sediment core. Environ. Earth Sci 75:565–571. https://doi.org/10.1007/s12665-016-5245-x
Manju MN, Ratheesh Kumar CS, Resmi P, Gireeshkumar TR, Joseph MM, Salas PM, Chandramohanakumar N (2020) Trace metal distribution in the sediment cores of mangrove ecosystem along northern Kerala coast, south-west coast of India. Mari Poll Bull 153:1–5. https://doi.org/10.1016/j.marpolbul.2020.110946
Marchand C (2017) Soil carbon stocks and burial rates along a mangrove forest chronosequence (French Guiana). For.Ecol.Manag 384:92–99. https://doi.org/10.1016/j.foreco.2016.10.030
Martin MV, Gebuhr C, Daniel O, Wiltshire KH (2014) Charecterization of a humic acids extracted from marine sediment and its influence on the growth of marine diatoms. J Mar Biol Assoc Uni King 94(5):895–906. https://doi.org/10.1017/S0025315414000368
Matsui N, Meepol W, Chukwamdee J (2015) Soil organic carbon in mangrove ecosystems with different vegetation and sedimentological conditions. J.Mar.Sci.Eng 3:1404–1424. https://doi.org/10.3390/jmse3041404
Meyers PA, Ishiwatari (1993) Lacustrine organic geochemistry: an overview of indicators of organic matter source and diagenesis in lake sediments. Org Geochem 20:867–900. https://doi.org/10.1016/0146-6380(93)90100-P
Mini M, Lekshmy S, Radhakrishnan T (2012) Kerala mangroves-pastures of estuaries-their present status and challenges. Inter J SciRes 3(11):2804–2280
Montoneri E, Savarino P, Adani F, Genevini PL, Ricca G, Zanetti F, Paletti S (2003) Polyalkylphenyl-sulphonic acid withgroups of variable strength from compost. Waste Manag 23(6):523–535
Moreda–Pineiro A, Bermejo–Barrea A, Bermejo–Barrea P (2004) New trends in involving the use of ultrasound energy for the extraction of humic substances from marine sediments. Anal Chim Acta 524:97–107. https://doi.org/10.1016/j.aca.2004.03.096
Muscolo A, Sidari M, Nardi S (2013) Humic substance: relationship between structure and activity. Deeper information suggests univocal findings. J Geochem Explor 159:57–63. https://doi.org/10.1016/j.gexplo.2012.10.012
Nair CK, Balchand AN, Chacko J (1993) Sediment characteristics in relation to changinghydrography of Cochin estuary. Ind JMariSci 22:33–36 http://nopr.niscair.res.in/handle/123456789/37754
Orlov DS (1985) Infrared spectra humus spectra. In: Orlov DS (ed) Humus acids of soils. Oxonian Press Pvt.Ltd, New Delhi
Pernetta JC (1993) Mangrove forest ,climate change and sea level rise: hydrological influences on community structure and survival ,with examples from the Indo-West Pacific .International Union for Conservation of Nature and Natural Resources (IUCN), pp45
Petrus AC, Ahmed OH, Muhammad AMN (2009) Chemical characteristics of compost and humic acid from sago waste. Ame J AppSci 6(11):1880–1884. https://doi.org/10.3844/ajassp.2009.1880.1884
Peuravuori J, Pihlaja K (1997) Molecular size distribution and spectroscopic properties on aquatic humic substances. Anal Chim Acta 337:133–149. https://doi.org/10.1016/S0003-2670(96)00412-6
Piccolo A, Nardi S, Concheri G (1996) Micelle-like conformation of humic substances as revealed by size exlusion chromatography. Chemosphere 4:595–602. https://doi.org/10.1016/0045-6535(96)00210-X
Prashob Peter KJ (2014) Modulation of hydrophobic partitioning affinity of humic acids from Mudbank sediments and Applications .PhD thesis. Cochin University of Science and Technology .pp49-54
Qi BC, Aldric C, Lorenzen L (2004) Effect of ultrasonication on the humic acid extracted from lignocellulose substrate decomposed by anaerobic digestion. Chem Eng J 98(1):153–163. https://doi.org/10.1016/j.cej.2003.07.002
Rani V, Bijoy Nandan S, Schwing PT (2021) Carbon source characterisation and historical carbon burial in the three mangrove ecosystems on the South West Coast Of India. Cantena 197:1–12. https://doi.org/10.1016/j.cantena.2020.104980
Ranjan RK, Routh J, Ramanathan AL (2010) Bulk organic matter characteristic in the Pichavam mangrove-esturinecomplex ,south –eastern India. Appl Geo Chem 25:1176–1186. https://doi.org/10.1016/j.apgeochem.2010.05.003
Rashid MA (1985) Geochemistry of marine humic compounds. Springer- Verlag, New York, p 30
Reddy S, Nagaraja MS, Raj TSP, Pattil ASP, Dhumgond P (2014) Elemental analysis ,E4/46 ratio and total acidity of soil humic acids and fulvic acids from different land use systems. Annals of Plant and Soil Research 16(2):89–92
Resmi TR (2004) Hydrogeochemical evaluation of inorganics and bioorganics in selected aquatic environments. PhD Thesis, Cochin University of Science and Technology
Rice JA, Mac Carthy P (1991) Statistical evaluation of the elemental composition of humic substances. Org Geochem 17:635–648. https://doi.org/10.1016/0146-6380(91)90006-6
Rigobello ES, Campos SX, Azevedo ER, Dantas ADB, Vieira EM (2017) Comparitive characterization of humic substances extracted from freshwater and peat of different apparent molecular sizes. Ambi Agua Interdisci Appl Sci 12(5):774–785. ISSN-1980-993X. https://doi.org/10.4136/ambi-agua.2022
Rini Sebastian (2002) Some Biogenic compound and their derivatives in selected mangrove eccosystems. PhD Thesis, Cohin University of Science and Techonology
Rocha JC, Oliveira LC, Sargentini EJ, Rosa AH, Simoes ML, Martin Netto L (2007) The influence of seasonalness on the structural characteristics of aquatic humic substances extracted from Negro River (Amazone state) waters: interaction with Hg (ll). J Braz Chem Soc 18:860–868. https://doi.org/10.1590/S0103200700400028
Sainto Y, Hayano S (1981) Characterisation of humic and fulvic acids isolated from marine sediments of Sagami and Saruga bays with C-13 and proton nuclear magnetic resonance. J Oceo SociJapan 36(6):286–292. https://doi.org/10.1007/BF02070157
Samios S, Lekkas T, Nikolaou A, Golfinopoulos S (2007) Structural investigations of aquatic humic substances from different water watersheds. Desalinanation 210:125–137. https://doi.org/10.1016/j.desal.2006.05.038
Sarala Devi K, Venugopal P, Sakarannarayanan V (1995) Organic matter and sediment characteristics of some estuaries of north Kerala,in Proc. Seventh Kerala Science Congress, (Kerala State Committee on Science Technology and Environment, Trivndrum).pp 113-114
Sarlaki E, Paghaleh AS, Kianmehr MH, Vakilian KA (2020) Chemical , spectral and morphological characterization of humic acids extracted and membrane purified from lignite. ChemChemTechnol 14(3):353–361. https://doi.org/10.23939/chcht14.03.353
Satisha GC, Devarajan L (2011) Composition and characterization of humic substances extracted from eluent –based pressmud composts. Agropedology 21(1):8–17
Schintzer M, Khan SU (1972) Humic substances in the Environment. Marcel Dekker, New York, p 327
Schnitzer M (1999) A life time perspective on the chemistry of soil organic matter. Advn Argo 68:1–58. https://doi.org/10.1016/S0065-2113(08)60842-1
Schnitzer M, LeVesque M (1979) Electron spin resonance as a guide to the degree of humification of peats. Soil Sci 127:140–145
Schnitzer M, Preston CM (1983) Effects of acid hydrolysis on the 13 CNMR spectra of humic substances. Plant Soil 75(2):201–211. https://doi.org/10.1007/BF02375565
Schnitzer M, Preston CM (1986) Analysis of humic acids by solution and solid state carbon-13 nuclear magnetic resonance. Soil Sci Soc AmJ 50:326–331
Schnitzer M, Skinner SIM (1974) The low temperature oxidation of humic substances. Can JChem 52:1072–1080. https://doi.org/10.1139/v74-171
Sein LT Jr, Varnum JM, Jansen SA (1999) Conformational modelling of a new building block of humic acid; approches to the lowest energy conformer. Environ SciTechnol 33:546–552. https://doi.org/10.1021/es9805324
Sensi N, Miano TM, Provenzano MR, Brunetti G (1989) Spectroscopic and compositional comparative characterization of IHSS reference and standard fulvic acid and humic acid of various origin. Sci Total Environ 81:143–146. https://doi.org/10.1016/0048-9697(89)90120-4
Sensi N, Miano TM, Provenzano MR, Brunetti G (1991) Charecterization ,differentiation, and classification of humic substances by fluorescence spectroscopy. SoilSci 152:259–271
Sierra MMD, Giovanela M, Parlanti E, Esteves VI, Duarte AC, Fransozo A, Soriano SEJ (2004) Structural description of humic substances from subtropical coastal environments using elemental analysis ,FT-IR and 13 CNMR data. J Coast Res 42:219–231 https://www.jstororg/stable /25737004
Silverstein RM, Bassler GC, Morrill TC (1991) Spectrometric identification of organic compounds. John Wiley and Sons, New York, p 419
Singh G, Ramanathan AL, Prasad MBK (2005) Nutrient cycling in mangrove ecosystem : a brief overview. Int J Ecol Environ Sci 30:231–244
Smid E, Eckhard KU, Lechner P, Schulten HR, Leinwebe P (2015) Characterization of different decomposition stages of biowaste using FTIR spectroscopy and pyrolysis –field ionization mass spectrometry. Biodegradation 16(1):67–79. https://doi.org/10.1007/s10531-004-0430-8
Srilatha M, Rao PC, Sharma SHK, Padmaja G (2013) Physico-chemical characterization of humic substances under long term application of fertilizers and manures in rice –rice cropping sequence in an Inceptisol. Inter JAdv Res 10(1):343–348
Stevenson FJ (1994) Humus chemistry, genesis, composition. Books.google.com, Wiley, New York
Stevenson FJ, Goh KM (1971) Infrared spectra of humic acids and related substances. Geochim Cosmochim Acta 35:471–483. https://doi.org/10.1016/0016-7037(71)90044-5
Stuemer DH, Peters KE, Kaplan IR (1978) Source indicators of humic substances and protokerogen: stable isotope ratios, elemental compositions and electron –spin resonance spectra. Geochim CosmochimActa 42:989–997. https://doi.org/10.1016/0016-7037(78)90288-0
Suthhof A, Jennerjahn TC, Schafer P, Ittekkott V (2000) Nature of organic matter in surface sediment from the Pakistan continental margin and the deep Arabian Sea:amino acids. Deep Sea Research Part II-Trop Res Oces 47:329–351. https://doi.org/10.1016/S0967-0645(99)00109-5
Swift MJ, Heal OW, Anderson JM (1979) Decomposition in terrestrial ecosystem .Studies in Eccology .5,University of Caliphornia
Tahiri A, Richel A, Destain J, Thonart P, Ongena M (2016) Comprehensive comparison of the chemical and structural characterization of leonardite humic fraction. Anal Bioanal Chem 408:1917–1928. https://doi.org/10.1007/s00216-016-9305-6
Tan KH (2014) Humic matter in soil and the environment : principles and controversies. 2nd EdnCRC Press ,NewYork. p495
Traina SJ, Novak J, Smeck NK (1990) An ultraviolet absorbance method of estimating the percent aromatic Carbon content of humic acids. J Environ Qual 19:151–162. https://doi.org/10.2134/jeq1990.00472425001900010023x
Uyguner CS, Bekbolet M (2004) Photocatalytic degradation of natural organic matter: kinetic consideration and light intencity dependence. Inter JPhot 6:73–80. https://doi.org/10.1155/S1110662X0400011X
Vergnoux A, Di Rocco R, Domeizel M, Theraulaz F, Doumenq P (2011) Quantitative and mid infrared changes of humic substances from burned soils. Environmental Reaserch 111:205–214. https://doi.org/10.1016/j.envres.2010.03.005
Vidyasagaran K, Madhusoodann VK (2014) Distribution and plant diversity of mangroves in the west coast of. Kerala IndiaJ Biodiv EnvirnStd 4(5):38–45 http://www.innspub.net/
Wersaw RL (1985) Application of nuclear magnetic resonance spectroscopy for determining functionality in humic substances. In Aiken GR,McKnight DM,WershawRL,MacCarthy,P(Eds). Humic substances in soil, sediment, water, geochemistry isolation and characterization. Wiley Inter science Publication,John Wiley and Sons, New York
Wilson MA (1981) Application of nuclear magnetic resonance spectroscopy to the study of the structure of soil organic matter. J Soil Sci 32(2):167–186. https://doi.org/10.1111/j.1365-2389.1981.tb01698.x
Yang J, Yu Z, Liang Z, Yue K, Changkun F, Ni X, Wu F (2019) The optical properties in alkali-soluble fractions extracted from newly shed litters in a subalpine forest. J Soils Sediments:1–9. https://doi.org/10.1007/s11368-019-02535-9
Yangi Y, Shindo H (2016) Assessment of long- term compost application on physical, chemical, and biological properties ,as well as fertility, of soil in a field subjected to double cropping. Agric Sci 7:30–43. https://doi.org/10.4236/as.2016.71004
Yasuda S, Hamaguchi E, Asano K (1999) Ready Chemical conversion of acid hydrolysis lignin into water soluble lignin sulphonates. III. Successive treatment of acid hydrolysis lignin and lignin model compounds by phenolation and aryl –sulfonation. J Wood Sci 45(3):245–249. https://doi.org/10.1007/BF01177733
Yavmetdinov IS, Stepanova EV, Gavrilova VP, Lokshin BV, Perminova IV, Koroleva OV (2003) Isolation and characterization of humin –like substances produced by wood-degrading white rot fungi. Appl Biochem Microbiol 39:257–264. https://doi.org/10.1023/A:1023571426331
Zeck W, Sensi N, Guggenberger G, Kaiser K, Lehmann J, Miano TM, Miltner A, Schroth GS (1997) Factors controlling humification and minarilization of soil organic matter in the tropics. Geoderma 79:117–161. https://doi.org/10.1016/S0016-7061(97)00040-2
Zhang Y, Jinzhou D, Zhang F, Yu Y, Zhang J (2011) Chemical characterization of humic substance isolated from mangrove swamp sediments: The Qinglan area of Hainan Island, China. Esturine, Coastal and Shelf Science 93:220–223. https://doi.org/10.1016/j.ecss2010.12025
Acknowledgements
We thank the Vice Chancellor and Director of Research, Kerala University of Fisheries and Ocean Studies, for providing all the support. We also acknowledge Sophisticated Training and Instrumentation Centre, Cochin University of Science and Technology for FTIR, NMR and CHNS analysis. We also thank Mr. Sanil V.L, Ms. Princy M.John, Mr. Vishnu Murali, and Ms. Greeshma K.S for their help in sampling and analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author(s) declare that they have no conflict of interest.
Additional information
Responsible Editor: Amjad Kallel
Rights and permissions
About this article

Cite this article
Mathew, J., Gopinath, A. & Vareed, R.A. Spectroscopic characterization of humic substances isolated from tropical mangrove sediments. Arab J Geosci 14, 668 (2021). https://doi.org/10.1007/s12517-021-06968-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12517-021-06968-w