
Abstract
Introduction
Triple-negative breast cancer (TNBC) is a highly heterogeneous disease. Mining differentially expressed genes of TNBC is helpful to explore new therapeutic targets. This study aimed to investigate diagnostic biomarker genes in TNBC compared to normal tissue. Additionally, we explored the functions and prognostic value of these key genes as well as potential targeted drugs that could affect these genes.
Methods
Differential gene expression analysis was conducted using the R software with data from the Gene Expression Omnibus (GEO) database. Then, the identified differentially expressed genes (DEGs) were used to construct a protein-protein interaction (PPI) network using the Search Tool for the Retrieval of Interacting Genes (STRING) database and Cytoscape software. The mRNA expression levels of key genes were analyzed using the UALCAN database with data from The Cancer Genome Atlas (TCGA). Enrichment and survival analyses were performed using R software. In addition, potential compounds showing sensitivity to key genes were identified by gene set cancer analysis (GSCA).
Results
Compared with normal tissues, a total of 203 DEGs were upregulated in TNBC. These DEGs participated in various biological processes including nuclear division, microtubule binding, cell cycle, and the p53 signaling pathway. Through the PPI network analysis, ten key genes were identified, among which four genes showed significant correlation with poor progression-free interval (PFI) in patients with TNBC. Moreover, the four survival-related genes were found to act as sensitive therapeutic targets.
Conclusion
The identified four key genes were considered new biomarkers for diagnosis and prognosis and also potential therapeutic targets for TNBC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Triple-negative breast cancer (TNBC) is a highly heterogeneous disease, and often some specific genes are highly expressed in TNBC, which can be regarded as biomarkers for TNBC. |
This study established a group of diagnostic and prognostic genes for TNBC based on breast invasive carcinoma (BRCA) samples of The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) database. The four identified key genes (CCNB2, DLGAP5, HJURP, and TTK) were highly expressed in TNBC and correlated with poor progression-free interval (PFI). |
The four identified key genes could be developed as potential therapeutic targets for TNBC. Nutlin-3a(-) was a potential compound targeting DLGAP5 and TTK, and the molecular docking showed positive results. |
Introduction
According to the Global Cancer Statistics 2020, breast cancer has now surpassed lung cancer as the most frequently diagnosed cancer, ranking first in terms of incidence in the majority of countries [1]. Among the various types of breast cancer, breast invasive carcinoma (BRCA) is the most common and is typically categorized into four groups: luminal A, luminal B, human epidermal growth factor receptor 2 (HER2) positive, and triple-negative breast cancer (TNBC) [2]. It is crucial to note that patients with different molecular subtypes of breast cancer exhibit varying clinical responses and survival times [3, 4]. In addition to chemotherapy, the systemic treatment of triple-negative breast cancer encompasses targeted therapy and immunotherapy [5, 6]. Targeted therapy options currently available include poly ADP-ribose polymerase (PARP) inhibitors, epidermal growth factor receptor(EGFR) inhibitors, and phosphoinositide 3-kinase (PI3K)/serine/threonine kinase (AKT) /mammalian target of rapamycin (mTOR) signaling pathway inhibitors, among others [7,8,9]. Immunotherapy options involve the use of programmed death-1(PD-1) antibody and programmed cell death-ligand 1 (PD-L1) antibody [10,11,12]. Nonetheless, it remains crucial to continually develop novel therapeutic targets and explore new drugs to enhance the efficacy and prognosis of treatment.
In this study, we aimed to investigate novel key genes in TNBC by utilizing multiple databases. By evaluating gene expression and its correlations with cancer-related pathways, survival rates, tumor cell immune infiltration, and sensitive drugs, we sought to identify potential targets for TNBC treatment.
Methods
Ethics and Data Acquisition
This study was based on publicly available data, and patients were not directly involved in the entire research process. Therefore, no Independent Review Board approval was needed. This study involved the use of several public databases, including: Search Tool for the Retrieval of Interacting Genes (STRING, http://string-db.org) [13], UALCAN (https://ualcan.path.uab.edu/index.html) [14], Gene Set Cancer Analysis (GSCA, http://bioinfo.life.hust.edu.cn/GSCA/#/) [15], Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) [16], and The Cancer Genome Atlas (TCGA, https://www.cancer.gov/about-NCI/organization/ccg/research/structural-genomics/TCGA) [17].
The GEO database, hosted by the National Biotechnology Information Center, is recognized as the most comprehensive and extensively used public data resource platform globally. For this study, we employed “TNBC” and “Homo sapiens” as the screening conditions. From the available microarray datasets, we selected GSE185645, GSE38959, and GSE167152, which included clinical or molecular typing information. Among these datasets, GSE38959 consisted of 30 cases of TNBC and 13 cases of normal tissue. GSE185645 comprised 15 cases of TNBC and 1 case of normal tissue, while GSE167152 had 14 cases of TNBC and 5 cases of normal tissue. The TCGA database is the largest existing database of cancer gene information. To assess the prognostic value and diagnostic sensitivity of the identified key genes, we utilized the TCGA database.
Identification of DEGs
Differential gene expression analysis between TNBC samples and normal samples was performed using GEO2R for each dataset. A screening threshold of |log2FC|> 1 and P < 0.05 was set to identify differentially expressed genes (DEGs). The DEGs were further filtered by obtaining the intersection of highly expressed genes from GSE185645, GSE38959, and GSE167152. These overlapping genes were defined as the final set of DEGs. To validate the expression of the identified key genes in TNBC, we utilized the UALCAN online database to analyze the data from TCGA.
PPI Network Analysis and Identification of Hub Genes
The protein-protein interaction (PPI) network was generated using the STRING database with an interaction score threshold set at > 0.4. The resulting interaction data were downloaded and visualized using the Cytoscape software (version 3.9.0). To identify key genes with high topological importance, Cytohubba, a plugin in Cytoscape, was employed. In this study, the Maximal Clique Centrality (MCC) algorithm was used to screen the top ten hub genes within the PPI network.
Gene Function Enrichment Analysis and Gene Set Enrichment Analysis (GSEA)
The R software (version 4.2.1) [18] was utilized to conduct gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses on all the genes present in the interaction network. The GO enrichment analysis provided predictions of gene function across three aspects: biological process (BP), cellular component (CC), and molecular function (MF).
For GSEA, the GSE38959 dataset was analyzed using the cluster Profiler package (version 4.4.4) of R software. The threshold for significance was set at |NES|> 2, FDR (q value) < 0.25, and p.adjust < 0.05. Additionally, an enrichment plot was generated to visualize the results. To validate the findings obtained from GSEA, the association between the set of hub genes and pathways was further examined using the GSCA tool. This online tool facilitates the exploration of correlations between gene sets and tumor-related pathways.
Immune Infiltration Analyses
The DEGs were imported into the GSCA database to calculate the gene set variation analysis (GSVA) score. We then analyzed the correlation between the GSVA score and the presence of immune cells in breast cancer. Subsequently, we performed an analysis to investigate the correlation between the expression levels of the key genes and immune cell infiltration using the BRCA data sourced from the TCGA database. To conduct this analysis, we utilized R software.
Diagnostic Value and Survival Analysis
The RNAseq data of STAR process of TCGA-BRCA project from TCGA database were downloaded, and the data in FPKM format and clinical data were extracted. The pROC package (version 1.18.0) of R software was utilized to assess the diagnostic value of the key genes. The area under the curve (AUC) values were calculated for each gene. A higher AUC value indicates that the gene has greater sensitivity and specificity in differentiating basal type from other types of breast cancer. AUC values ranging from 0.7 to 0.9 suggest moderate diagnostic efficiency, while AUC values > 0.9 indicate good diagnostic efficiency.
To investigate the relationship between key gene expression and progression-free interval (PFI), the survival package (version 3.3.1) of R software was employed. The survminer package and ggplot2 package were used to visually present the survival curves. All the BRCA data source was from the TCGA database. Prognostic data were from Liu’s research [19]. A significance level of P < 0.05 was considered statistically significant. Genes with prognostic value were defined as key genes.
In addition, further analysis was conducted using the GSEA datasets to investigate the association between the expression levels of the ten hub genes and important clinical outcomes in BRCA, including disease-free interval (DFI), disease-specific survival (DSS), overall survival (OS), and progression-free survival (PFS).
Sensitive Drug Mining and Molecular Docking with the Key Genes
GSCA is a platform that integrates genomic, pharmacogenomic, and immunogenomic gene set cancer analysis. In GSEA, users can combine clinical information and small molecular drugs to identify biomarkers and drugs for experimental design. GSCA integrates 10,000 + genomic data across 33 cancer types from TCGA and > 750 small molecule drugs from the Genomics of Drug Sensitibity in Cancer (GDSC) and the Cancer Therapeutics Response Portal (CTRP). In the “drug” module, the key genes were inputted to analyze and identify the specific drugs that target these genes with high sensitivity.
The crystal structures of the proteins encoded by the four key genes were obtained by PDB database (http://www.rcsb.org/) or AlphaFold2 [20]. The two-dimensional sdf structure file of compound Nutlin-3a(-) was processed by LigPrep module in Schrodinger suit software, and all its three-dimensional chiral conformations were generated. First, the SiteMap module in Schrodinger suit software was used to predict the best binding site, and then the Receptor Grid Generation module in Schrodinger suit software was used to set the most suitable Enclosing box to wrap the predicted binding site perfectly. On this basis, the active sites of four proteins were obtained. The treated ligand compound Nutlin-3a(-) was molecularly docked with the active sites of four proteins (using XP docking with the highest accuracy). The ligand compound Nutlin-3a(-) and the active sites of four proteins were calculated and analyzed by MM-GBSA [21].
Statistical Analysis
R software was primarily employed for various bioinformatics and statistical analyses in this study. These analyses included normalizing and transforming RNA-seq data, conducting survival analyses, performing receiver-operating characteristic curve (ROC) analysis, and carrying out enrichment analysis. All statistical p values were bilateral, and a threshold of < 0.05 was deemed statistically significant.
Results
Identification of DEGs and Key Genes in TNBC
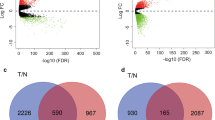
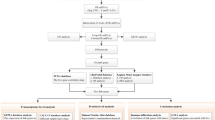
The whole study flow chart is presented in Fig. 1. Analysis of three GEO datasets (GSE185645, GSE38959, and GSE167152) showed that 2904, 372, and 3818 genes were overexpressed, respectively. A Venn chart was utilized to illustrate the overlap among the three datasets, leading to the identification of 203 DEGs (Fig. 2A, B). The 203 DEGs were then subjected to analysis using the STRING online database, resulting in the visualization of the PPI network (Supplementary Material Fig. 1). Further utilizing the cytohubba tool, ten hub genes were identified. These key genes include cyclin B1 (CCNB1), cyclin B2 (CCNB2), DLG-associated protein 5 (DLGAP5), Holliday junction recognition protein (HJURP), kinesin family member 15 (KIF15), kinesin family member 23 (KIF23), non-SMC condensin I complex subunit G (NCAPG), nucleolar and spindle-associated protein 1 (NUSAP1), sperm-associated antigen 5 (SPAG5), and threonine and tyrosine kinase (TTK) (Fig. 2C). These ten key genes were subsequently subjected to additional investigation.

Flow chart of the study. TNBC triple-negative breast cancer, DEG differentially expressed gene, PPI protein-protein interaction

Identification of DEGs and hub genes. A DEGs in the three GSE datasets. B Intersections of the three datasets. C PPI network of the DEGs. D Hub genes screened by cytohubba. TNBC triple-negative breast cancer, DEG differentially expressed gene, PPI protein-protein interaction
Enrichment Analyses
GO and KEGG enrichment analyses were performed on all 203 genes within the interaction network. The results revealed a total of 320 biological process (BP) terms, 50 cellular component (CC) terms, 44 molecular function (MF) terms, and 11 KEGG pathways. Notable terms included nuclear division, microtubule binding, cell cycle, and the p53 signaling pathway, among others. The top five terms from each GO subtype and KEGG are presented in Table 1 and Fig. 3A. The GSEA results analyzed by R software are shown in Fig. 3B. The hub genes were most related to cell cycle and mitotic cell cycle. The pathway correlation analyzed by GSCA tool showed strong positive correlation with cell cycle, apoptosis, and DNA damage and negative correlation with hormone ER, RASMAPK pathways, etc. (Fig. 3C).

Function enrichment analyses of DEGs. A GO and KEGG analyses of DEGs. B Gene set enrichment analysis. C Relationship of the key genes and tumor related pathways. DEG differentially expressed gene, GO gene ontology, KEGG Kyoto Encyclopedia of Genes and Genomes, BP biological process, CC cellular component, MF molecular function
Survival Analyses
A single-factor Cox regression analysis was conducted on the ten hub genes. The results of the univariate Cox regression analysis are presented in Table 2. The findings indicated that the differential expression of CCNB2, DLGAP5, HJURP, and TTK had a significant impact on the prognosis. Kaplan-Meier survival analysis was performed to assess the expression of these key genes in TNBC and its association with patient outcomes. The results demonstrated that high expression levels of CCNB2, DLGAP5, HJURP, and TTK in TNBC were indicative of poor PFI (Fig. 4A) while the other hub genes did not show a significant difference on PFI. Additionally, the GSEA database was utilized to further analyze the prognostic capabilities of these ten key genes. The results obtained through this analysis were consistent with the previous findings (Fig. 4B). These findings collectively suggest that the group of four genes, which includes CCNB2, DLGAP5, HJURP, and TTK, could serve as a prognostic biomarker group for TNBC. These four genes were identified as key genes.

Survival analyses of the hub genes. A–D PFI difference between high expression group and low expression group of CCNB2, DLGAP5, HJURP, and TTK. E Survival analyses conducted by GSCA tool. PFI progression-free interval, CCNB2 cyclin B2, DLGAP5 DLG-associated protein 5, HJURP Holliday junction recognition protein, TTK threonine and tyrosine kinase, GSCA gene set cancer analysis, DFI disease-free interval, DSS disease-specific survival, OS overall survival, PFS progression-free survival
Diagnostic Value and Expression of the Four Key Genes
The pROC package in R software was utilized to analyze the diagnostic value of different PAM50 subtypes of breast cancer using the BRCA data from the TCGA database. Classification data of PAM50 were from Berger’s study [22]. The findings demonstrated that CCNB2, DLGAP5, HJURP, and TTK exhibited good discriminative capabilities in distinguishing the basal-like subtype from the normal-like and luminal A subtypes, with area under the curve (AUC) values > 0.9 (Fig. 5A). These genes were also able to differentiate the basal-like subtype from the Her2 + and luminal B subtypes, with AUC values ranging from 0.6 to 0.8.

Diagnostic value and expression of the key genes. A ROC curve of the four key genes. B Expression of each key genes in different BRCA subtypes in UALCAN database. ROC receiver-operating characteristic curve, BRCA breast invasive carcinoma, CCNB2 cyclin B2, DLGAP5 DLG-associated protein 5, HJURP Holliday junction recognition protein, TTK threonine and tyrosine kinase
The expression levels of these four key genes were further investigated using the GEPIA2 database. The results consistently showed that these genes were highly expressed in TNBC compared to other subtypes of breast cancer as well as normal tissues. These differences in expression levels were found to be statistically significant (Fig. 5B). These four key genes exhibited differential expression not only between TNBC and normal tissues but also between TNBC and other clinical subtypes, with the highest expression observed in TNBC. Consequently, utilizing this set of four genes as a collective diagnostic marker possesses considerable specificity, potentially enhancing the accuracy of identifying TNBC and distinguishing it from other breast cancer subtypes.
Immune Infiltration Analyses
The GSCA online tool was utilized to calculate the GSVA score of the hub genes in BRCA. The results indicated that the GSVA score in BRCA samples was significantly higher compared to normal tissues. Based on this GSVA score, an analysis of immune cell infiltration in BRCA was conducted. The findings demonstrated that the GSVA score exhibited positive correlations with dendritic cells (DC) and B cells while showing negative correlations with helper T cells 17 (Th17) and CD4 T cells, among others (Fig. 6). These results suggest that targeting these immune cells through appropriate immunotherapy strategies may be beneficial.

GSVA score and immune infiltration of the hub genes. A GSVA score of the hub genes was higher in BRCA than in normal tissue. B GSVA score of the hub genes was correlated with immune cells. C–F Correlation among GSVA score and B cells, DC cells, Th17 cells, and CD4 T cells in BRCA. GSVA gene set variation analysis, BRCA breast invasive carcinoma, DC dendritic cell, Th17 helper T cell 17
Furthermore, using the ssGSEA algorithm from the R package GSVA (version 1.46.0) [23], the correlation between the infiltration of 24 immune cell types in breast cancer and the key genes was analyzed based on data from the TCGA database (specifically for BRCA). The results were visualized using the ggplot2 package (version 3.3.6). The findings revealed that the four key genes exhibited positive correlations with immune cells such as Th2 cells, activated DC, Th1 cells, and Treg, while exhibiting negative correlations with immune cells such as NK cells, mast cells, eosinophils, and others (Fig. 7).

Immune infiltration analyses of the key genes (A–D CCNB2, DLGAP5, HJURP, and TTK). CCNB2 cyclin B2, DLGAP5 DLG-associated protein 5, HJURP: Holliday junction recognition protein, TTK threonine and tyrosine kinase
Therapeutic Targets and Sensitive Drug Mining
The ten hub genes were analyzed using the GSCA database to identify drugs that exhibit the strongest correlation with these targets. From this analysis, it was found that RDEA119, selumetinib, and Nutlin-3a(-) displayed strong positive correlations with CCNB2, DLGAP5, HJURP, and TTK (Fig. 8A, B). As a result, it is speculated that these three compounds could serve as effective drugs for treating TNBC. Targeting these four genes may potentially improve the prognosis for patients with TNBC.

Small-molecule drugs from the GDSC (A) and CTRP (B) targeting the hub genes. GDSC Genomics of Drug Sensitibity in Cancer, CTRP Cancer Therapeutics Response Portal
Next, the compound Nutlin-3a(-) was molecularly docked with these proteins encoded by the four key genes. XP score and MM-GBSA dG Bind methods were used to evaluate the binding free energy and the binding stability. The lower the XP docking score and MM-GBSA score were, the lower the binding free energy and the higher the binding stability of the compound with protein. The results show that Nutlin-3a(-) did not bind with HJURP and CCNB2. However, the binding force between Nutlin-3a(-) and DLGAP5 and TTK was quite low, suggesting that Nutlin-3a(-) can target these two targets (Table 3; Fig. 9) with high stability.

Molecular docking of Nutlin-3a(-) and the key genes of DLGAP5 (A) and TTK (B). LGAP5 DLG-associated protein 5, TTK threonine and tyrosine kinase
Discussion
TNBC, an aggressive subtype of breast cancer, differs from luminal and Her2 + types because of its strong heterogeneity. Currently, to guide prognosis, TNM staging and clinical pathological indicators are commonly employed. In recent years, with the development of high-throughput sequencing technologies, multi-gene prediction has emerged as a new approach in this regard. Several polygenic tests have been suggested for use, including the 21-gene OncotypeDx assay [24], 70-gene MammaPrint [25], and PAM50 assay, among others. However, in view of the limitations of these methods, such as insufficient discrimination for TNBC and long detection time, there is still an urgent need to develop new markers. Therefore, identifying differentially expressed genes (DEGs) specific to TNBC is crucial for discovering new diagnostic biomarkers and potential therapeutic targets. This study aimed to analyze three GEO datasets to establish a group of DEGs highly expressed in TNBC. After identifying ten hub genes (CCNB1, CCNB2, DLGAP5, HJURP, KIF15, KIF23, NCAPG, NUSAP1, SPAG5, and TTK) from the DEGs, four of the key genes were identified as diagnostic markers that can distinguish TNBC from other breast cancer subtypes. Although this four-gene model has not been comprehensively compared with the 21-gene model and the 70-gene model in the real world, this model may have some practical value because of its simplicity. Alternatively, the four genes can be added to other models. Next, the survival analysis showed a significant difference of PFI between the high- and low-expression group of the four genes. Thus, these four genes (CCNB2, DLGAP5, HJURP, and TTK) were considered to be a group of prognostic markers. While CCNB2 has been extensively studied in the past, DLGAP5 and HJURP have had limited research conducted on them. As the previous studies reported, downregulation of DLGAP5 repressed the proliferation of breast cancer MDA-MB-231 cells and induced cell cycle arrest [26]. DLGAP5 may prompt HCC cellular proliferation, invasion and metastasis [27]. There are also some studies on HJURP. HJURP is expressed at a higher level in breast cancer and regulates cell proliferation [28]. Hu’s study suggests that HJURP mRNA level is a prognostic factor for disease-free and overall survival in patients with breast cancer [29]. These studies were conducted in breast cancer, not specifically for TNBC. To the best of our knowledge, this study marks the first instance where these two genes have been identified as potential prognostic markers for TNBC. Based on the findings of this study, we propose that these four genes can serve as a set of diagnostic and prognostic markers for TNBC.
In addition, this study aimed to identify potential therapeutic targets and potentially effective compounds for TNBC. By conducting GO and KEGG analyses, as well as GSEA analysis, the main biological enrichments were found to involve cell cycle and p53 pathways. This correlation emphasizes the importance of exploring therapeutic strategies targeting these pathways in TNBC. In previous studies, the development of p53-targeted therapies for use as medicine has encountered challenges. However, recent advancements in research have shown promising results with certain compounds demonstrating effectiveness in TNBC treatment. For instance, the MDM2 inhibitor idasanutlin has gained approval for treating acute myeloid leukemia (AML) and is currently being assessed in clinical trials for other types of tumors. Nutlin-3a(-), another MDM2 inhibitor identified in this study, has also been investigated in preclinical studies involving breast cancer cells [30]. It was also reported that nutlin-3a(-) can enhance carboplatin-mediated DNA damage [31]. In this study, we screened three compounds that can target the four key genes based on GSCA database, and nutlin-3a(-) was one of them. We ran a molecular docking analysis based on nutlin-3a(-) and the four targets, and the results showed that nutlin-3a(-) could be closely connected with TTK and DLGAP5. Besides Nutlin-3a(-), two other compounds, namely RDEA119 and selumetinib, have shown potential sensitivity to key genes implicated in TNBC. RDEA119 is an effective, non-ATP-competitive, and highly selective inhibitor of MEK1 and MEK2, and selumetinib (AZD 6244, ARRY-142886) is an inhibitor mainly targeting MEK1. Previous studies [32,33,34] have shown that selumetinib can inhibit the proliferation and metastasis of breast cancer cells and promote apoptosis, but this study has not yet entered the clinical stage. Further research is needed to determine its efficacy specifically in triple-negative breast cancer, both in vitro and in vivo.
This study has some limitations. First, it is limited to analyzing data from databases and does not validate the findings through biological experiments. Second, there may be slight diversity between different GSE data sets, thus causing slight bias of the results.
Conclusion
In this study, the analysis of three TNBC datasets led to the identification of ten hub genes. Subsequent investigations into the diagnostic and prognostic values of these genes revealed a group of gene sets (CCNB2, DLGAP5, HJURP, and TTK) that can serve as diagnostic and prognostic markers for TNBC. Furthermore, the enrichment analysis of differentially expressed genes in TNBC highlighted significant biological functions such as the cell cycle and the p53 signaling pathway. Additionally, three compounds that exhibited sensitivity towards these differentially expressed genes were explored, suggesting their potential as novel therapeutic options for TNBC. These findings provide valuable insights for further research in the field of TNBC and hold promise for the development of effective diagnostic tools and targeted treatments.
Data Availability
All data generated or analyzed during this study are included in this published article/as supplementary information files.
References
Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49.
Nagini S. Breast cancer: current molecular therapeutic targets and new players. Anticancer Agents Med Chem. 2017;17(2):152–63.
Poorolajal J, Nafissi N, Akbari ME, et al. Breast cancer survival analysis based on immunohistochemistry subtypes (ER/PR/HER2): a retrospective cohort study. Arch Iran Med. 2016;19(10):680–6.
Plevritis SK, Munoz D, Kurian AW, et al. Association of screening and treatment with breast cancer mortality by molecular subtype in US Women, 2000–2012. JAMA. 2018;319(2):154–64.
Wang J, Wu SG. Breast cancer: an overview of current therapeutic strategies, challenge, and perspectives. Breast Cancer (Dove Med Press). 2023;15:721–30.
Kotsifaki A, Alevizopoulos N, Dimopoulou V, et al. Unveiling the immune microenvironment’s role in breast cancer: a glimpse into promising frontiers. Int J Mol Sci. 2023;24(20):15332.
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34.
Ueno NT, Zhang D. Targeting EGFR in triple negative breast cancer. J Cancer. 2011;2:324–8.
Blanco E, Sangai T, Wu S, et al. Colocalized delivery of rapamycin and paclitaxel to tumors enhances synergistic targeting of the PI3K/Akt/mTOR pathway. Mol Ther. 2014;22(7):1310–9.
Gibson J. Anti-PD-L1 for metastatic triple-negative breast cancer. Lancet Oncol. 2015;16(6): e264.
Narayan P, Wahby S, Gao JJ, et al. FDA approval summary: atezolizumab plus paclitaxel protein-bound for the treatment of patients with advanced or metastatic TNBC whose tumors express PD-L1. Clin Cancer Res. 2020;26(10):2284–9.
Nanda R, Chow LQ, Dees EC, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 study. J Clin Oncol. 2016;34(21):2460–7.
Szklarczyk D, Kirsch R, Koutrouli M, et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638–46.
Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–58.
Liu CJ, Hu FF, Xie GY, et al. GSCA: an integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief Bioinform. 2023;24(1):bbac558.
Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10.
Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45(10):1113–20.
Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Liu J, Lichtenberg T, Hoadley KA, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400–16.
Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9.
Mulakala C, Viswanadhan VN. Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies? J Mol Graph Model. 2013;46:41–51.
Berger AC, Korkut A, Kanchi RS, et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell. 2018;33(4):690–705.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7.
Sparano JA, Paik S. Development of the 21-gene assay and its application in clinical practice and clinical trials. J Clin Oncol. 2008;26(5):721–8.
van ‘t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6.
Xu T, Dong M, Li H, et al. Elevated mRNA expression levels of DLGAP5 are associated with poor prognosis in breast cancer. Oncol Lett. 2020;19(6):4053–65.
Tang N, Dou X, You X, et al. Pan-cancer analysis of the oncogenic role of discs large homolog associated protein 5 (DLGAP5) in human tumors. Cancer Cell Int. 2021;21(1):457.
Mao M, Jia Y, Chen Y, et al. HJURP regulates cell proliferation and chemo-resistance via YAP1/NDRG1 transcriptional axis in triple-negative breast cancer. Cell Death Dis. 2022;13(4):396.
Hu Z, Huang G, Sadanandam A, et al. The expression level of HJURP has an independent prognostic impact and predicts the sensitivity to radiotherapy in breast cancer. Breast Cancer Res. 2010;12(2):R18.
Das M, Dilnawaz F, Sahoo SK. Targeted nutlin-3a loaded nanoparticles inhibiting p53-MDM2 interaction: novel strategy for breast cancer therapy. Nanomedicine (Lond). 2011;6(3):489–507.
Tonsing-Carter E, Bailey BJ, Saadatzadeh MR, et al. Potentiation of carboplatin-mediated DNA damage by the Mdm2 modulator Nutlin-3a in a humanized orthotopic breast-to-lung metastatic model. Mol Cancer Ther. 2015;14(12):2850–63.
Maiello MR, D’Alessio A, Bevilacqua S, et al. EGFR and MEK blockade in triple negative breast cancer cells. J Cell Biochem. 2015;116(12):2778–85.
Zhou Y, Lin S, Tseng KF, et al. Selumetinib suppresses cell proliferation, migration and trigger apoptosis, G1 arrest in triple-negative breast cancer cells. BMC Cancer. 2016;16(1):818.
Demirtas Korkmaz F, Dogan Turacli I, Esendagli G, et al. Effects of thiostrepton alone or in combination with selumetinib on triple-negative breast cancer metastasis. Mol Biol Rep. 2022;49(11):10387–97.
Acknowledgements
We sincerely acknowledge TCGA and GEO database for providing transcriptomic and clinical data. We also thank all individuals who participated in this study and donated samples.
Funding
No funding or sponsorship was received for this study. The journal’s Rapid Service Fee was paid by Xinghe Wang.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation and data collection were performed by Yan Li and Shengjie Yang. Analysis and interpretation of data: Yan Li, Lu Qi and Shengjie Yang. The first draft of the manuscript was written by Yan Li. Revising the article critically: Yan Li, Yinjuan Li and Xinghe Wang. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Yan Li, Lu Qi, Shengjie Yang, Yinjuan Li, and Xinghe Wang declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical Approval
This study was based on publicly available data, and patients were not directly involved in the entire research process. Therefore, no Independent Review Board approval was needed. This study involved the use of several public databases, including: Search Tool for the Retrieval of Interacting Genes (STRING, http://cn.string-db.org), UALCAN (https://ualcan.path.uab.edu/index.html), Gene Set Cancer Analysis (GSCA, http://bioinfo.life.hust.edu.cn/GSCA/#/), Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and The Cancer Genome Atlas (TCGA, https://www.cancer.gov/about-NCI/organization/ccg/research/structural-genomics/TCGA).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Y., Yang, S., Qi, L. et al. Identification of a Group of Therapeutic Targets and Prognostic Biomarker for Triple Negative Breast Cancer. Adv Ther 41, 1621–1636 (2024). https://doi.org/10.1007/s12325-024-02806-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-024-02806-z