
Abstract
Metabolic reprogramming is a newly emerged hallmark of cancer attaining a recent consideration as an essential factor for the progression and endurance of cancer cells. A prime event of this altered metabolism is increased glucose uptake and discharge of lactate into the cells surrounding constructing a favorable tumor niche. Several oncogenic factors help in promoting this consequence including, pyruvate kinase M2 (PKM2) a rate-limiting enzyme of glycolysis in tumor metabolism via exhibiting its low pyruvate kinase activity and nuclear moon-lightening functions to increase the synthesis of lactate and macromolecules for tumor proliferation. Not only its role in cancer cells but also its role in the tumor microenvironment cells has to be understood for developing the small molecules against it which is lacking with the literature till date. Therefore, in this present review, the role of PKM2 with respect to various tumor niche cells will be clarified. Further, it highlights the updated list of therapeutics targeting PKM2 pre-clinically and clinically with their added limitations. This upgraded understanding of PKM2 may provide a pace for the reader in developing chemotherapeutic strategies for better clinical survival with limited resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer, a dynamic disease which develops one in three people during lifetime and considered as a major health burden to the society across the worldwide. According to the National Cancer Institute (NIH), roughly 1.7 million new cases will be identified and 0.6 million people will perish with cancer in the USA [1]. Typically, throughout the progression of a disease, cancers become more variant due to their somatic evolutionary changes [2] and this is referred as tumor heterogeneity which was explained by a pathologist, Rudolf Virchow in 1800s [3]. Moreover, it has been considered as a key element for the failure of classical and modern therapeutics and offers multiple hindrances like chemotherapeutic resistance [4] cancer pharmacogenomics [5] and development of cancer stem-like cells [6]. It means the tumor shows the existence of divergent tumors cells in terms of genetic and phenotypic patterns within the subpopulations of same histopathological types of cancer in different patients or within tumors that show divergent biological behavior and metastatic potential [7, 8]. This heterogeneity may be spatial and temporal in which former describes the uneven distribution of cancers subclones in various sites of primary tumors due to genetic instability whereas the latter describes the variation in molecular markers expression in same sites of tumors over a period of time [9]. However, a new perspective of heterogeneity has been explained recently based on the metabolic demands of cancer cells termed as metabolic heterogeneity [10]. Clinical studies of various cancers like breast cancer, acute myeloid leukemia, and Barrett's esophagus showed a positive correlation with tumors heterogeneity and displayed a high risk for cancer progression [11,12,13]. Four inter-dependent drivers like epigenetic regulation, cellular differentiation hierarchies, gene expression stochasticity, and microenvironment have direct control of cancer cells resulting in the tumor heterogeneity [14]. Many researchers found a high spatial and metabolic heterogeneity under different microenvironments such as developing more vascularate in the hypoxic region [15], aberrant expression of glycolytic enzymes [16], lactate mediated acidification [17], and autonomous cell proliferation without stimuli [18]. Consequently, these relationships between cancer cells with its microenvironment might contribute the cancers cells to adopt various conditions existed within the tumor niche [19].
Despite these heterogeneities, cancer cells even display some basic analogous characteristics of various cancers which are often described as “hallmarks of cancer” required for their growth and multiplication [20]. Amongst all, dysregulated cellular energetics has a starring role in supporting the tumor development by providing the desired necessities to the cells for fulfilling their metabolic demands by increasing aerobic glycolysis. This fulfillment gets accomplished by modifying the crucial metabolic enzymes (Hexokinase II, PKM2, LDH-A, and enolase, etc.) [21, 22], transporters (GLUT1, MCT-1, and MCT-4 etc.) [23] involved in different biosynthetic pathways through various oncogenic stimuli (HIF-1α, c-Myc, PI3K/mTOR signaling) [24, 25]. Amongst all, PKM2 has a major role in shifting the tumor towards the aerobic glycolysis and even remodels the tumor microenvironment (TME) cells towards the tumor development, invasion, and metastasis. Lately, the studies on cancer cell metabolism greatly expanded the understanding of cancer pathology and even revealed the presence of a widespread metabolic heterogeneity in tumors compared to spatial and temporal heterogeneities [10]. This disclosure exhilarated the researchers to focus more on recognizing the root cause of the phenotype and even the genetic anomalies facilitating these progressions in the TME. Even though the aerobic glycolysis is understood in many aspects, yet the role of a glycolytic enzyme PKM2 which catalyzes the last step of glycolysis has not been understood properly with respect to its role in various cells of TME. In addition, a study even suggested the differential requirement of PKM2 among the TME population [26] and has a positive correlation with cancer growth, metastasis, and resistance [27, 28]. Therefore, this contemporary review in detail highlights the role of PKM2 in the metabolic reprogramming of different cell population involved with the TME. Further, we discussed the preclinical and clinical studies with respect to PKM2 and the limitations included by aiming its activation or inhibition for modulating the metabolic conditions of cancer cells.
Glycolysis and its Metabolic Reprogramming
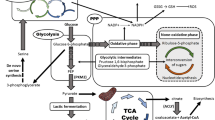
In both prokaryotes and eukaryotes, glycolysis is a central metabolic pathway for the energy generation and anabolism with coupled TCA [29]. In the 1920s, Dr. Otto Warburg and his colleagues observed an alteration in the glycolysis of the tumor. Huge amounts of glucose were consumed by the cancer cells in comparison to normal cells, and the consumed glucose was further fermented to lactate in aerobic conditions characterizing it as aerobic glycolysis or Warburg effect [30,31,32]. For the detailed process of aerobic glycolysis and the upregulated enzymes involved with it were mentioned in Fig. 1. Uptake of glucose is considered as a rate-limiting step of glycolysis in both cancer and fast dividing cells. Its uptake above the threshold level gradually increases aerobic glycolysis and slightly decreases mitochondrial respiration. This relationship between metabolic switching form mitochondrial oxidation to aerobic glycolysis improves the cancer cell proliferation and progression. Even the dysfunction of mitochondrial respiration and dysregulated glycolysis promoted the transformation of cells to form tumor [33].

Aerobic glycolysis in cancer cells: Illustration represents that cancer cells undergo aerobic glycolysis by inflowing the glucose molecules and converts the produced pyruvate to lactate instead of allowing it to enter TCA cycle in the aerobic conditions. Oncogenic proteins involved with the tumorigenesis upregulate many enzymes and transporters (indicated by *) of glycolysis further supporting this process
In the conditions of malignancies, increased expression of GLUT1 occurs due to the different signaling molecules (HIF-1α, c-Myc) preparing the cell to uptake more glucose. A study displayed the involvement of transporter (GLUT1) in the drug resistance of EGFR inhibitors (Gefitinib and Erlotinib) by increasing the glucose metabolism in lung cancer suggesting that glucose uptake by GLUT1 is a critical factor for the EGFR resistance [34]. Even the enzymes entailed with this process are upregulated and are modified post-translationally [35,36,37] by which the intermediates of the pathway like glucose-6-phosphate (G6P), fructose-6-phosphate (F6P), glyceraldehyde-3-phosphate (G3P), dihydroxyacetone phosphate (DHAP) are fluxed for the biosynthesis of various macromolecules (nucleic acids, fatty acids, phospholipids, amino acids, and sphingolipids) and maintains the redox system against ROS in cancer cells by making NADPH [38]. Recently, researchers are more interested to target the increased expression of various signaling molecules (β-catenin, c-Myc, HIF-1α, STAT), glycolytic pathway enzymes (HK, PKM2, PDK1, enolase, LDH) and glucose transporters (GLUT1, GLUT2) using siRNAs, small molecules treatment and formulation based strategies. From the above-mentioned proteins, PKM2 has a major role in assisting the progression of cancer by shifting the cellular metabolism to aerobic glycolysis by its nuclear moonlighting functions [39, 40].
PKM2
In the glycolytic pathway, a rate restrictive enzyme termed as pyruvate kinase (PK) catalysis the phopho-transfer reaction of phosphoenolpyruvate (PEP) to pyruvate with the generation of one molecule of ATP. In mammals, it exists in four isoforms namely PKM1 in high energy demanding tissues (skeletal muscle, heart, and brain), PKM2 (proliferating more cells and most cancer cells), PKL (liver and intestine), and PKR (erythrocytes) [41]. PKL and PKR isoforms are encoded from the gene PKLR, whereas PKM1 and PKM2 isoforms are encoded from a mutually exclusive exon of a gene PKM through alternative splicing [42,43,44]. Heterogeneous nuclear ribonucleoproteins (hnRNPs) - hnRNPA1 and hnRNPA2 controls this alternative splicing of PKM gene and inclusion of either the exon 9 or exon 10 into the pyruvate kinase mRNA results in PKM1, a constitutively active tetramer, and PKM2 respectively. Low expression of hnRNPs allows, exon 9 recognition by splicing machinery and favors the exon 10 inclusion into the PKM mRNA resulting in the upregulated PKM2 protein expression. Apart from hnRNPs some oncogenic factors like HIF-1α and c-Myc were reported to enhance the splicing of PKM2 [45, 46]. In addition, both the PKM isoforms possess a pyruvate kinase activity towards the PEP. But, due to the presence of 22 amino acid residues difference in mRNA of PKM2 exon 10 with respect to PKM1 exon 9, it offers an FBP binding pocket and letting it dependent on the FBP for the allosteric regulation and induction of tetrameric PKM2 [47]. Moreover, PKM2 exists in tetrameric and dimeric forms and the ratio of these isoforms in the cancer cells depends on the influence of different oncogenic proteins and has no fixed value. The dimeric form has the less affinity towards its substrate PEP at physiological conditions (Km, 0.46mM) and acts as an active nuclear protein kinase whereas the tetrameric form has a high affinity toward PEP (Km, 0.03mM) [48]. Post-translational modifications of PKM2 with signaling molecules (EGF, β-catenin, and FGFR), glucose and ROS mediate the phosphorylation at Y105 [49], acetylation at K305, and oxidation at C358 respectively promoting the dimeric form instead of a tetrameric form [50]. While, in-built molecules like serine, FBP and SAICAR were recognized allosteric PKM2 activators. Recently, small molecules like DASA-58 and TEPP-46 were found to be the reported synthetic PKM2 activators which act by steadying or stabilizing the tetrameric form [51].
This peculiar enzyme has both canonical and non-canonical roles in both glycolysis and cancer development respectively. It executes the canonical role by upholding the metabolic program of cancer cells. PKM2 contains a nuclear localization sequence (NLS) similar to importin α-5, by which it takes the assistance of importin α-5 for its translocation to the nucleus [52]. In addition, in the nucleus, this dimer PKM2 possess the protein kinase activity and phosphorylates STAT3 at Y705 [53] resulting in the upregulation of N-cadherin and metalloproteases like MMP-2, MMP-9 promoting colorectal cancer (CRC) and metastasis [54]. Further, by acting as a transcription factor, it phosphorylates histone at threonine 11 (T11) and upregulates the cell cycle proliferation markers (cyclin D and c-Myc) promoting brain tumorigenesis [55]. Interaction with the phosphotyrosine-containing proteins and JMJD5 [56] inhibits the pyruvate kinase activity of PKM2 and supports the accumulation of the glycolytic metabolite. Apart from this PKM2 also upregulates expression of various tumor proliferation (β-catenin) and metastasis markers (E-cadherin, N cadherin and vimentin). So, these findings indicated that PKM2 has both protein kinase and pyruvate kinase role. Butyrate (short-chain fatty acid) found in the gut lumen at higher concentration decreases glycolytic intermediates and nucleotide synthesis in HCT-116 cells by inhibiting Warburg effect due to dephosphorylation and tetramerization activating PKM2 [57]. Many researchers reported that small molecules activating PKM2 tetrameric form attenuated PKM2 mediated tumor growth and metastasis, although some reports suggested that inhibition of PKM2 with its inhibitors like shikonin and lapachol inhibits tumor growth [58, 59]. Structural studies revealed that these small molecules bind at the subunit interface of PKM2 site but not at the endogenous activator FBP binding sites [60]. However, understanding the mechanism of PKM2 in the metabolic reprogramming of cells (cancer cells and TME cells) is an important criterion for targeting it in anticipation to inhibit the cancer progression.
The Interplay between PKM2 and the Metabolic Reprogramming of Tumor Niche
Metabolic reprogramming is one of the newly emerged hallmarks of cancer for the fast and continuous proliferation of a cancer cell. It is an important feature in satisfying the biosynthetic and redox requirement of a cell by reprogramming the important biosynthetic cycles like glycolysis, OXPHOS, pentose phosphate pathway, and glutaminolysis [61, 62]. Signaling molecules (Akt, β-catenin, Nrf2, c-Myc, Ras, and HIF-1α) along with the specific isoforms of metabolic enzymes (dimeric PKM2, hexokinase, and LDH) can enhance the reprogramming of cells in tumor niche in accordance to their needs [63].
Tumor Microenvironment (TME)
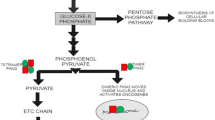
Formation of the tumor niche involves, the cancer cell communication with its surrounding cells necessary for the metastasis and resistance to cancer therapeutics. It acts like an ocean with embedded tumor cells encircled by extracellular matrix and stromal cells. It consists of non-malignant (TME cells) and malignant (cancer) cells and exists a bidirectional interaction between these cell components creating an environment known as TME, which partakes a role in tumor development and metastasis of cancer in all stages of carcinogenesis. Consistency in these interactions is maintained by paracrine and juxtracrine signalings involving different pathways like Notch, Foxp3, TGFβ, and IFN-γ [64, 65]. Early stages of tumorigenesis show an inflammatory microenvironment with a co-operative effect of COX-2/PGE2 and TLR/MyD88 pathways [66]. Hypoxia, altered pH, condensed extracellular matrix and immunosuppression are the features of TME which helped in developing different strategies like physical based targeting (thermal), pulsed based targeting, permeability based targeting, and chemical based targeting based on pH and hypoxia for targeting TME. Further, nano-therapeutic targeting (nanomedicines, nanoparticles, nanocarriers and ultra-small gold nanosatellite bearing nanoparticles), molecular-based targeting (siRNA, anti-PD-1, anti-VEGF, anti-HIFα) and cancer vaccines were developed focusing the tumor niche [67,68,69]. TME consist of different types of components namely non-cellular and cellular components surrounded by stromal cells Fig. 2 [70, 71].

Components of tumor microenvironment: Illustration represents the cellular and non-cellular composition of the tumor microenvironment
Activation of PKM2 in TME cells results in different physiological functions like reversed metabolic shift, increased OXPHOS, reduced lactate production, inhibition of tumor growth [72], and even provides immunity depending on the cell specificity [73]. Till date many reviews suggested the role of PKM2 with respect to the cancer cells [74,75,76], therefore in the next sections of the review, we will focus on discussing the importance of PKM2 in TME cells for their modulation and their maintenance Fig. 3.

Regulation of TME cells in the tumor microenvironment: Illustration represents the regulatory molecules modulated by PKM2 in different TME cells. This modulation helps in their activation and progression
T cells
T cell plays an important role in cell-based immunity against the pathogen. Naive T cell requires low energy requirement to prevent cells from atrophy and to keep cell survival and migration into circulation. However, activated T cell has high metabolic demand for rapid proliferation, differentiation and massive synthesis of cytokines and interleukins. The purpose of activated T cells to increase metabolic demand is either for its self-development or to erect the pathogens. To achieve this high metabolic demand, T-cell subtypes like TH1, TH2, and TH17 starts the massive influx of nutrients like glucose and amino acids via increased expression of GLUT1 and glycolytic enzymes shifting the cell to aerobic glycolysis. In controversy, regulatory T-cells downregulate GLUT1 increasing fatty acid oxidation. This phenomenon of metabolic shifting was even observed in CD4+ inflammatory T-cells but not with the CD4+ regulatory cells [77]. Moreover, aerobic shifting initiated by T-cell receptor (TCR) was sustained by cytokines like interleukin-2 and PEP concentration [78, 79]. But, TME shows glucose limiting condition which suppresses the tumouricidal function of T cell, compromising TCR-induced proliferation and growth increasing the expression of effectors molecules such as IFN-γ.
Many preclinical studies have shown the relation of PKM2 with the T-cell activity. Programmed death ligand 1 (PD-L1), a transmembrane protein present on the normal and cancer cells interacts with PD-1 checkpoint receptor existing on T-cells. This interaction plays a crucial role in autoimmunity and cancer by acting as “off-switch”. It means this interaction halts the T-cell activity towards the PD-L1 representing cells [80]. A report suggested that PKM2 dimer upregulates the PD-L1 expression by binding to the PD-L1 promoter region, and either the RNA silencing of PKM2 or treatment with TEPP-46 (PKM2 activator) inhibits LPS induced PD-L1 expression showing the significant role of PKM2 in limiting the T-cell response against tumors [81]. Even the increased expression of c-Myc proteins also resulted in modulating T inflammatory cell metabolic reprogramming contributing to the inflammation of tumor [82, 83]. c-Myc signaling has a role in post-transcriptional modification levels of PKM2 where it controls the splicing pattern by upregulating hnRNPAI (primary transcript) expression resulting in the high expression of PKM2 in glioma cells [84]. In controversy to the above study, the haploid efficiency of a transcriptional repressor of c-Myc gene, FUSE-binding protein (FBP)-interacting repressor (FIR) resulted in the alternative splicing of PKM2 by inhibiting hnRNPAI expression [85]. Apart from these signaling molecules, some co-stimulatory molecules found on T-cell surface like CD43 also affect PKM2 expression and proliferation, activation and migration of T cells [86]. CD43 and TCR co-signaling stimulates the phosphorylation of Y105 of PKM2 and Y705 of Signal transducer and activator of transcription 3 (STAT3) transducer molecules resulting in the activation of MEPK5/ERk5 pathway which activates nuclear localization kappa B (NF-kB), Myc as well as Bcl-2-associated death promoter (BAD) phosphorylation promoting the survival [87]. Abnormal expression of this protein is also observed in some non-hematologic neoplasms such as lung, colon, and salivary gland cancers [86]. Even the siRNA silencing of CD43 increased the vulnerability of breast and lung cancer cells to NK cells and apoptosis [88, 89].
Nuclear factor of activated T cells (NFAT1), a member of transcription factors family act as a tumor repressor by inhibiting the growth and differentiation of cancer cells [90]. Moreover, the substrate of PKM2, PEP was involved in the activation of calcium-dependent NFAT1 signaling by repressing the calcium uptake in the endoplasmic reticulum by Sarco/ER Ca2+-ATPase (SERCA) in T-cell. So, glucose deficient TME may diminish the anti-tumor activity of T-cell by downregulating NFTA1 signaling [91]. Even, NFAT1 was found to regulate MDM2 oncogene in-vitro and in-vivo in MCF-7 and MDA-MB-231 cells in which JapA showed inhibitory activity [92]. Interacting proteins of PKM2, like PTPB1, promotes cell proliferation and colony formation in ALCL cells by facilitating pY105 of PKM2 and nuclear STAT3 activation [93]. Further, a sulfur-containing amino acid homocysteine found to regulate the T-cell glycolytic reprogramming by upregulating PKM2 expression through PI3K/AKT/mTOR signaling in the conditions of hyperhomocysteinemia mediated inflammation in Apo-/-mice (apolipoprotein E-deficient mice) promoting atherosclerosis [94]. Moreover, high serum levels of homocysteine show a positive correlation with risk of cancer [95]. So, an additional study is required to know the possibility of homocysteine in dysregulating PKM2 levels of T-cells within TME in promoting the inflammation induced cancer progression.
B cells
B-cells are the part of the immune system and are the subtypes of lymphocytes, which gets differentiated into plasma cells and memory cells. Plasma cells finally get differentiated into antibodies and memory cells that help in keeping a track record of attacked antigens. These are derived from bone marrow stem cells and contribute a major role in immune response and immune system related disease such as autoimmunity and alloimmunity [96]. B-cell infiltration is common in draining of lymph nodes and lymphoid structures and also in the prognosis of some cancers like breast and ovarian cancers which are associated with TME [97].
A study disclosed the role of PKM2 in B-cell activation, in which homocysteine upregulated the expression of enzymes involved in both glycolysis and oxidative phosphorylation activating B-cell by shifting the glycolysis towards pentose pathway. PKM2 inhibitor like shikonin or knockdown of PKM2 attenuated Hcy mediated metabolic changes, B-cell proliferation and antibody synthesis (IgM & IgG) in-vitro and in-vivo implies the role of PKM2 in B-cell activation. The in-depth analysis showed the involvement of Akt dependent mTOR signaling, whereas the treatment with mTOR inhibitor (rapamycin) and shikonin attenuated Hcy induced metabolic changes and ultimately diminished atherosclerotic lesion [98]. These findings conclude that PKM2 activation is involved with the B-cell activation which is necessary for the immune response.
Macrophages
Macrophages are considered as a protective and pathogenic driver for an immune response [99]. Their activation is classified as classic vs. alternative or also M1 and M2. M1 stimuli e.g. Granulocyte-macrophage colony-stimulating factor (GM-CSF), Toll-like receptors (TLRs), IFN-γ, LPS or TNF that show Th1 dependent response whereas M2 stimuli e.g. macrophage colony-stimulating factor (M-CSF), IL-4, IL-10, IL-13, and glucocorticoids show Th2 dependent response [100]. They perform wide range of functions involving some receptors and different molecules like TLR (Toll-like receptors, intracellular pattern recognition receptors), nitric oxide (NO), reactive oxygen species (ROS), various types of inflammatory markers like TNFα, cytokines, chemokines, tissue-damaging proteases, and different types of interferon [101].
In macrophages, PKM2 act as a critical modulator for the production of cytokines as well as inflammatory markers. An endotoxin named lipopolysaccharide (LPS), which is a Toll-like receptors 4 (TLR4) agonist activates M1 macrophages by increasing the three-fold expression of PKM2. These activated macrophages induce the inhibition of HIF-1α and modulate the differentiation of T-helper cells. Activators of PKM2 like DASA-58 and TEPP-46 promote tetramer form, inhibits the IL-1β production and boost up the production of IL-10 in-vivo [102, 103]. Already studies had proved that over-production of ROS by glucose metabolism has a positive correlation with various types of pathologies like diabetes, cancer and neurodegenerative diseases. Glucose metabolism increases the cellular ROS by means of several mechanisms like glucose autoxidation, polyol pathway, and glycation [104]. Overconsumption of glucose drives up the glucose-ROS-PKM2-STAT3 pathway which led to the increased expression of various cytokines (IL-6, IL-1β, TNF-α). Thus, ultimately resulting in the hyper-inflammatory macrophages leading to the host damage [105]. So, the glucose-rich tumor niche can even activate the macrophages in the TME and may encourage the tumor inflammation by releasing cytokines promoting tumor progression. 5' AMP-activated protein kinase or AMPK (5' adenosine monophosphate-activated protein kinase) is an enzyme that has a role in energy metabolism, inflammation, and cell homeostasis. Like p53 and liver 9 kinase B1 (LKB1), activation of AMPK was also closely related to tumor suppressor function [106]. However, deficiency of AMPK upregulates PKM2 expression which results in the release of HMGB1 and lactate production in macrophages. But, administration of the shikonin reduces the lactate production and HMGB1 release [107]. Even, AMPK shows the protective effect by modulating PKM2 expression. Plumbagin plant-derived naphthoquinone also showed the similar actions of PKM2 inhibitors decreasing the lactate production and pro-inflammatory cytokines expression in lipopolysaccharide (LPS) activated macrophages [108].
Apart from oxidation, acetylation, and phosphorylation of PKM2 dimeric form, succinylation is another factor that promotes PKM2 dimer transition. SIRT5 overexpression predicts the poor survival of non-small cell lung cancer patients [109]. In LPS activated macrophages, PKM2 act as a substrate for SIRT5 and regulates the hyper succinylation and inhibit the PKM2 activity by upregulating its transition from tetrameric to dimeric form. It promotes PKM2 nuclear accumulation where it complexes with HIF1α and promotes transcription of inflammatory cytokines like IL-1β. But, PKM2 activators like TEPP-46 attenuates IL-1β production and macrophages activation in SIRT5 deficient mice [110] indicating the role of nuclear PKM2 in the activation of macrophages. Further, the ω-Alkynyl arachidonic acid treatment decreased the expression of macrophages M1 biomarkers (TNF-α, CXCL10, iNOS, and IL-6) and displayed a cardio protective effect by upregulating PKM2 expression in a mouse model of myocardial infarction. But downregulation of nuclear expression of PKM2 in LPS activated macrophages was observed. [111]. Thus, a further investigation is needed to understand the effect of ω-alkynyl arachidonic acid on the nuclear accumulation of PKM2 in cancer cells as its nuclear accumulation has a major oncogenic role in cancer progression. Altogether, PKM2 acts as a pivotal regulator in cytokines production, tumor inflammation and macrophage activation involved with the maintenance of TME.
Dendritic Cells
Dendritic (DC) cell is a bone marrow-derived leukocytes, as well as a potent antigen-presenting cell type. Paul Langerhans in 1968, was the first person in observing DC cell in human skin. Major subtypes in human DC includes myeloid DCs (e.g. Dectin1 and Dectin2), Plasmacytoid DCs (e.g. CD304 (neurophilin), CD123 (IL-3R)) and monocytes related (e.g. CD+16 monocyte, CD16 negative and inflammatory DC) [112]. These are mainly involved in antigen presenting and processing of antigen. These cells are also found in the TME but are not sufficient to stimulate an immune response due to their disturbed function by hypoxia and inflammation in TME [113].
Aqueous extract of Artemisia iwayomogi (AIP1) (a member of the Compositae family) inhibited the differentiation and maturation of bone marrow-derived dendritic cells that consequences the suppression of immunological responses. Proteomic analysis and RT-PCR profiling revealed that AIP1 treatment downregulated the expression of various proteins like PKM2, TRAF 5-like protein, coactosin-like protein 1, and allogeneic stimulation ability of T cell by dendritic cell (CLP1) in BALB/c mice [114]. Thus, this drug can have the potential in reversing the aerobic glycolysis by targeting PKM2. SRSF3 is a Splicing factor encoded by SFRS3gene was found to aberrantly overexpress in various cancers like ovarian cancer, triple-negative breast cancer and malignancy of mammary epithelial cells [115]. Treatment with trichostatin A, a broad range of histone deacetylase possesses immunomodulatory and protective function by upregulating the expression of SRSF3-PKM2-dependent glycolytic pathway in DC2.4 cells. Silencing of PKM2 by shRNA effects the cytokines expression and lactate production but trichostatin re-treatment restores partially the DC cell function [116]. This shows that PKM2 is involved in the immune response and the proper functioning of DC during antigen processing.
Myeloid-Derived Suppressor Cells (MDSCs)
Myeloid-derived suppressor cell is a myeloid originated type of cell and are considered to be a potent suppressor of the various function of T-cell as well as macrophages. These are the population of immune cells which are classified under the inhibitory immune cells influencing the development of T regulatory cell and inhibit CD8+ T-cell activation by controlling nitric oxide synthase in arginase dependent manner [117].
Wei-R et al., studied the non-canonical role of PKM2 in MDSCs in hepatocellular carcinoma in which PKM2 upregulates the recruitment of Gr-1+CD11b+ granulocytic MDSCs, F4/80+CD11b+ macrophages, and Ly6C+CD11b+ monocytic MDSCs in the TME. High levels of MDSC in bone marrow, peripheral blood and spleen have a role in the formation of a pre-metastatic niche in pulmonary foci. [118]. This shows that PKM2 has a role in designing the metastatic niche. Concanavalin-A induced immune-dependent liver injury involving TNF and IL-6 pathways triggering liver proliferation [119]. A similar study demonstrated the role of PKM2 in immune-mediated liver injury induced by Concanavalin A in mice. There was a significant correlation with increased plasma levels of PKM2, ALT, IL-1β, and IL-6 and hepatic myeloid cell infiltration and necrosis which was reversed by TEPP-46 [120]. This further gives an additional role to dimeric PKM2 in inducing liver injury.
NK Cells
NK cell is a type of lymphocytes derived from a common progenitor cell as B and T cells. NK cells are part of the innate immune system which spontaneously responded to cytolytic effect toward cancer and virus-infected cells. Activation of NK cell results in the release of various cytokines (IFN-γ, TNF-α, GM-CSF) and chemokines (CCL1-5 and CXCL8) [121]. In Humans, NK cells functionally categories according to the expression of CD56 and CD16 marker. CD56lowCD16+ NK cell type kill by means of produce cytokines by specific manners of recognition of target cells while CD56hiCD16− NK cells type (major NK cell subtype of peripheral lymphoid organs) produce a large number of cytokines (IFN-γ), TNF and granulocyte–macrophage colony-stimulating factor (GM-CSF) stimulation by pro-inflammatory cytokines and attain cytotoxicity for a sustained period [122]. Innate cytotoxic lymphocytes or natural killer cells also infiltrate towards TME.
NK cell functionally exists as licensed and unlicensed NK cells based on the presence of MHC-1 molecule. Licensed NK cell in which cell-specific Ly49 receptor interaction exists with self MHC-1 while unlicensed NK cell lacks Ly49 receptor and show impairment in MHC-1 cell recognition [123]. PKM2 is a crucial enzyme which gets upregulated during the differentiation of licensed NK cells and cross-links with KIR (killer cell immunoglobulin-like receptor) increasing the expression of phosphorylated metabolic modulators like p38-α and AMPKα. Moreover, licensed NK cell showed high metabolic reprogramming in glycolysis and OXPHOS dependent glutaminolysis while the unlicensed NK cells are exclusively reliant on OXPHOS for cytolytic function [124]. Evolutionary, HMGB1 protein was conserved as a regulator for cell death and survival process. It is a nuclear chromatin protein [125] derived from the NK cells was found to be an allosteric inhibitor of tetramer PKM2 by competing with its FBP binding sites thus blocking the Warburg effect in cancer cells. But, by amassing the glycolysis through dimer PKM2 and glutaminolysis, tumor cells can acquire resistance to HMGB1 induced metabolic cell death [126].
Fibroblasts
Cancer-associated fibroblasts (CAFs) in tumor environment has a very well familiar role in tumorigenesis which includes tumor initiation and progression, metastasis, stimulate angiogenesis, and chemotherapy resistance [127]. These include matrix components (collagen I-II, fibronectin, tenascin C and periostin), enzymes (MMP, LOX, and TIMPs), cytokines, growth factor receptors (GFβ, VEGF, PGE2, CTGF, SDF-1), and cytoplasmic proteins (desmin, vimentin, α-SMA). Recently, a new phenotypic marker of CAF like caveolin-1 (Cav-1) and MCT4 were documented [128, 129]. Normally, myofibroblasts get activated in the wound healing process. But, in TME, residential fibroblasts gets differentiated to myofibroblasts known as CAFs by direct or indirect signaling crosstalk between fibroblast and cancerous cells using paracrine signals. Signaling molecules that involved in activation of fibroblast include various cytokines like TGFβ, IL-1β [130]. Activated myofibroblasts show deregulated MMPs and increased the level of ROS that increases high chances of organs fibrosis and development of tumorigenesis and malignancy [131].
Primary culture of MCKH (mammary carcinoma from KH) cells shows the similar properties of CAFs like EMT, cancer stem cell-like features and also displayed many upregulated glycolytic enzyme genes including GAPDH, LDH, PGAM1, HIF1-α, and PKM2. This enrichment of CAF features suggested to occur via the dependence of FGF signaling [132]. US28 signaling activates HIF-1α/PKM2 axis, in turn promoting proliferation, angiogenesis, and energy reprogramming in cancer cells. It upregulated the expression of many proteins including VEGF, HIF1α and many glycolytic enzymes like GAPDH, PKM2 in fibroblasts and glioblastoma cells [133]. FGFR1 expression also has a positive correlation with the p-PKM2 and p-LDHA which are having the most suitable diagnostic potential in thyroid malignancies in thyroglobulin negative thyroid cancer [134]. A contradictory study displayed that PKM1 expression in fibroblasts induced inflammation and increase lactate production (~1.5) fold higher than PKM2 containing fibroblast when co-injected with MDA-MB-231 in a xenograft model. PKM2 expressed fibroblasts triggered pseudo-starvation in stromal cells with NF-kB dependent upregulation of Beclin-1 and Cathepsin-B. They also found the loss of Cav-1 expression, which is a marker of autophagy and stromal glycolysis [135]. Dysregulated expression of caveolin-1 was reported to be associated with various cancers due to the aberrant activation of JAK/STAT, JNK, estrogen and src signalings in pancreatic cancer [136]. It is even reported that acute loss of caveolin-1 results in mitochondrial dysfunction, increased ROS, and stimulation of aerobic glycolysis. Oxidative stress in fibroblasts is capable of genetic instability and creates an oncogenic/mutagenic effect to reprograming fibroblast. Treatment of fibroblasts with anti-oxidants likes N-acetyl-cysteine and quercetin) or NO inhibitors (L-NAME) reversed the aggressive behavior of CAFs in MCF-7 breast cancer cells [137].
A study found that cancer-associated fibroblasts have the ability to transfer high quantity proteins (PKM2, SOD2, thio0redoxin, malate dehydrogenase, and galectin-1) and lipids to human prostate cancer cells (DU145) through ectosome mediated delivery. But, a reverse transfer from cancer cell to fibroblast is negligible or absent and even the rate of protein transfer depends upon the donor cell and recipient cell line co-culture ratio [138]. miR-21 is a single-stranded micro-RNA (miRNAs) in human encode by MIR21gene present on chromosome number 17q23.2. Various experimental data show that overexpression of miR-21 is a feature of pathological cell growth or cell stress which target various survival signaling pathway like STAT3, MAPK, AKT, and AP-1 [139]. miR-21 mediated remodeling of CAFs metabolic changes was also reported in the pancreatic cancer cell. CAF showed high expressions of LHDA, PKM2, and miR-21 during the co-culture of CAFs with human pancreatic cancer cells (BxPc-3). Treatment (miR-21 inhibitor) of CAFs and co-culture showed a significant reduction in the expressions of PKM2, LDHA, and MCT [140]. Similarly, a study report suggested miR-21 regulation of PTBP1 in the fibroblasts of pulmonary arterial walls. Knockdown of PTBP1, treatment with PKM2 activator (TEPP-46) and histone deacetylase inhibitors reversed glycolytic phenotypes and decrease cell proliferation [141]. An additional study is needed to explore the role of PTBP1 knockdown to target PKM2 during angiogenesis. Another miRNA named miR-205 regulates development, apoptosis, and cancer. It is found that miR-205 has the ability to suppress EMT by directly targeting VEGF-A and ErbB3 in breast cancer [142]. CAFs upregulate PKM2 and allows its nuclear accumulation of PKM2 that form complex with HIF1α that recruits transcriptional Chondrocytes-1 (DEC1). Chondrocytes-1 (DEC1) that downregulate the expression of miR205. Treatment with metformin and DASA-58 decreased the metastatic potential of the prostate cancer cell in SCID mice [143]. These all findings suggest the role of PKM2 in promoting glycolysis, proliferation, and remodeling of CAFs.
Endothelial Cells
Endothelial cells are specialized types of cells that form the inner lining of blood vessels and play a crucial role in many physiological processes including to regulate vasomotor tone, blood cells, and WBCs trafficking, to uphold blood fluidity, angiogenesis, and both innate and adaptive immunity [144]. Vascular endothelial cells are the main key players in cellular homeostasis, normal tracking of leukocytes and in tumor development. In normal and cancer cells, soluble factors like chemokines, platelets derived factor, vascular endothelial factor (VEGF), EGF, and HIF1-α are important to provoke the endothelial proliferation, differentiation into neovascularisation process [145, 146].
Hypoxia-induced PKM2 expression in tumor results in the activation of HIF1-α and NF-kB signaling subsequently triggering the secretion of VEGF-A factor. Deletion of PKM2 impaired tumor proliferation, induced apoptosis and decrease blood vessel formation in in-vitro (pancreatic cancer cell lines) and in-vivo (tumor xenograft on chicken chorioallantoic membranes) [147]. Even the circulating PKM2 stimulates angiogenesis, proliferation, migration and cell-ECM junctions in endothelial cells of colon xenograft cells (SW620) [146]. A positive correlation was observed with PKM2 upregulation and aberrant expression of VEGF-C in breast cancer. The study confirmed that knockdown of PKM2 downregulates the expression of VEGF-C and inhibit proliferation, metastasis and lymphovascular invasion [148]. Further, analysis of 65 patients with lung adenocarcinoma found a significant relationship between PKM2 expression and proliferation, invasion and metastasis. siRNA mediated knockdown of PKM2 result in decreased glucose uptake (25%), ATP formation (20%), synthesis of fatty acids and down-regulated MMP-2 and VEGF expressions in A549 cell lines and patient specimens [149]. PKM2/HIF-1/vGPCR signaling pathway promotes the angiogenesis and tumor growth in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV8). PKM2 controls the paracrine angiogenesis (VEGR dependent) and proliferation through vGPCR in KSHV infection of an endothelial cell. Upregulation of HIF-1 results in increased expression of angiogenic promoting factors like VEGF, PDGF, TGFα, TGFβ, ANGPT2, and ANGPTL4 [150].
A study found that TCM treated with lymphatic endothelial cells (LEC) promotes the growth of tumor in-vitro and in-vivo by releasing the excessive amount of EGF and PDGF-BB. PDGF-BB, in turn, stimulates the pericyte infiltration and angiogenesis in breast cancer [151]. Even the accumulation of the end product of glycolysis, i.e. lactate, due to the low enzyme activity of PKM2 contributes to the formation of neovessels by the inhibition of prolyl hydroxylase 2, activation of HIF-1-NF-kB signaling pathway and stimulation of interleukin (IL-8/CXCL8) synthesis which enhances the endothelial proliferation and maturation of mature blood vessels [146]. miR-143, a short non-coding mRNA regulates various genes and has a role in various cancers like oesophageal squamous cell carcinoma, gastric cancer, and gall bladder and acts as a tumor suppressor [152]. Its overexpression results in a significant decrease of glycolytic enzymes like PKM2, LDHA, and HK2 in atherosclerotic plaque forming endothelial cell lines [153]. Similarly, miRNA-124 was found to be a tumor suppressor by acting through diverse mechanisms in various cancers like breast, bone and colorectal cancer [154]. It was examined that, there exists a positive correlation between miRNA-124 and a splicing factor, polypyrimidine-tract-binding protein (PTBP1) which is a PKM2 alternative splicing factor. They found that overexpression of miRNA-124 silences the PTBP1 expression resulting in the restoration of dysregulated glycolysis and mitochondrial respiration [155]. These suggest that PKM2 is involved with the proliferation and metastasis of endothelial cells.
Adipocytes
Adipocytes are mainly involved in storing the energy as fats and triglycerides in lipid droplets. In mammals, two types of Adipocytes are present i.e. brown adipocytes which have a large number of mitochondria and involved in maintaining the body temperature while white adipocytes have large lipid droplets and are mainly involved in the storage of fats. A proper balance between brown and white fat is required for maintaining the energy homeostasis [156]. Adipocytes play a role in the development of some cancers like intra-abdominal tumors by means of providing homing to cancer cells, by releasing adipokines which includes interleukins-8 (IL-8) and provide direct transfer of fatty acids to cancer cells. Obesity is considered as one of the risk factors for breast cancer development by chronic low-grade inflammation, macrophages recruitment and upregulation of several signaling pathways like AMPK, p53 and HIF, etc. [157].
Proteomic data by 8-plexi TRAQ-2DLC-MS/MS analysis shows the involvement of 549 proteins in adipogenesis. Among them, PKM2, C/EBPβ, and PIN1 are found to have a link with the PI3K/AKT pathway. But, knockdown of C/EBPβ downregulated the PKM2 expression at RNA and protein level displaying the role of PKM2 in adipogenesis [158]. Leptin promotes the EMT of breast cancer cells by targeting the PI3K/AKT pathway via PKM2 over-expression. But, leptin-induced PKM2 expression and metastasis markers were abolished by PI3K/AKT inhibitor LY294002 and PKM2 knockdown in-vitro and in-vivo [159]. Overexpression of PTP1B, a member of protein tyrosine phosphatase (PTP) family was observed in 70% of TNBC cases [160]. Reports suggest that PKM2 acts as a novel substrate for PTP1B, and downregulated PTP1B expression increased Tyr105 phosphorylation of PKM2 in in-vivo and cultured adipocytes [161]. All these findings strengthen the role of PKM2 in adipogenesis.
Therapeutics Targeting PKM2
In recent years, apart from other glycolytic enzymes (LDH, hexokinase, and enolase), PKM2 comes out as an emerging and potential target for anticancer therapy. In fact cancer cells utilize more amounts of glucose and due to the existence of dimeric PKM2, it accumulates the upstream substrates of glycolysis, activating one carbon metabolism a feature that assists the cancer progression [162]. As discussed earlier, even the nuclear PKM2 controls and modulates various signaling pathway mediators (STAT3, histone H3, and HIF-1α) in various diseases like cancer, inflammatory disease, and diabetes. Still, a study lacked the evidence in displaying the protein kinase activity of PKM2 [163]. mTOR is a central factor in the warburg effect in inducing PKM2 expression [164]. Further, activation of PKM2 with the small molecules induced serine auxotrophy by allowing the cells not to get adapt with the nutrient stress [165]. So, PKM2 targeting shows a synergistic effect in cancer treatment by means of suppressing PKM2 dependent cancer proliferation and metastasis as well as in decreasing the macromolecules synthesis and lactate production. While some contradictory studies reported that PKM2 deletion accelerated mammary tumors formation in Brca1-drive model of breast cancer by disrupting PKM1 expression [166]. Even knockdown of PKM2 resulted in modest inhibition of tumor growth [167], developed hepatic cancer, metabolic stress [168] and soft tissue sarcoma [169] whereas the nuclear PKM2 induced the cell death in a caspase-independent manner [170] indicating PKM2 is not needed primarily for cancer proliferation [171]. Besides, its requirement is not needed in APC lost colon cancer model [172]. Based on all these studies, various drugs have been developed and recognized in stabilizing or promoting the PKM2 in its tetramer form as well as to inhibit the PKM2 activity directly. But, the drugs inhibiting PKM2 expression directly may have limitations, particularly the functional consequences associated with kinase inhibition and may even give other possible drawbacks. Firstly, there will be a suppressed immune response in the TME, as the immune cells (T-cell, B-cell) needs PKM2 for their activation and proliferation. Secondly, it influences the glycolytic pathway globally which may hamper the processes in which PKM2 plays a major role. So, it is desired to develop PKM2 activators rather than its inhibitors to limit these drawbacks. Some of the reported drugs targeting PKM2 at preclinical and clinical setup are discussed in Table 1.
It is clear from Table 1 that several molecules having an activator or inhibitory action on PKM2 have been reported. Some of them being more drug like and promising have moved to clinical phases while several others could not be taken further either because of poor drug likeness or lack of in vivo efficacies. Some metal based (2, Table 1), phenolics (4, 5, 7, 1, Table 1), and heterocyclics (6, 8-15, Table 1) are known to cause PKM2 inhibition and in terminating tumor progression. Some of the molecules like 8 and 13 which are heterocyclics have more likelihood to cross the valley of death because of better drug like features and it will be interesting to find them in the list of clinical candidates in near future. The clinical candidates have been successful in several ways and their future promise to lessen the burden of cancer is yet to be elucidated [187, 188].
Concluding Remarks
In diverse cancers, PKM2 is known to act as a gatekeeper in regulating the tumor metabolism, proliferation, and its niche maintenance. Till date, the clear cut role of PKM2 involvement in the TME cells is not established appropriately. So, a comprehensive discussion on the role of PKM2 in the TME cells was clarified in this review. As this enzyme is a key metabolic conduit in the metabolism and proliferation of various body cells including immune cells, aiming it creates a major challenge. In this context, targeting PKM2 with activators rather than its inhibitors may provide a therapeutic effect with limited resistance, lesser immune suppression by consequently avoiding the functional consequences associated with kinase inhibitors. Further, these activators can be used in combination with the existed anti-cancer agents to achieve a synergistic effect inhibiting the tumor growth and proliferation. In addition, its involvement with the inflammatory cells was discussed in this report which may provide a new application for the PKM2 activators in drug repurposing. The upcoming approaches and investigations should be to improve the molecular studies on PKM2 with respect to its role in tumor niche cells and to understand the effect of therapeutics against PKM2 in developing resistance and immune suppression. For all these whys and wherefores, PKM2 signifies as a potential target in metabolic reprogramming of tumor.
Abbreviations
- ANGPT2:
-
Angiopoietin 2
- AKT:
-
Serine/threonine-specific protein kinase
- ALCL:
-
Anaplastic large cell lymphoma
- ALT:
-
Alanine aminotransferase
- AMPK:
-
5' AMP-activated protein kinase
- ANGPTL4 :
-
Angiopoietin-like 4
- AP-1:
-
Activator protein 1
- C/EBPβ :
-
CCAAT-enhancer binding protein β
- CD4:
-
Cluster of differentiation 4
- COX-2:
-
Cyclooxygenase-2
- CTGF:
-
Connective tissue growth factor
- CXCL8:
-
C-X-C motif chemokine 8
- DASA-58:
-
Dihydro benzodioxin sulfonyl hexahydro diazepin sulfonyl benzenamine
- EGF:
-
Epidermal growth factor
- EMT:
-
Epithelial–mesenchymal transition
- ErbB3:
-
Erb-B2 Receptor tyrosine kinase 3
- ERK-5:
-
Extracellular-signal-regulated kinase 5
- FBP:
-
Fructose bisphosphate
- FGF:
-
Fibroblast growth factor
- FGFR1 :
-
Fibroblast growth factor receptor 1
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GLUT-1:
-
Glucose transporter -1
- HCT-116:
-
Human colon cancer cell line
- HIF-1α:
-
Hypoxia induced factor-1 alpha
- HK-2:
-
Hexokinase-2
- HMGB1:
-
High mobility group box-1
- IFN-ϒ:
-
Interferon-gamma
- IL-10:
-
Interleukin-10
- IL-13:
-
Interleukin-13
- IL-1β:
-
Interleukin-1β
- IL-4:
-
Interleukin-4
- IL-6:
-
Interleukin-6
- JAK:
-
Janus kinase
- JNK:
-
c-Jun N-terminal kinases
- LDH:
-
Lactate dehydrogenase
- LDH-A:
-
Lactate dehydrogenase-A
- LOX:
-
Lysyl oxidase
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinases
- MAP K-5:
-
Mitogen activated protein kinase-5
- MCT-1:
-
Monocarboxylase transporter -1
- MCT4:
-
Monocarboxylate transporter 4
- MDM2:
-
Mouse double minute-2 homolog
- miRNA-124:
-
MicroRNA-124
- MMP:
-
Matrix metalloproteinase
- MMP-2 :
-
Matrix metalloproteinase 2
- mTOR:
-
Mechanistic target of rapamycin
- NF-kB :
-
Nuclear factor kappa B
- NK cells:
-
Natural killer cells
- OXPHOS:
-
Oxidative phosphorylation
- p53:
-
Tumor protein p53
- PD-1:
-
Programmed cell death-1
- PD-4:
-
Programmed cell death-4
- PDGF:
-
Platelet-derived growth factor
- PDGF-BB:
-
Ligand for platelet derived growth factor receptor
- PGAM1:
-
Phosphoglycerate Mutase 1
- PGE2:
-
Prostaglandin E2
- PI3K:
-
Phosphoinositide-3-kinase
- PIN1:
-
Peptidyl-prolyl cis-trans isomerase
- PTP1B:
-
Tyrosine-protein phosphatase non-receptor type 1
- ROS:
-
Reactive oxygen species
- RT-PCR:
-
Real time-polymerase chain reaction
- SAICAR:
-
Succino 5-amino-4-imidazole-N-succinocarboxamide ribonucleotide
- SDF-1:
-
Stromal cell-derived factor 1
- shRNA:
-
Small hairpin ribonucleic acid
- siRNA:
-
Small interfering ribonucleic acid
- SOD2:
-
Superoxide dismutase 2
- STAT3:
-
Signal transducer and activator of transcription 3
- TCA:
-
Tricarboxylic acid
- TCR:
-
T cell receptor
- TEPP-46:
-
Thieno-pyrrole-pyridazine
- TGFα:
-
Transforming growth factor alpha
- TGFβ:
-
Transforming growth factor beta
- TH-1:
-
T helper cell-1
- TIMP:
-
Tissue inhibitor of metalloproteinases
- TNF-α:
-
Tumor necrosis factor-alpha
- TRAF-5:
-
TNF-receptor associated factor-5
- US28:
-
G-protein coupled receptor homolog US28
- VEGF:
-
Vascular endothelial growth factor
- vGPCR :
-
Viral G protein-coupled receptor
References
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA: A Cancer Journal for Clinicians 68 (1):7-30. https://doi.org/10.3322/caac.21442
Rübben A, Araujo A (2017) Cancer heterogeneity: converting a limitation into a source of biologic information. J Transl Med 15(1):190
Brown TM, Fee E (2006) Rudolf Carl Virchow: medical scientist, social reformer, role model. Am J Public Health 96(12):2104–2105
Seth S, Li C-Y, Ho I-L, Corti D, Loponte S, Sapio L, Del Poggetto E, Yen E-Y, Robinson FS, Peoples M (2019) Pre-existing Functional Heterogeneity of Tumorigenic Compartment as the Origin of Chemoresistance in Pancreatic Tumors. Cell Rep 26(6):1518–1532. e1519
Kang J, Chen HJ, Zhang XC, Su J, Zhou Q, Tu HY, Wang Z, Wang BC, Zhong WZ, Yang XN (2018) Heterogeneous responses and resistant mechanisms to crizotinib in ALK-positive advanced non-small cell lung cancer. Thorac Cancer 9(9):1093–1103
Nalla LV, Kalia K, Khairnar A (2019) Self-renewal signaling pathways in breast cancer stem cells. Int J Biochem Cell Biol 107:140–153
Marusyk A, Polyak K (2010) Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 1805(1):105–117. https://doi.org/10.1016/j.bbcan.2009.11.002
Fisher R, Pusztai L, Swanton C (2013) Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer 108:479. https://doi.org/10.1038/bjc.2012.581
Dagogo-Jack I, Shaw AT (2017) Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 15:81. https://doi.org/10.1038/nrclinonc.2017.166
Neugent ML, Goodwin J, Sankaranarayanan I, Yetkin CE, Hsieh M-H, J-w K (2018) A new perspective on the heterogeneity of cancer glycolysis. Biomol Ther 26(1):10
Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS, Reid BJ (2006) Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet 38(4):468–473. https://doi.org/10.1038/ng1768
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M (2011) Tumour evolution inferred by single-cell sequencing. Nature 472(7341):90–94. https://doi.org/10.1038/nature09807
Bochtler T, Stolzel F, Heilig CE, Kunz C, Mohr B, Jauch A, Janssen JW, Kramer M, Benner A, Bornhauser M, Ho AD, Ehninger G, Schaich M, Kramer A (2013) Clonal heterogeneity as detected by metaphase karyotyping is an indicator of poor prognosis in acute myeloid leukemia. J Clin Oncol Off J Am Soc Clin Oncol 31(31):3898–3905. https://doi.org/10.1200/jco.2013.50.7921
Almendro V, Marusyk A, Polyak K (2013) Cellular heterogeneity and molecular evolution in cancer. Annu Rev Pathol 8:277–302. https://doi.org/10.1146/annurev-pathol-020712-163923
Bidkhori G, Benfeitas R, Klevstig M, Zhang C, Nielsen J, Uhlen M, Boren J, Mardinoglu A (2018) Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc Natl Acad Sci 115(50):E11874–E11883
Lamb R, Ozsvari B, Bonuccelli G, Smith DL, Pestell RG, Martinez-Outschoorn UE, Clarke RB, Sotgia F, Lisanti MP (2015) Dissecting tumor metabolic heterogeneity: Telomerase and large cell size metabolically define a sub-population of stem-like, mitochondrial-rich, cancer cells. Oncotarget 6(26):21892
Robertson-Tessi M, Gillies RJ, Gatenby RA, Anderson AR (2015) Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Res 75(8):1567–1579
Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L (2016) Metabolic heterogeneity in human lung tumors. Cell 164(4):681–694
Yuan Y (2016) Spatial heterogeneity in the tumor microenvironment. Cold Spring Harb Perspect Med 6(8):a026583
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Chen G, Zhang Y, Liang J, Li W, Zhu Y, Zhang M, Wang C, Hou J (2018) Deregulation of Hexokinase II Is Associated with Glycolysis, Autophagy, and the Epithelial-Mesenchymal Transition in Tongue Squamous Cell Carcinoma under Hypoxia. BioMed research international 2018
Kachel P, Trojanowicz B, Sekulla C, Prenzel H, Dralle H, Hoang-Vu C (2015) Phosphorylation of pyruvate kinase M2 and lactate dehydrogenase A by fibroblast growth factor receptor 1 in benign and malignant thyroid tissue. BMC Cancer 15(1):140
Mogi A, Koga K, Aoki M, Hamasaki M, Uesugi N, Iwasaki A, Shirakusa T, Tamura K, Nabeshima K (2013) Expression and role of GLUT-1, MCT-1, and MCT-4 in malignant pleural mesothelioma. Virchows Archiv 462(1):83–93
Miranda-Gonçalves V, Granja S, Martinho O, Honavar M, Pojo M, Costa BM, Pires MM, Pinheiro C, Cordeiro M, Bebiano G (2016) Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 7(29):46335
Zha X, Sun Q, Zhang H (2011) mTOR upregulation of glycolytic enzymes promotes tumor development. Taylor & Francis
Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155(2):397–409
Sounni NE, Noel A (2013) Targeting the tumor microenvironment for cancer therapy. Clin Chem 59(1):85–93
Yang L, Lin PC (2017) Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. In: Seminars in cancer biology. Elsevier, pp 185–195
Schowen RL (1993) Principles of biochemistry 2nd ed. (Lehninger, Albert L.; Nelson, David L.; Cox, Michael M.). Journal of Chemical Education 70 (8):A223. https://doi.org/10.1021/ed070pA223.1
Locasale JW, Cantley LC (2010) Altered metabolism in cancer. BMC Biology 8(1):88. https://doi.org/10.1186/1741-7007-8-88
Phan LM, Yeung S-CJ, Lee M-H (2014) Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med 11(1):1–19. https://doi.org/10.7497/j.issn.2095-3941.2014.01.001
Warburg O (1925) The Metabolism of Carcinoma Cells. J Cancer Res 9(1):148–163. https://doi.org/10.1158/jcr.1925.148
Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H, Chiao PJ, Keating MJ, Garcia-Manero G, Huang P (2012) K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res 22(2):399–412. https://doi.org/10.1038/cr.2011.145
Suzuki S, Okada M, Takeda H, Kuramoto K, Sanomachi T, Togashi K, Seino S, Yamamoto M, Yoshioka T, Kitanaka C (2018) Involvement of GLUT1-mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells. Oncotarget 9(66):32667–32679. https://doi.org/10.18632/oncotarget.25994
Šmerc A, Sodja E, Legiša M (2011) Posttranslational modification of 6-phosphofructo-1-kinase as an important feature of cancer metabolism. PloS one 6(5):e19645
Wiese EK, Hitosugi T (2018) Tyrosine kinase signaling in cancer metabolism: PKM2 paradox in the warburg effect. Front Cell Dev Biol 6
Hallows WC, Yu W, Denu JM (2012) Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem 287(6):3850–3858
Ganapathy-Kanniappan S, Geschwind JF (2013) Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 12:152. https://doi.org/10.1186/1476-4598-12-152
Li H, Xu H, Xing R, Pan Y, Li W, Cui J, Lu Y (2019) Pyruvate kinase M2 contributes to cell growth in gastric cancer via aerobic glycolysis. Pathology-Research and Practice
Boukouris AE, Zervopoulos SD, Michelakis ED (2016) Metabolic enzymes moonlighting in the nucleus: metabolic regulation of gene transcription. Trends Biochem Sci 41(8):712–730
Dayton TL, Jacks T, Vander Heiden MG (2016) PKM2, cancer metabolism, and the road ahead. EMBO Rep 17(12):1721–1730
Mazurek S, Boschek CB, Hugo F, Eigenbrodt E (2005) Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol 15(4):300–308. https://doi.org/10.1016/j.semcancer.2005.04.009
Altenberg B, Greulich KO (2004) Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 84(6):1014–1020. https://doi.org/10.1016/j.ygeno.2004.08.010
Iqbal MA, Gupta V, Gopinath P, Mazurek S, Bamezai RN (2014) Pyruvate kinase M2 and cancer: an updated assessment. FEBS Letters 588(16):2685–2692. https://doi.org/10.1016/j.febslet.2014.04.011
David CJ, Chen M, Assanah M, Canoll P, Manley JL (2010) HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463(7279):364
Semenza G (2011) Regulation of metabolism by hypoxia-inducible factor 1. In: Cold Spring Harbor symposia on quantitative biology. Cold Spring Harbor Laboratory Press, pp 347–353
Dombrauckas JD, Santarsiero BD, Mesecar AD (2005) Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 44(27):9417–9429
Mazurek S (2011) Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 43(7):969–980
Hitosugi T, Kang S, Vander Heiden MG, Chung T-W, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2(97):ra73-ra73
Yang W, Lu Z (2013) Regulation and function of pyruvate kinase M2 in cancer. Cancer Lett 339(2):153–158. https://doi.org/10.1016/j.canlet.2013.06.008
Warner SL, Carpenter KJ, Bearss DJ (2014) Activators of PKM2 in cancer metabolism. Future Med Chem 6(10):1167–1178. https://doi.org/10.4155/fmc.14.70
Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z (2012) ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol 14(12):1295
Gao X, Wang H, Yang JJ, Liu X, Liu ZR (2012) Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell 45(5):598–609. https://doi.org/10.1016/j.molcel.2012.01.001
Yang P, Li Z, Fu R, Wu H, Li Z (2014) Pyruvate kinase M2 facilitates colon cancer cell migration via the modulation of STAT3 signalling. Cell Signal 26(9):1853–1862. https://doi.org/10.1016/j.cellsig.2014.03.020
Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z (2012) PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150(4):685–696. https://doi.org/10.1016/j.cell.2012.07.018
Wang H-J, Hsieh Y-J, Cheng W-C, Lin C-P, Lin Y-s, Yang S-F, Chen C-C, Izumiya Y, Yu J-S, Kung H-J (2014) JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α–mediated glucose metabolism. Proc Natl Acad Sci 111 (1):279-284
Li Q, Cao L, Tian Y, Zhang P, Ding C, Lu W, Jia C, Shao C, Liu W, Wang D, Ye H, Hao H (2018) Butyrate Suppresses the Proliferation of Colorectal Cancer Cells via Targeting Pyruvate Kinase M2 and Metabolic Reprogramming. Mol Cell Proteomics 17(8):1531–1545. https://doi.org/10.1074/mcp.RA118.000752
Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X (2011) Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 30(42):4297
Babu MS, Mahanta S, Lakhter AJ, Hato T, Paul S, Naidu SR (2018) Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PloS one 13(2):e0191419
Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, Austin CP, Inglese J, Harris MH, Asara JM, Stephanopoulos G, Salituro FG, Jin S, Dang L, Auld DS, Park HW, Cantley LC, Thomas CJ, Vander Heiden MG (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8(10):839–847. https://doi.org/10.1038/nchembio.1060
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20. https://doi.org/10.1016/j.cmet.2007.10.002
Sun L, Suo C, Li S-t, Zhang H, Gao P (2018) Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim Biophys Acta 1870(1):51–66. https://doi.org/10.1016/j.bbcan.2018.06.005
Tennant DA, Durán RV, Gottlieb E (2010) Targeting metabolic transformation for cancer therapy. Nat Rev Cancer 10:267. https://doi.org/10.1038/nrc2817
Li K, Huang S-h, X-m L, Yang L, G-q L, Y-j L (2018) Interaction of cancer cell-derived Foxp3 and tumor microenvironment in human tongue squamous cell carcinoma. Exp Cell Res 370(2):643–652. https://doi.org/10.1016/j.yexcr.2018.07.029
Meurette O, Mehlen P (2018) Notch Signaling in the Tumor Microenvironment. Cancer Cell. https://doi.org/10.1016/j.ccell.2018.07.009
Echizen K, Oshima H, Nakayama M, Oshima M (2018) The inflammatory microenvironment that promotes gastrointestinal cancer development and invasion. Adv Biol Regul 68:39–45. https://doi.org/10.1016/j.jbior.2018.02.001
Ding Z-Y, Zou X-L, Wei Y-Q (2012) Cancer Microenvironment and Cancer Vaccine. Cancer Microenviron 5(3):333–344. https://doi.org/10.1007/s12307-012-0107-x
Taube JM, Klein AP, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA (2014) Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res
Ivey JW, Bonakdar M, Kanitkar A, Davalos RV, Verbridge SS (2016) Improving cancer therapies by targeting the physical and chemical hallmarks of the tumor microenvironment. Cancer Lett 380(1):330–339. https://doi.org/10.1016/j.canlet.2015.12.019
Koontongkaew S (2013) The tumor microenvironment contribution to development, growth, invasion and metastasis of head and neck squamous cell carcinomas. J Cancer 4(1):66–83. https://doi.org/10.7150/jca.5112
Subramaniam R, Mizoguchi A, Mizoguchi E (2016) Mechanistic roles of epithelial and immune cell signaling during the development of colitis-associated cancer. Cancer Res Front 2(1):1–21. https://doi.org/10.17980/2016.1
Buetti-Dinh A, O’Hare T, Friedman R (2016) Sensitivity Analysis of the NPM-ALK Signalling Network Reveals Important Pathways for Anaplastic Large Cell Lymphoma Combination Therapy. PLoS ONE 11(9):e0163011. https://doi.org/10.1371/journal.pone.0163011
Liang J, Cao R, Wang X, Zhang Y, Wang P, Gao H, Li C, Yang F, Zeng R, Wei P (2017) Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res 27(3):329
Li Y-H, Li X-F, Liu J-T, Wang H, Fan L-L, Li J, Sun G-P (2018) PKM2, a potential target for regulating cancer. Gene 668:48–53
Wong N, Ojo D, Yan J, Tang D (2015) PKM2 contributes to cancer metabolism. Cancer Lett 356(2):184–191
Dayton TL, Jacks T, Vander Heiden MG (2016) PKM2, cancer metabolism, and the road ahead. EMBO Rep 17:1721–1730
Wang R, Green DR (2012) The immune diet: meeting the metabolic demands of lymphocyte activation. F1000 Biol Rep 4:9. https://doi.org/10.3410/b4-9
Finlay DK (2013) mTORC1 regulates CD8+ T-cell glucose metabolism and function independently of PI3K and PKB. Biochem Soc Trans 41(2):681–686. https://doi.org/10.1042/bst20120359
MacIver NJ, Michalek RD, Rathmell JC (2013) Metabolic regulation of T lymphocytes. Annu Rev Immunol 31:259–283
Gong J, Chehrazi-Raffle A, Reddi S, Salgia R (2018) Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer 6(1):8. https://doi.org/10.1186/s40425-018-0316-z
Palsson-McDermott EM, Dyck L, Zaslona Z, Menon D, McGettrick AF, Mills KHG, O'Neill LA (2017) Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front Immunol 8:1300. https://doi.org/10.3389/fimmu.2017.01300
Yang W, Lu Z (2013) Nuclear PKM2 regulates the Warburg effect. Cell Cycle (Georgetown, Tex) 12(19):3154–3158. https://doi.org/10.4161/cc.26182
Filipp FV (2013) Cancer metabolism meets systems biology: Pyruvate kinase isoform PKM2 is a metabolic master regulator. J Carcinog 12:14. https://doi.org/10.4103/1477-3163.115423
Luan W, Wang Y, Chen X, Shi Y, Wang J, Zhang J, Qian J, Li R, Tao T, Wei W, Hu Q, Liu N, You Y (2015) PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget 6(15):13006–13018. https://doi.org/10.18632/oncotarget.3514
Kimura A, Kitamura K, Ailiken G, Satoh M, Minamoto T, Tanaka N, Nomura F, Matsushita K (2017) FIR haplodeficiency promotes splicing to pyruvate kinase M2 in mice thymic lymphoma tissues revealed by six-plex tandem mass tag quantitative proteomic analysis. Oncotarget 8(40):67955
Batdorf B, Kroft S, Hosking P, Harrington A, Mackinnon A, Olteanu H (2014) Evaluation of CD43 Expression in Non-Hematologic Malignancies. Am J Clin Pathol 142(suppl_1):A244–A244. https://doi.org/10.1093/ajcp/142.suppl1.244
Bravo-Adame ME, Vera-Estrella R, Barkla BJ (2017) An alternative mode of CD43 signal transduction activates pro-survival pathways of T lymphocytes. 150 (1):87-99. https://doi.org/10.1111/imm.12670
Fu Q, Cash S, Andersen J, Kennedy C, Madadi A, Raghavendra M, Dietrich L, Agger W, Shelley C (2014) Intracellular patterns of sialophorin expression define a new molecular classification of breast cancer and represent new targets for therapy. British J Cancer 110(1):146
Fu Q, Cash SE, Andersen JJ, Kennedy CR, Oldenburg DG, Zander VB, Foley GR, Simon Shelley C (2013) CD43 in the nucleus and cytoplasm of lung cancer is a potential therapeutic target. Int J Cancer 132(8):1761–1770
Ranger AM, Gerstenfeld LC, Wang J, Kon T, Bae H, Gravallese EM, Glimcher MJ, Glimcher LH (2000) The nuclear factor of activated T cells (NFAT) transcription factor NFATp (NFATc2) is a repressor of chondrogenesis. J Exp Med 191(1):9–22
Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, Kleinstein SH, Abel ED, Insogna KL, Feske S, Locasale JW, Bosenberg MW, Rathmell JC, Kaech SM (2015) Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162(6):1217–1228. https://doi.org/10.1016/j.cell.2015.08.012
Qin J-J, Wang W, Voruganti S, Wang H, Zhang W-D, Zhang R (2015) Inhibiting NFAT1 for breast cancer therapy: new insights into the mechanism of action of MDM2 inhibitor JapA. Oncotarget 6(32):33106
Hwang SR, Murga-Zamalloa C, Brown N, Basappa J, McDonnell SR, Mendoza-Reinoso V, Basrur V, Wilcox R, Elenitoba-Johnson K, Lim MS (2017) Pyrimidine tract-binding protein 1 mediates pyruvate kinase M2-dependent phosphorylation of signal transducer and activator of transcription 3 and oncogenesis in anaplastic large cell lymphoma. Lab Investig 97(8):962
Lu S, Deng J, Liu H, Liu B, Yang J, Miao Y, Li J, Wang N, Jiang C, Xu Q, Wang X, Feng J (2018) PKM2-dependent metabolic reprogramming in CD4(+) T cells is crucial for hyperhomocysteinemia-accelerated atherosclerosis. J Mol Med (Berlin, Germany) 96(6):585–600. https://doi.org/10.1007/s00109-018-1645-6
Lin J, Lee I-M, Song Y, Cook NR, Selhub J, Manson JE, Buring JE, Zhang SM (2010) Plasma Homocysteine and Cysteine and Risk of Breast Cancer in Women. Cancer Res 70(6):2397–2405. https://doi.org/10.1158/0008-5472.can-09-3648
Hoffman W, Lakkis FG, Chalasani G (2016) B Cells, Antibodies, and More. Clin J Am Soc Nephrol: CJASN 11(1):137–154. https://doi.org/10.2215/CJN.09430915
Sugio T, Miyawaki K, Kato K, Sasaki K, Yamada K, Iqbal J, Miyamoto T, Ohshima K, Maeda T, Miyoshi H, Akashi K (2018) Microenvironmental immune cell signatures dictate clinical outcomes for PTCL-NOS. Blood Adv 2(17):2242–2252. https://doi.org/10.1182/bloodadvances.2018018754
Deng J, Lü S, Liu H, Liu B, Jiang C, Xu Q, Feng J, Wang X (2017) Homocysteine Activates B Cells via Regulating PKM2-Dependent Metabolic Reprogramming. J Immunol Author Choice 198(1):170–183. https://doi.org/10.4049/jimmunol.1600613
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11(11):723–737. https://doi.org/10.1038/nri3073
Martinez FO, Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6:13. https://doi.org/10.12703/p6-13
Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8(12):958–969. https://doi.org/10.1038/nri2448
Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O'Neill LA (2015) Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21(1):65–80. https://doi.org/10.1016/j.cmet.2014.12.005
Corcoran SE, O'Neill LA (2016) HIF1alpha and metabolic reprogramming in inflammation. J Clin Invest 126(10):3699–3707. https://doi.org/10.1172/jci84431
Liou G-Y, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44(5):479–496. https://doi.org/10.3109/10715761003667554
Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, Goronzy JJ, Weyand CM (2016) The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med 213(3):337–354. https://doi.org/10.1084/jem.20150900
Li W, Saud SM, Young MR, Chen G, Hua B (2015) Targeting AMPK for cancer prevention and treatment. Oncotarget 6(10):7365–7378. https://doi.org/10.18632/oncotarget.3629
Huang J, Liu K, Zhu S, Xie M, Kang R, Cao L, Tang D (2018) AMPK regulates immunometabolism in sepsis. Brain Behav Immun 72:89–100. https://doi.org/10.1016/j.bbi.2017.11.003
Zhang Z, Deng W, Kang R, Xie M, Billiar T, Wang H, Cao L, Tang D (2016) Plumbagin Protects Mice from Lethal Sepsis by Modulating Immunometabolism Upstream of PKM2. Molecular medicine. Cambridge, Mass. https://doi.org/10.2119/molmed.2015.00250
Lu W, Zuo Y, Feng Y, Zhang M (2014) SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumour Biol 35(11):10699–10705. https://doi.org/10.1007/s13277-014-2372-4
Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, Xu Y, Zhou L, Chu Y, Zhang C, Qin X, Yang P, Yu H (2017) SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1beta Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep 19(11):2331–2344. https://doi.org/10.1016/j.celrep.2017.05.065
Cheng Y, Feng Y, Xia Z, Li X, Rong J (2017) omega-Alkynyl arachidonic acid promotes anti-inflammatory macrophage M2 polarization against acute myocardial infarction via regulating the cross-talk between PKM2, HIF-1alpha and iNOS. Biochim Biophys Acta 1862(12):1595–1605. https://doi.org/10.1016/j.bbalip.2017.09.009
Collin M, McGovern N, Haniffa M (2013) Human dendritic cell subsets. Immunology 140(1):22–30. https://doi.org/10.1111/imm.12117
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12(4):253–268. https://doi.org/10.1038/nri3175
Lee JA, Sung HN, Jeon CH, Gill BC, Oh GS, Youn HJ, Park JH (2008) AIP1, a carbohydrate fraction from Artemisia iwayomogi, modulates the functional differentiation of bone marrow-derived dendritic cells. Int Immunopharmacol 8(4):534–541. https://doi.org/10.1016/j.intimp.2007.12.005
Vannini F, Kashfi K, Nath N (2015) The dual role of iNOS in cancer. Redox Biol 6:334–343. https://doi.org/10.1016/j.redox.2015.08.009
Jiang H, Zhang S, Song T, Guan X, Zhang R, Chen X (2018) Trichostatin a Protects Dendritic Cells Against Oxygen-Glucose Deprivation via the SRSF3/PKM2/Glycolytic Pathway. Front Pharmacol 9:612. https://doi.org/10.3389/fphar.2018.00612
Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P (2003) L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol 24(6):301–305. https://doi.org/10.1016/S1471-4906(03)00132-7
Liu WR, Tian MX, Yang LX, Lin YL, Jin L, Ding ZB, Shen YH, Peng YF, Gao DM, Zhou J, Qiu SJ, Dai Z, He R, Fan J, Shi YH (2015) PKM2 promotes metastasis by recruiting myeloid-derived suppressor cells and indicates poor prognosis for hepatocellular carcinoma. Oncotarget 6(2):846–861. https://doi.org/10.18632/oncotarget.2749
Trautwein C, Rakemann T, Malek NP, Plümpe J, Tiegs G, Manns MP (1998) Concanavalin A-induced liver injury triggers hepatocyte proliferation. J Clin Invest 101(9):1960–1969. https://doi.org/10.1172/JCI504
Weltzsch JP, Krech T, Vander Heiden MG, Tiegs G, Horst A (2018) Activation of pyruvate kinase isoform M2 (PKM2) in myeloid cells protects from Concanavalin A-mediated liver injury. J Hepatol 68:S448. https://doi.org/10.1016/S0168-8278(18)31136-X
Paul S, Lal G (2017) The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front Immunol 8:1124. https://doi.org/10.3389/fimmu.2017.01124
Shi F-D, Ljunggren H-G, La Cava A, Van Kaer L (2011) Organ-specific features of natural killer cells. Nat Rev Immunol 11(10):658–671. https://doi.org/10.1038/nri3065
Tu MM, Mahmoud AB, Makrigiannis AP (2016) Licensed and Unlicensed NK Cells: Differential Roles in Cancer and Viral Control. Front Immunol 7:166–166. https://doi.org/10.3389/fimmu.2016.00166
Schafer JR, Salzillo TC, Chakravarti N, Kararoudi MN, Trikha P, Foltz JA, Wang R, Li S, Lee DA (2019) Education-dependent activation of glycolysis promotes the cytolytic potency of licensed human natural killer cells. J Allergy Clin Immunol 143:346–358. e6
Tang D, Kang R, Zeh HJ 3rd, Lotze MT (2010) High-mobility group box 1 and cancer. Biochim Biophys Acta 1799(1-2):131–140. https://doi.org/10.1016/j.bbagrm.2009.11.014
Gdynia G, Sauer SW, Kopitz J, Fuchs D, Duglova K, Ruppert T, Miller M, Pahl J, Cerwenka A, Enders M, Mairbaurl H, Kaminski MM, Penzel R, Zhang C, Fuller JC, Wade RC, Benner A, Chang-Claude J, Brenner H, Hoffmeister M, Zentgraf H, Schirmacher P, Roth W (2016) The HMGB1 protein induces a metabolic type of tumour cell death by blocking aerobic respiration. Nat Commun 7:10764. https://doi.org/10.1038/ncomms10764
Tao L, Huang G, Song H, Chen Y, Chen L (2017) Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol Lett 14(3):2611–2620. https://doi.org/10.3892/ol.2017.6497
LeBleu VS, Kalluri R (2018) A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech 11(4):dmm029447. https://doi.org/10.1242/dmm.029447
Martinez-Outschoorn UE, Lisanti MP, Sotgia F (2014) Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol 25:47–60. https://doi.org/10.1016/j.semcancer.2014.01.005
Huang L, Xu AM, Liu S, Liu W, Li T-J (2014) Cancer-associated fibroblasts in digestive tumors. World J Gastroenterol: WJG 20(47):17804–17818. https://doi.org/10.3748/wjg.v20.i47.17804
Radisky DC, Kenny PA, Bissell MJ (2007) Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem 101(4):830–839. https://doi.org/10.1002/jcb.21186
Ishikawa M, Inoue T, Shirai T, Takamatsu K, Kunihiro S, Ishii H, Nishikata T (2014) Simultaneous expression of cancer stem cell-like properties and cancer-associated fibroblast-like properties in a primary culture of breast cancer cells. Cancers 6(3):1570–1578. https://doi.org/10.3390/cancers6031570
de Wit RH, Mujic-Delic A, van Senten JR, Fraile-Ramos A, Siderius M, Smit MJ (2016) Human cytomegalovirus encoded chemokine receptor US28 activates the HIF-1alpha/PKM2 axis in glioblastoma cells. Oncotarget 7(42):67966–67985. https://doi.org/10.18632/oncotarget.11817
Kachel P, Trojanowicz B, Sekulla C, Prenzel H, Dralle H, Hoang-Vu C (2015) Phosphorylation of pyruvate kinase M2 and lactate dehydrogenase A by fibroblast growth factor receptor 1 in benign and malignant thyroid tissue. BMC Cancer 15:140. https://doi.org/10.1186/s12885-015-1135-y
Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE, Witkiewicz AK, Birbe R, Howell A, Pestell RG, Smith J, Daniel R, Sotgia F, Lisanti MP (2011) Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther 12(12):1101–1113. https://doi.org/10.4161/cbt.12.12.18703
Chatterjee M, Ben-Josef E, Thomas DG, Morgan MA, Zalupski MM, Khan G, Andrew Robinson C, Griffith KA, Chen C-S, Ludwig T, Bekaii-Saab T, Chakravarti A, Williams TM (2015) Caveolin-1 is Associated with Tumor Progression and Confers a Multi-Modality Resistance Phenotype in Pancreatic Cancer. Sci Rep 5:10867–10867. https://doi.org/10.1038/srep10867
Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, Flomenberg N, Howell A, Pestell RG, Knudsen ES, Sotgia F, Lisanti MP (2010) Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle (Georgetown, Tex) 9(16):3256–3276. https://doi.org/10.4161/cc.9.16.12553
Santi A, Caselli A, Ranaldi F, Paoli P, Mugnaioni C, Michelucci E, Cirri P (2015) Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim Biophys Acta 1853(12):3211–3223. https://doi.org/10.1016/j.bbamcr.2015.09.013
Feng Y-H, Tsao C-J (2016) Emerging role of microRNA-21 in cancer. Biomed Rep 5(4):395–402. https://doi.org/10.3892/br.2016.747
Chen S, Chen X, Shan T, Ma J, Lin W, Li W, Kang Y (2018) MiR-21-mediated Metabolic Alteration of Cancer-associated Fibroblasts and Its Effect on Pancreatic Cancer Cell Behavior. Int J Biol Sci 14(1):100–110. https://doi.org/10.7150/ijbs.22555
Zhang H, Wang D, Li M, Plecita-Hlavata L, D'Alessandro A, Tauber J, Riddle S, Kumar S, Flockton A, McKeon BA, Frid MG, Reisz JA, Caruso P, El Kasmi KC, Jezek P, Morrell NW, Hu CJ, Stenmark KR (2017) Metabolic and Proliferative State of Vascular Adventitial Fibroblasts in Pulmonary Hypertension Is Regulated Through a MicroRNA-124/PTBP1 (Polypyrimidine Tract Binding Protein 1)/Pyruvate Kinase Muscle Axis. Circulation 136(25):2468–2485. https://doi.org/10.1161/circulationaha.117.028069
Wu H, Zhu S, Mo Y-Y (2009) Suppression of cell growth and invasion by miR-205 in breast cancer. Cell Res 19(4):439–448. https://doi.org/10.1038/cr.2009.18
Giannoni E, Taddei ML, Morandi A, Comito G, Calvani M, Bianchini F, Richichi B, Raugei G, Wong N, Tang D, Chiarugi P (2015) Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget 6(27):24061–24074. https://doi.org/10.18632/oncotarget.4448
Aird WC (2012) Endothelial Cell Heterogeneity. Cold Spring Harb Perspect Med 2(1):a006429. https://doi.org/10.1101/cshperspect.a006429
Lopes-Bastos BM, Jiang WG, Cai J (2016) Tumour-Endothelial Cell Communications: Important and Indispensable Mediators of Tumour Angiogenesis. Anticancer Res 36(3):1119–1126
Dudley AC (2012) Tumor Endothelial Cells. Cold Spring Harb Perspect Med 2(3):a006536. https://doi.org/10.1101/cshperspect.a006536
Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, Genze F, Simmet T, Seufferlein T (2016) PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol Cancer 15:3. https://doi.org/10.1186/s12943-015-0490-2
Lin Y, Liu F, Fan Y, Qian X, Lang R, Gu F, Gu J, Fu L (2015) Both high expression of pyruvate kinase M2 and vascular endothelial growth factor-C predicts poorer prognosis in human breast cancer. Int J Clin Exp Pathol 8(7):8028–8037
Sun H, Zhu A, Zhang L, Zhang J, Zhong Z, Wang F (2015) Knockdown of PKM2 Suppresses Tumor Growth and Invasion in Lung Adenocarcinoma. Int J Mol Sci 16(10):24574–24587. https://doi.org/10.3390/ijms161024574
Ma T, Patel H, Babapoor-Farrokhran S, Franklin R, Semenza GL, Sodhi A, Montaner S (2015) KSHV induces aerobic glycolysis and angiogenesis through HIF-1-dependent upregulation of pyruvate kinase 2 in Kaposi’s sarcoma. Angiogenesis 18:477–488
Lee E, Pandey NB, Popel AS (2014) Lymphatic endothelial cells support tumor growth in breast cancer. Sci Rep 4:5853. https://doi.org/10.1038/srep05853 https://www.nature.com/articles/srep05853#supplementary-information
He Z, Yi J, Liu X, Chen J, Han S, Jin L, Chen L, Song H (2016) MiR-143-3p functions as a tumor suppressor by regulating cell proliferation, invasion and epithelial–mesenchymal transition by targeting QKI-5 in esophageal squamous cell carcinoma. Mol Cancer 15(1):51. https://doi.org/10.1186/s12943-016-0533-3
Xu RH, Liu B, Wu JD, Yan YY, Wang JN (2016) miR-143 is involved in endothelial cell dysfunction through suppression of glycolysis and correlated with atherosclerotic plaques formation. Eur Rev Med Pharmacol Sci 20(19):4063–4071
Zhang L, Chen X, Liu B, Han J (2018) MicroRNA-124-3p directly targets PDCD6 to inhibit metastasis in breast cancer. Oncol Lett 15(1):984–990. https://doi.org/10.3892/ol.2017.7358
Caruso P, Dunmore BJ, Schlosser K, Schoors S, Dos Santos C, Perez-Iratxeta C, Lavoie JR, Zhang H, Long L, Flockton AR, Frid MG, Upton PD, D'Alessandro A, Hadinnapola C, Kiskin FN, Taha M, Hurst LA, Ormiston ML, Hata A, Stenmark KR, Carmeliet P, Stewart DJ, Morrell NW (2017) Identification of MicroRNA-124 as a Major Regulator of Enhanced Endothelial Cell Glycolysis in Pulmonary Arterial Hypertension via PTBP1 (Polypyrimidine Tract Binding Protein) and Pyruvate Kinase M2. Circulation 136(25):2451–2467. https://doi.org/10.1161/circulationaha.117.028034
Esteve Rafols M (2014) Adipose tissue: cell heterogeneity and functional diversity) Endocrinologia y nutricion : organo de la Sociedad Espanola de. Endocrinol Nutr 61(2):100–112. https://doi.org/10.1016/j.endonu.2013.03.011
Zahid H, Simpson ER, Brown KA (2016) Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr Opin Pharmacol 31:90–96. https://doi.org/10.1016/j.coph.2016.11.003
Jiang Y, Guo L, Xie LQ, Zhang YY, Liu XH, Zhang Y, Zhu H, Yang PY, Lu HJ, Tang QQ (2014) Proteome profiling of mitotic clonal expansion during 3T3-L1 adipocyte differentiation using iTRAQ-2DLC-MS/MS. J Proteome Res 13(3):1307–1314. https://doi.org/10.1021/pr401292p
Wei L, Li K, Pang X, Guo B, Su M, Huang Y, Wang N, Ji F, Zhong C, Yang J, Zhang Z, Jiang Y, Liu Y, Chen T (2016) Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J Exp Clin Cancer Res : CR 35(1):166. https://doi.org/10.1186/s13046-016-0446-4
Liao SC, Li JX, Yu L, Sun SR (2017) Protein tyrosine phosphatase 1B expression contributes to the development of breast cancer. J Zhejiang Univ Sci B 18(4):334–342. https://doi.org/10.1631/jzus.B1600184
Bettaieb A, Bakke J, Nagata N, Matsuo K, Xi Y, Liu S, AbouBechara D, Melhem R, Stanhope K, Cummings B, Graham J, Bremer A, Zhang S, Lyssiotis CA, Zhang ZY, Cantley LC, Havel PJ, Haj FG (2013) Protein tyrosine phosphatase 1B regulates pyruvate kinase M2 tyrosine phosphorylation. J Biol Chem 288(24):17360–17371. https://doi.org/10.1074/jbc.M112.441469
Xie J, Dai C, Hu X (2016) Evidence That Does Not Support Pyruvate Kinase M2 (PKM2)-catalyzed Reaction as a Rate-limiting Step in Cancer Cell Glycolysis. J Biol Chem 291(17):8987–8999. https://doi.org/10.1074/jbc.M115.704825
Hosios AM, Fiske BP, Gui DY, Vander Heiden MG (2015) Lack of evidence for PKM2 protein kinase activity. Mol Cell 59(5):850–857
Cheng H, Qin Y, Fan H, Su P, Zhang X, Zhang H, Zhou G (2013) Overexpression of CARM1 in breast cancer is correlated with poorly characterized clinicopathologic parameters and molecular subtypes. Diagn Pathol 8:129–129. https://doi.org/10.1186/1746-1596-8-129
Kung C, Hixon J, Choe S, Marks K, Gross S, Murphy E, DeLaBarre B, Cianchetta G, Sethumadhavan S, Wang X (2012) Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem Biol 19(9):1187–1198
Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, Burga LN, Xie J, Jurczak MJ, DePinho RA, Clish CB, Jacks T, Kibbey RG, Wulf GM, Di Vizio D, Mills GB, Cantley LC, Vander Heiden MG (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155(2):397–409. https://doi.org/10.1016/j.cell.2013.09.025
Cortes-Cros M, Hemmerlin C, Ferretti S, Zhang J, Gounarides JS, Yin H, Muller A, Haberkorn A, Chene P, Sellers WR, Hofmann F (2013) M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proc Natl Acad Sci U S A 110(2):489–494. https://doi.org/10.1073/pnas.1212780110
Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, Davidson SM, Luengo A, Bronson RT, Jacks T (2016) Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev
Dayton TL, Gocheva V, Miller KM, Bhutkar A, Lewis CA, Bronson RT, Vander Heiden MG, Jacks T (2018) Isoform-specific deletion of PKM2 constrains tumor initiation in a mouse model of soft tissue sarcoma. Cancer & metabolism 6, 6(1)
Stetak A, Veress R, Ovadi J, Csermely P, Keri G, Ullrich A (2007) Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer research 67(4):1602–1608. https://doi.org/10.1158/0008-5472.can-06-2870
Hillis AL, Lau AN, Devoe CX, Dayton TL, Danai LV, Di Vizio D, Vander Heiden MG (2018) PKM2 is not required for pancreatic ductal adenocarcinoma. Cancer Metab 6(1):17
Lau AN, Israelsen WJ, Roper J, Sinnamon MJ, Georgeon L, Dayton TL, Hillis AL, Yilmaz OH, Di Vizio D, Hung KE (2017) PKM2 is not required for colon cancer initiated by APC loss. Cancer Metab 5(1):10
Curless BP, Uko NE, Matesic DF (2018) Modulator of the PI3K/Akt oncogenic pathway affects mTOR complex 2 in human adenocarcinoma cells. Investig New Drugs:1–10
Li Y, Bao M, Yang C, Chen J, Zhou S, Sun R, Wu C, Li X, Bao J (2018) Computer-aided identification of a novel pyruvate kinase M2 activator compound. Cell Prolif 51(6):e12509
Li R, Ning X, Zhou S, Lin Z, Wu X, Chen H, Bai X, Wang X, Ge Z, Li R (2018) Discovery and structure-activity relationship of novel 4-hydroxy-thiazolidine-2-thione derivatives as tumor cell specific pyruvate kinase M2 activators. Eur J Med Chem 143:48–65
Ning X, Qi H, Li R, Li Y, Jin Y, McNutt MA, Liu J, Yin Y (2017) Discovery of novel naphthoquinone derivatives as inhibitors of the tumor cell specific M2 isoform of pyruvate kinase. Eur J Med Chem 138:343–352
Zhang Y, Liu B, Wu X, Li R, Ning X, Liu Y, Liu Z, Ge Z, Li R, Yin Y (2015) New pyridin-3-ylmethyl carbamodithioic esters activate pyruvate kinase M2 and potential anticancer lead compounds. Bioorg Med Chem 23(15):4815–4823
Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X (2011) Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 30(42):4297–4306. https://doi.org/10.1038/onc.2011.137
Palsson-McDermott EM, Dyck L, Zasłona Z, Menon D, McGettrick AF, Mills KH, O’Neill LA (2017) Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors. Front Immunol 8:1300
Yacovan A, Ozeri R, Kehat T, Mirilashvili S, Sherman D, Aizikovich A, Shitrit A, Ben-Zeev E, Schutz N, Bohana-Kashtan O (2012) 1-(sulfonyl)-5-(arylsulfonyl) indoline as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg Med Chem Lett 22(20):6460–6468
Guo C, Linton A, Jalaie M, Kephart S, Ornelas M, Pairish M, Greasley S, Richardson P, Maegley K, Hickey M (2013) Discovery of 2-((1H-benzo [d] imidazol-1-yl) methyl)-4H-pyrido [1, 2-a] pyrimidin-4-ones as novel PKM2 activators. Bioorg Med Chem Lett 23(11):3358–3363
Xu Y, Liu X-H, Saunders M, Pearce S, Foulks JM, Parnell KM, Clifford A, Nix RN, Bullough J, Hendrickson TF (2014) Discovery of 3-(trifluoromethyl)-1H-pyrazole-5-carboxamide activators of the M2 isoform of pyruvate kinase (PKM2). Bioorg Med Chem Lett 24(2):515–519
Walsh MJ, Brimacombe KR, Anastasiou D, Yu Y, Israelsen WJ, Hong B-S, Tempel W, Dimov S, Veith H, Yang H (2013) ML265: A potent PKM2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft model. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. National Center for Biotechnology Information (US)
Brimacombe KR, Anastasiou D, Hong B-S, Tempel W, Dimov S, Veith H, Auld DS, Vander Heiden MG, Thomas CJ, Park H-W (2013) ML285 affects reactive oxygen species’ inhibition of pyruvate kinase M2.
Kim DJ, Park YS, Kim ND, Min SH, You Y-M, Jung Y, Koo H, Noh H, Kim J-A, Park KC (2015) A novel pyruvate kinase M2 activator compound that suppresses lung cancer cell viability under hypoxia. Mol Cells 38(4):373
Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P (2011) Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol 2:49–49. https://doi.org/10.3389/fphar.2011.00049
Martins P, Jesus J, Santos S, Raposo L, Roma-Rodrigues C, Baptista P, Fernandes A (2015) Heterocyclic anticancer compounds: recent advances and the paradigm shift towards the use of nanomedicine’s tool box. Molecules 20(9):16852–16891
Spilioti E, Jaakkola M, Tolonen T, Lipponen M, Virtanen V, Chinou I, Kassi E, Karabournioti S, Moutsatsou P (2014) Phenolic acid composition, antiatherogenic and anticancer potential of honeys derived from various regions in Greece. PloS One 9(4):e94860
Acknowledgments
We acknowledge the support from National Institute of Pharmaceutical Education and Research (NIPER), Ahmedabad.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rihan, M., Nalla, L.V., Dharavath, A. et al. Pyruvate Kinase M2: a Metabolic Bug in Re-Wiring the Tumor Microenvironment. Cancer Microenvironment 12, 149–167 (2019). https://doi.org/10.1007/s12307-019-00226-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-019-00226-0