
Abstract
Background
Triple-negative breast cancer (TNBC), defined by lack of expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), is characterized by early recurrence of disease and poor survival.
Objective
Here, we sought to identify genes associated with TNBC that could provide new insight into gene dysregulation in TNBC and, at the same time, provide additional potential therapeutic targets for breast cancer treatment.
Methods
Gene expression profiles from accession series GSE76275 were downloaded from the Gene Expression Omnibus database (GEO). The Cancer Genome Atlas (TCGA) was used to validate potential hub genes in the TCGA database. Protein–protein interaction (PPI) networks were identified using STRING (Search Tool for the Retrieval of Interacting Genes/Proteins). Finally, overall survival (OS) and relapse-free survival (RFS) analysis of hub genes was performed using a Kaplan–Meier plotter online tool.
Results
A total of 750 genes were identified after analysis of GSE76275. After validation with the TCGA database, a total of 155 differentially expressed genes (DEGs) were consistent with those identified by GSE76275. Based on the STRING database, we constructed a PPI network using the DEGs obtained from GSE76275 datasets. Furthermore, in the prognostic analysis of the 155 DEGs, we found that there were 10 genes associated with OS and 33 genes associated with RFS. Combined with the degree scores from the PPI network, a total of ten genes with the highest degree scores were selected as hub genes pertaining to TNBC.
Conclusion
Our research provides new insight into the subnetwork of biomarkers connected with TNBC, which could be useful for prognostication and risk stratification of TNBC patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Triple-negative breast cancer (TNBC) is a particular type of breast cancer that does not express estrogen receptor (ER), progesterone receptor (PR), or human epidermal growth factor receptor 2 (HER2). TNBC accounts for approximately 15–20% of breast cancer cases and is characterized by early recurrence and poor survival, as compared with other types of breast cancer [1]. TNBC is a heterogeneous disease, not only at the molecular level, but also in terms of its pathology and clinical manifestation. Due to its high prevalence, lack of effective targeted therapies, poor prognosis, and its tendency to affect younger women, TNBC has become an intractable problem for clinical treatment.
Due to the difficulty in treating TNBC, current research scholars are focusing on targeted gene therapy for TNBC based on the recent discovery of TNBC-related genes. A computational approach, through the analysis of complex biological networks and the identification of novel genes associated with TNBC, could help researchers cope with various confounding biological issues.
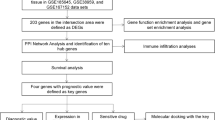
In this study, we sought to identify differentially expressed genes (DEGs) through analysis of the gene expression omnibus (GEO) database for TNBC combined with survival analysis. The Cancer Genome Atlas (TCGA) database was used to validate identified DEGs. The Search Tool for the Retrieval of Interacting Genes (STRING) database and protein–protein interaction (PPI) database were used to identify candidate genes. Combined with survival analysis, we sought to identify key genes associated with TNBC.
Materials and methods
Microarray data
GSE76275 expression profiles were obtained from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo), which is a free public repository for data storage, including microarray data and next-generation sequencing. The array data available for GSE76275 (Affymetrix Human Genome U133 Plus 2.0 Array) consist of 198 triple-negative breast cancer tissue samples and 67 non-triple-negative breast cancer tissue samples. In TNBC samples, the expression of ER and PgR was less than 1% using immunohistochemical assays; in non-TNBC samples, a total of 22 HER2 positive patients were included.
Data processing
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) is an interactive tool that can be used to compare two groups of samples to identify DEGs in almost any GEO series [2]. GEO2R performs comparisons on original submitter-supplied processed data tables using the GEO query and limma R packages (linear models for microarray analysis) [3, 4] from the Bioconductor project. The GEO query R package parses GEO data into R data structures that can be used by other packages. The limma R package has emerged as one of the most widely used statistical tests for identifying DEGs, which handles a wide range of experimental designs and data types and applies multiple testing corrections on P values to help correct for the occurrence of false positives. We used GEO2R to screen for DEGs between TNBC and non-TNBC samples in the datasets. P < 0.05 and |log FC| > 1 were used as the cutoff criteria for the identification of DEGs.
Integration of protein–protein interaction (PPI) networks
We evaluated the protein–protein interaction (PPI) data using the Search Tool for the Retrieval of Interacting Genes (STRING, http://string.embl.de/) database, an online tool designed to evaluate the protein–protein interaction (PPI) information. STRING (version 9.0) includes 5,214,234 proteins from 1133 organisms [5]. To identify connections among DEGs, the protein products of DEGs were matched to STRING using Cytoscape software. Only experimentally validated interactions with a combined score > 0.4 were considered to be significant.
Validation of DEGs in TCGA database
The cancer genome atlas (TCGA, https://cancergenome.nih.gov/publications/publicationguidelines) is a collaboration between the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI). TCGA has been used to generate comprehensive, multi-dimensional maps of the key genomic changes in 33 types of cancer [6]. We downloaded the genomic data through TCGA biolinks and a total of 71 cases of TNBC were selected. We utilized the edgeR data processing package [7] from Bioconductor; P < 0.05 was considered a significant difference.
Kaplan–Meier plotter
Kaplan–Meier plotter, a software available online that specializes survival analysis, was used to estimate prognosis information for the ten key candidate genes we had previously identified. The software can not only effectively evaluate the effect of 54,675 genes on survival drawing on data from 10,461 cancer samples [8], but also uses the log-rank test to generate survivorship curves. We were able to stratify 618 basal subtype patients based on RFS and 241 basal subtype patients based on OS. The hazard ratio with 95% confidence intervals and the log-rank P < 0.05 were calculated.
Results
Identification of DEGs between TNBC and NTNBC samples and verification via the TCGA
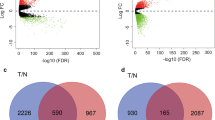
We analyzed a total of 198 TNBC tissue samples and 67 non-triple-negative breast cancer tissue (NTNBC) samples. The series from each chip was analyzed separately using GeneSpring software, finally resulting in the list of DEGs. A total of 750 genes were identified after analyzing GSE76275, of which 491 were upregulated and 259 were downregulated. The first ten most upregulated and downregulated genes are shown in Table 1. Then, we screened the DEGs for TNBC using the TCGA database; we found that a total of 155 DEGs were consistent with those identified in the GSE76275 series.
PPI network construction
A PPI network was constructed using the STRING database. A total of 619 nodes and 2255 edges were mapped in the PPI network, with a local clustering coefficient of 0.352 and a PPI enrichment P value < 1.0e−16. Based on the results from the STRING database, all the genes were ranked by degree scores; epidermal growth factor receptor (EGFR) exhibited the highest node degree with forty-six. And the PPI network is shown in Fig. 1.

Visualization of the protein–protein interaction (PPI) network of the identified differentially expressed genes (DEGs)
The association between DEGs and prognosis of TNBC
Prognostic data associated with the 155 DEGs overlapped both in GSE7625 datasets and the TCGA database were evaluated using www.kmplot.com. Overall survival (OS) and relapse-free survival (RFS) for patients with TNBC were insufficient to adequately analyze prognosis, therefore we evaluated the prognosis for breast cancer patients expressing these genes by basal subtype. Ten genes were found to be associated with OS and 33 genes were associated with RFS. Combining the degree scores of the PPI network yielded a total of ten genes with higher degree scores, which were then selected as hub genes for TNBC. The ten hub genes were epidermal growth factor receptor (EGFR), keratin 16 (KRT16), ret proto-oncogene (RET), sex-determining region Y-box 10 (SOX10), PDZ domain-containing 1 (PDZK1), X-box binding protein 1 (XBP1), trefoil factor 3 (TFF3), prostaglandin E receptor 3 (PTGER3), NME/NM23 family member 5 (NME5) and interleukin 6 signal transducer (IL6ST) (Figs. 2, 3). Among them, EGFR, KRT16 and SOX10 were of high expression in TNBC, and the other seven genes were of low expression in TNBC (Table 2).

Prognostic value of three high-expression hub genes in TNBC patients. Prognostic value of EGFR, KRT16 and SOX10 were obtained from www.kmplot.com. The desired Affymetrix IDs are valid: 1565483_at (EGFR), 209800_at (KRT16), 209842_at (SOX10). HR hazard ratio, CI confidence interval

Prognostic value of seven low-expression hub genes in TNBC patients. Their prognostic value was obtained from www.kmplot.com. The desired Affymetrix IDs are valid: 205879_x_at (RET), 205380_at (PDZK1), 200670_at (XBP1), 204623_at (TFF3), 208169_s_at (PTGER3), 206197_at (NME5), 204863_s_at (IL6ST). HR hazard ratio, CI confidence interval
Discussion
The term TNBC was first introduced in 2005 to refer a certain type of breast cancer for which conventional chemotherapy is the only pharmacological treatment available, as patients with TNBC lack expression of ER, PR, and HER2 and are, therefore, not eligible to receive hormonal therapy or anti-HER2 agents [9]. In addition, two major subgroups of TNBC characterized on the basis of gene ontologies and differential gene expression profile have been reported: basal-like TNBC driven by genes associated with the cell cycle, cell division, and DNA damage response; and mesenchymal-like TNBC driven by genes involved in cell motility, cell differentiation, and growth factor pathways [10]. Therefore, TNBC is actually highly heterogeneous. Since TNBC has a high mortality rate, early molecular diagnosis plays a critical role achieving a favorable prognosis. To date, numerous genes have been found to participate in breast cancer formation and can act as specific diagnostic indicators with potential clinical applications. Although dozens of such genes have been identified, the exact mechanism of tumorigenesis in TNBC remains to be fully elucidated. Therefore, it is of great importance to identify more candidate genes for the diagnosis and treatment of TNBC.
In our analysis, 750 genes obtained from GSE76275 were investigated; of these, 491 were upregulated and 259 were downregulated. Then, these genes were further verified using the TCGA database, which showed that a total of 155 genes were consistent with those identified in the analysis of GSE76275. Next, we constructed a PPI network using the DEGs obtained from GSE76275 database. Furthermore, via Kaplan–Meier analysis, we sought to determine prognoses for patients from the 155 genes. However, there were an insufficient number of TNBC cases available to accurately utilize the Kaplan–Meier plotter; therefore, we broadened the search to include all patients with basal-like breast cancer instead of simply TNBC. At the molecular level, TNBC has significant overlap with the basal-like subtype with approximately 80% of patients with TNBCs also being classified as having the basal-like subtype [1]. Therefore, we used the Kaplan–Meier plotter to identify genes related to the prognosis of basal subtype breast cancer patients; we found there were 10 genes associated with OS and 33 genes associated with RFS. Combining the degree scores of the PPI network allowed us to assemble a list of 10 genes, including EGFR, KRT16, RET, SOX10, PDZK1, XBP1, TFF3, PTGER3, NME5, and IL6ST, that we identified as hub genes for TNBC.
The protein encoded by EGFR is a transmembrane glycoprotein that is a member of the protein kinase superfamily and acts as a receptor for members of the epidermal growth factor family. The human epidermal growth factor receptor (EGFR) family consists of four EGFRs: EGFR (ErbB2 or HER1), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) [11]. EGFR activation results in cell proliferation, motility, and survival by activating downstream signaling pathways such as the c-Jun N-terminal kinase, phosphatidylinositol-3-kinase, and mitogen-activated protein kinase pathways [12]. As an important regulator of epithelial cell biology, the function of EGFR in breast tumors is complex and may vary according to some clinical features like estrogen receptor (ER) and HER2 status [13]. EGFR has been reported to be overexpressed, at both the mRNA and protein level, in approximately in 50% of TNBC patients and has been observed to be an independent predictor of poor prognosis [14, 15]. Significant evidence shows that EGFR overexpression in TNBC makes TNBC more difficult to treat and significantly lowers the 10-year survival rate in breast cancer patients [16, 17].
The protein encoded by KRT16 is a member of the keratin gene family. The keratins are intermediate filament proteins responsible for the structural integrity of epithelial cells. Studies have established that KRT16 is significantly downregulated in luminal cell lines, but upregulated in basal-like breast cancer cell lines as indicated by Western blot [18]. Patients with high expression of KRT16 in primary breast cancer exhibited a shorter RFS [19, 20]. These results imply that KRT16 expression may be associated with poor prognosis in breast cancer.
RET, located on human chromosome 10q11.2, encodes a transmembrane receptor and member of the tyrosine protein kinase family of proteins [21]. Binding of ligands such as GDNF (glial cell line-derived neurotrophic factor) to the encoded receptor can lead to RET dimerization and activation of downstream signaling pathways, which in turn regulate cell differentiation, growth, migration, and survival. RET is highly expressed in subsets of hormone receptor-positive breast cancer. Moreover, cell line studies indicate that estrogen seems to be able to stimulate high expression of RET and that RET activation can lead to increased incidence of oncogenicity [22, 23]. RET expression differed significantly between breast cancer subtypes, with lowest RET expression in the basal-like subtype and the highest expression in the luminal A subtype; these data may indicate that RET is a favorable prognostic indicator [24].
The protein encoded by SOX10 may act as a transcriptional activator, which plays an important role in embryonic development and determination of cell fate in different tissues and at different stages of development [25]. Transcriptional activity of SOX10 has been shown to be sufficient to transform pluripotent cells into multipotent state; specifically, it can regulate the stem/progenitor activity of mammary epithelial cells [26, 27]. Researchers have shown that SOX10 has the capacity to promote both stem-like and EMT-like behaviors, indicating that SOX10 may be directly responsible for tumor initiation and progression [28].
PDZK1 encodes a PDZ domain-containing scaffolding protein. PDZ domain-containing molecules bind to and mediate the subcellular localization of target proteins. Studies showed that expression of PDZK1 is limited to epithelial cells and its overexpression has been reported in a variety of cancer types, including breast cancer particularly in young patients [29, 30]. PDZK1 is induced by estradiol in hormone-responsive breast cancer, which may be important when considering estrogen treatment [31].
XBP1 encodes a transcription factor that participates in the unfolded protein response (UPR). XBP1 is a novel protein involved in cancer progression and outcome, including the development of breast cancer [32,33,34]. Several studies have shown that XBP1 is upregulated in breast cancer cell lines and is associated with metastasis [34]. Another analysis revealed that XBP1 gene expression was closely related to poor prognosis in a cohort of patients with TNBC [33].
TFF3 belongs to the trefoil family, found on chromosome 21 with two other related trefoil family member genes, TFF1 and TFF2 [35]. TFF3 has been reported as a biomarker for gastric cancer, lung cancer, prostate cancer, and cholangiocarcinoma [35, 36]. In breast cancer patients, TFF3 levels were significantly higher than in normal individuals, thus suggesting that TFF3 may become an effective marker for breast cancer screening [37].
The protein encoded by PTGER3 is a member of the G-protein coupled receptor family and is one of four receptors identified for prostaglandin E2 (PGE2). EP1-4 receptor expression has been identified in a variety of cancers, including breast cancer [38]. However, unlike the other receptors, PG3 acts as a protective factor in the progression of breast cancer; its expression is closely related to better prognosis, although the exact mechanisms remain unclear [39].
The NME family of proteins consists of 10 isoforms, NME1 to NME10, which are diverse in their enzymatic activities and patterns of subcellular localization. As a member of the NME family, NME5 also participates in exonuclease activity, implying that it plays a role in DNA proofreading and repair [40]. The protein encoded by IL6ST functions as a part of the cytokine receptor complex, a signal transducer shared by many cytokines including interleukin 6 (IL6), ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), and oncostatin M (OSM). IL-6 secreted in the breast cancer microenvironment can regulate cancer progression by enhancing migration and invasion as well as inducing EMT [41]. Blockade of IL-6 signaling decreases proliferation, migration, and invasion capacity of breast cancer cells [42].
Here, we sought to identify the differentially expressed genes in TNBC using bioinformatics analyses. The newly identified ten DEGs may help elucidate the underlying molecular mechanisms of TNBC development and provide potential new avenues for personalized therapy. Two limitations should be acknowledged in this investigation. One limitation is that the TNBC sample number was too low for adequate prognostic analysis; therefore, we utilized basal subtype breast cancer samples, not just the TNBC subset. We can use the analysis of the effects of the ten genes on prognosis in patients with basal subtype breast cancer to speculate on the effects of the genes on prognosis in patients with TNBC. Besides, the selection of the hub genes was only validated in TCGA database. Further validation using tissue or cultured cell is needed to better confirm the findings of the identified genes in TNBC of our investigation.
In summary, in this article, we identified ten novel DEGs that have not previously been associated with triple-negative breast cancer. Our results will guide us toward identifying TNBC-related therapeutic targets in future investigation. Thus, bioinformatics analysis may give new insight into the mechanisms of the development of TNBC and other types of breast cancers.
Abbreviations
- TNBC:
-
Triple-negative breast cancer
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- HER2:
-
Human epidermal growth factor receptor 2
- DEG:
-
Differentially expressed genes
- GEO:
-
Gene expression omnibus
- PPI:
-
Protein–protein interaction
- TCGA:
-
The Cancer genome atlas
- OS:
-
Overall survival
- RFS:
-
Relapse-free survival
- FC:
-
Log fold control
- EGFR:
-
Epidermal growth factor receptor
- KRT16:
-
Keratin 16
- RET:
-
Ret proto-oncogene
- SOX10:
-
Sex-determining region Y-box 1
- PDZK1:
-
PDZ domain-containing 1
- XBP1:
-
X-box binding protein 1
- TFF3:
-
Trefoil factor 3
- PTGER3:
-
Prostaglandin E receptor 3
- NME5:
-
NME/NM23 family member 5
- IL6ST:
-
Interleukin 6 signal transducer
References
Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–48.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41:991–5.
Davis Sean, Meltzer Paul S. GEO query: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–7.
Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3(1):Article3.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta- Cepas J, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:447–52.
Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012;19(2):197–208.
Pareja F, Geyer FC, Marchiò C, Burke KA, Weigelt B, Reis-Fiho JS. Triple-negative breast cancer: the importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer. 2016;2:16036.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–67.
Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136:331.
Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res. 2016;6:1609–23.
Hoadley KA, Weigman VJ, Fan C, Sawyer LR, He X, Troester MA, et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genom. 2007;8:258.
Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–74.
Burness ML, Grushko TA, Olopade OI. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker? Cancer J. 2010;16:23–32.
Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14(5):1368–76.
Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30(21):2615–23.
Joosse Simon A, Hannemann Juliane, Spotter Julia, Bauche Andreas, Andreas Antje, Muller Volkmar, et al. Changes in keratin expression during metastatic progression of breast cancer: impact on the detection of circulating tumor cells. Hum Cancer Biol. 2012;18(4):993–1003.
Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–9.
Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9.
Morandi A, Plaza-Menacho I, Isacke CM. RET in breast cancer: functional and therapeutic implications. Trends Mol Med. 2011;17(3):149–57.
Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M, et al. The ret receptor tyrosine kinase pathway functionally interacts with the ERalpha pathway in breast cancer. Cancer Res. 2008;68(10):3743–51.
Plaza-Menacho I, Morandi A, Robertson D, Pancholi S, Drury S, Dowsett M, et al. Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene. 2010;29(33):4648–57.
Mechera R, Soysal SD, Piscuoglio S, Ng CKY, Zeindler J, Mujagic E, et al. Expression of RET is associated with Oestrogen receptor expression but lacks prognostic significance in breast cancer. BMC Cancer. 2019;19:41.
Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15–30.
Kim YJ, Lim H, Li Z, Oh Y, Kovlyagina I, Choi IY, Dong X, et al. Generation of multipotent induced neural crest by direct reprogramming of human postnatal fibroblasts with a single transcription factor. Cell Stem Cell. 2014;15:497–506.
Dravis C, Spike BT, Harrell JC, Johns C, Trejo CL, Southard-Smith EM, et al. Sox10 regulates stem/progenitor and mesenchymal cell states in mammary epithelial cells. Cell Rep. 2015;12:2035–48.
Dravis C, Spike BT, Harrell JC, Johns C, Trejo CL, Southard-Smith EM, et al. Sox10 regulates stem/progenitor and mesenchymal cell states in mammary epithelial cells. Cell Rep. 2015;12(12):2035–48.
Kocher O, Comella N, Tognazzi K, Brown LF. Identification and partial characterization of PDZK1: a novel protein containing PDZ interaction domains. Lab Invest. 1998;78:117–25.
Kim H, Abd Elmageed ZY, Ju J, Naura AS, Abdel-Mageed AB, Varughese S, et al. PDZK1 is a novel factor in breast cancer that is indirectly regulated by estrogen through IGF-1R and promotes estrogen-mediated growth. Mol Med. 2013;19:253–62.
Vic P, Vignon F, Derocq D, Rochefort H. Effect of estradiol on the ultrastructure of the MCF7 human breast cancer cells in culture. Cancer Res. 1982;42:667–73.
Shajahan AN, Riggins RB, Clarke R. The role of X-box binding protein-1 in tumorigenicity. Drug News Perspect. 2009;22(5):241–6.
Chen X, Iliopoulos D, Zhang Q, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature. 2014;508(7494):103–7.
Li H, Chen X, Gao Y, Wu J, Zeng F, Song F. XBP1 induces snail expression to promote epithelial-to-mesenchymal transition and invasion of breast cancer cells. Cell Signal. 2015;27(1):82–9.
Kjellev S. The trefoil factor family—small peptides with multiple functionalities. Cell Mol Life Sci. 2009;66(8):1350–69.
May FE, Church ST, Major S, Westley BR. The closely related estrogen-regulated trefoil proteins TFF1 and TFF3 have markedly different hydrodynamic properties, overall charge, and distribution of surface charge. Biochemistry. 2003;42(27):8250–9.
Ishibashi Yuko, Ohtsu Hiroshi, Ikemura Masako, Kikuchi Yasuko, Niwa Takayoshi, Nishioka Kotoe, et al. Serum TFF1 and TFF3 but not TFF2 are higher in women with breast cancer than in women without breast cancer. Sci Rep. 2017;7:4846.
Cheuk IW, Shin VY, Siu MT, Tsang JY, Ho JC, Chen J, et al. Association of EP2 receptor and SLC19A3 in regulating breast cancer metastasis. Am J Cancer Res. 2015;5(11):3389–99.
Semmlinger A, von Schoenfeldt V, Wolf V, Meuter A, Kolben TM, Kolben T, et al. EP3 (prostaglandin E2 receptor 3) expression is a prognostic factor for progression-free and overall survival in sporadic breast cancer. BMC Cancer. 2018;18:431.
Puts GS, Leonard MK, Pamidimukkala NV, Snyder DE, Kaetzel DM. Nuclear functions of NME proteins. Lab Invest. 2018;98(2):211–8.
Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X, et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget. 2017;8(13):20741–50.
Gyamf Jones, Lee Yun-Hee, Eom Minseob, Choi Junjeong. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci Rep. 2018;8:8859.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments and revised the manuscript: XMW, YY. Performed the experiments: QXZ, LPS, HL. Analyzed the data: QXZ, HL. Responsible for bioinformatics and bio-statistics analysis: QXZ, HL. Wrote the paper: QXZ.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Zhai, Q., Li, H., Sun, L. et al. Identification of differentially expressed genes between triple and non-triple-negative breast cancer using bioinformatics analysis. Breast Cancer 26, 784–791 (2019). https://doi.org/10.1007/s12282-019-00988-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12282-019-00988-x