
Abstract
Prevalence of diabetes mellitus, a chronic metabolic disease characterized by hyperglycemia, is growing worldwide. The majority of the cases belong to type 2 diabetes mellitus (T2DM). Globally, India ranks second in terms of diabetes prevalence among adults. Currently available classes of therapeutic agents are used alone or in combinations but seldom achieve treatment targets. Diverse pathophysiology and the need of therapeutic agents with more favourable pharmacokinetic-pharmacodynamics profile make newer drug discoveries in the field of T2DM essential. A large number of molecules, some with novel mechanisms, are in pipeline. The essence of this review is to track and discuss these potential agents, based on their developmental stages, especially those in phase 3 or phase 2. Unique molecules are being developed for existing drug classes like insulins, DPP-4 inhibitors, GLP-1 analogues; and under newer classes like dual/pan PPAR agonists, dual SGLT1/SGLT2 inhibitors, glimins, anti-inflammatory agents, glucokinase activators, G-protein coupled receptor agonists, hybrid peptide agonists, apical sodium-dependent bile acid transporter (ASBT) inhibitors, glucagon receptor antagonists etc. The heterogeneous clinical presentation and therapeutic outcomes in phenotypically similar patients is a clue to think beyond the standard treatment strategy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is a chronic metabolic disease characterized and diagnosed by increased blood glucose levels [1]. Pathophysiology of type 2 diabetes mellitus (T2DM), the more common type, is collectively governed by eight factors (ominous octet) with peripheral insulin resistance and pancreatic β-cell failure being the core defects [2]. Approximately 415 million people worldwide are estimated to have diabetes, and about 75% of these inhabit low- and middle-income countries. Trends of urbanization, increasing proportion of the elderly population; alongside adverse lifestyle changes (reduced physical activity, increased refined carbohydrate with low fruit and vegetable intake) contributed to rise in T2DM prevalence. Globally India ranks second after China in terms of diabetes prevalence among adults [1]. Currently, available T2DM therapeutic agents are usually continued in combinations, as monotherapy seldom suffices for long.
Currently Approved Drugs
Several drug classes have been approved by various drug regulatory authorities, around the world, for T2DM treatment (Table 1). Drug-related adverse effects have been reported as the leading cause for non-adherence to anti-diabetic medications [3, 4]. Non-adherences to treatment results in inefficient glycemic control and a higher risk of hospitalization and mortality [5, 6]. Also, even combination therapies may not target all the pathways involved in pathophysiology and fall short in halting the progression of the disease.
The above-mentioned facts make newer drug discoveries in the field of T2DM essential. Researchers around the world are working to develop new agents, either with known or novel mechanisms, which could deliver optimum or at least better benefit- risk ratio and tolerability than the existing antidiabetic drugs. The success rate of lead candidates from phase 2 to phase 3 is the least while it improves post phase-3 [7]. Thus, the focus of this review is to bring forth promising drug candidates either in phase-2 or those progressed to phase 3 or further in clinical development.
Newer/Improved Agents Under Approved Classes
Insulins
Insulin has always been the antidiabetic medication with the highest efficacy. Currently, available insulin analogs are “injectable only”, needing “multiple administrations”. To overcome these limitations, dosage forms other than injections or longer acting injectable analogs are being worked upon.
Injectable Insulin
FIAsp (NN1218) is an insulin analog having a quicker onset and shorter duration of action than Lispro [10] and thus, is more suitable for post prandial glycemic control. FIAsp is currently submitted for marketing approval with United States Food and Drug Administration (US-FDA) [11] and European Medical Agency (EMA) [12]. Linjeta (VIAject 25) was a similar analog which made it to phase 3 but was rejected by FDA due to doubtful efficacy and tolerability [13].
The basal insulin analog LY2605541 was a pegylated insulin Lispro (pegLispro), where the PEGylation resulted in a longer duration of action. It showed more sustained glycemic control than insulin Glargine in T2DM patients. Its development was halted later as it was associated with unfavorable lipid profile and raised hepatic enzymes [14,15,16].
Oral Insulin
Oral insulin, like endogenous insulin, would undergo first-pass metabolism in the liver; thereby reducing hepatic gluconeogenesis and glycogenolysis which equates to 65-80% suppression of basal glucose production [17, 18]. Thus, oral insulin is considered as a viable option because of its physiologic nature and better acceptability.
Several molecules are in pipeline but only IN-105 reached phase 3 clinical trial. Unfortunately, the phase 3 trial conducted in India did not meet its primary efficacy endpoint; although several secondary efficacy endpoints significantly improved and no serious adverse event was reported [19].
Inhaled Insulins
Post unfortunate and discouraging withdrawal of Exubera within 2 years of its marketing approval, the US FDA approved Afrezza in June 2014. It is ultra- rapid-acting inhaled insulin i.e. has shorter onset time and a more rapid elimination as compared to subcutaneous (SC) insulin [20]. Afrezza showed good postprandial blood glucose control with a low risk of hypoglycemia [21].
A meta-analysis favored SC insulin over inhaled in terms of HbA1c reduction but the good glycemic control criteria were achieved in similar fraction of subjects in both groups [22].
Dipeptidyl-Peptidase 4 (DPP-4) Inhibitors
Currently, several molecules, with plasma half-lives longer than the existing agents are in pipeline. The longer duration of action will be helpful in improving adherence and sustaining the glycemic control.
Japanese Pharmaceuticals and Medical Devices Agency (PMDA) approved Trelagliptin, the first once-weekly DPP-4 inhibitor, for the treatment of T2DM in March 2015 [23]. However anticipating high marketing approval costs, the company discontinued development of Trelagliptin in USA and Europe [24]. In clinical trials, Trelagliptin produced significant dose-dependent HbA1c reductions compared with placebo in Phase 2 [25] and was found to be non-inferior to Alogliptin in phase 3 [26]. No hypoglycemic episode was reported in both the studies [25, 26].
Omarigliptin (Marizev), another oral once-weekly DPP-4 inhibitor, was approved by PMDA Japan in 2015. Its efficacy and side effect profile was found similar to the currently marketed DPP-4 inhibitors [27]. However, the company decided not to file the marketing application for Omarigliptin in the United States or Europe [28]. According to a review of the Omarigliptin trial data, it was found to have, at least in two studies, more serious side effects than Sitagliptin [29].
Glucagon-Like Peptide (GLP-1) Agonists
GLP-1 analogs restore β-cells glucose sensitivity in T2DM patients [30]. Studies have also shown that GLP-1 promotes β-cell proliferation and inhibits β-cell apoptosis. This class of drugs also slow gastric emptying and suppression of appetite causing weight loss [31]. Administration via injection is the biggest drawback of marketed GLP-1 analogs. There is a need to develop drugs with either longer plasma half-life or with non-invasive route of administration.
Several oral GLP-1 analogs are under development but till date, only Semaglutide (NN9924), oral once-daily drug, has successfully completed phase 2 trials [32] and is ready for phase 3 [33] (NCT02607865).
Meanwhile, the same company has completed phase 3a trials with Semaglutide (NN9535), a once-weekly injectable GLP-1 analog. The results present it as a potential molecule for broad usage in T2DM patients [34].
Another approach experimented for better adherence with GLP-1 analogs is continuous subcutaneous delivery. Extended release Exenatide is already in the market (Bydureon), which uses drug filled microspheres enabling weekly administration. Next in pipeline with much longer plasma half-life is ITCA 650. ITCA 650 utilizes DUROS technology of drug delivery, capable of delivering a wide range of therapeutic molecules for durations ranging from 3 to 12 months [35]. It has shown good patient acceptance and sustained reductions in HbA1c in a 48-week phase 2 study [36]. Further a 52-week phase 3 study proved its superiority over Sitagliptin in terms of sustained glucose control and weight reduction [37]. In U.S. regulatory filing of ITCA 650 is targeted for end of 3rd quarter of 2016 [38].
Drug Classes With Newer Mechanism of Action (Molecules in Phase 3 Development)
Peroxisome Proliferator-Activated Receptors (PPAR) Agonists
PPARs forms a family of ligand-activated nuclear hormone receptors (NRs) [39] that regulate transcription of genes involved in various metabolic processes, especially lipid, and glucose homeostasis. PPARs exist as three isoforms: PPARα, PPARβ/δ, and PPARγ; which have different but overlapping tissue distributions, ligands and physiological roles.
PPAR-γ has insulin sensitizing effect and serves target to thiazolidinediones (TZDs). The major disadvantage of using PPAR-γ ligands is they promote adipogenesis and edema. Partial PPAR-γ agonists or selective PPAR-γ modulators (SSPARMs) are under development to address these issues.
PPAR-δ’s are mainly present in skeletal muscles and adipose tissues where they regulate fatty acid catabolism and increased insulin sensitivity; and thus can be utilised as dual target for glycemic and lipid metabolism disorders.
PPAR-γ Agonists/Modulators
Inlacin (DLBS 3233), a combined bioactive fraction of Cinnamomum burmanii and Lagerstroemia speciosa, is the first to be approved in this class by Philippines FDA. Balaglitazone, a novel insulin sensitizer, showed clinically meaningful improvements in glucose levels and HbA1c, in the phase 3 BALLET trial [40]. Further developments were halted due to lack of promoting partners.
PPAR-α/γ Co-agonists
Lobeglitazone (CDK-501), a dual PPARα and PPARγ agonist, was approved by the Ministry of Food and Drug Safety (Korea) on July 4, 2013. In various phase 3 trials, Lobeglitazone has shown a favorable balance in the efficacy and safety profile [41]. It was also found to be non-inferior to Pioglitazone as an add-on to Metformin in terms of their efficacy and safety [42].
Prior attempts to develop dual PPAR-activating drugs for T2DM have been unsuccessful. Muraglitazar showed increased cardiac ischemic events [43]; while Tesaglitazar showed renal toxicity [44] thus, resulting in termination of their development. Aleglitazar development was also stopped following regular safety review of phase 3 trial by US FDA [45]. Similar was the fate of Ragaglitazar and Naveglitazar.
PPAR Pan-agonists
Chiglitazar, an oral drug, activates all three PPAR isoforms [46] and has shown to improve the impaired insulin and glucose tolerance in a preclinical study. The study also suggests that Chiglitazar may prove to have better lipid control in diabetic patients, than selective PPARγ agonist [47]. Phase 2 results of the molecule are promising [48] and the drug is currently in phase 3 of development (NCT02121717, NCT02173457).
Sodium Glucose Cotransporter Type 1 (SGLT1) and Dual SGLT1/SGLT2 Inhibitors
The kidney plays role in glucose metabolism primarily by regulating glucose reabsorption from the urinary filtrate [49]. Carrier proteins SGLTs and the facilitated glucose transporters (GLUTs) are involved in the process of glucose reabsorption in the proximal convoluted tubule [50, 51]. Approximately 90% of filtered renal glucose is reabsorbed by SGLT2, and the remaining 10% is removed by SGLT1 [50, 51]. These are insulin independent processes.
SGLT2 is expressed to a lower extent in organs other than renal, whereas SGLT1 is located in the small intestine also, where it has a significant role in glucose absorption [51]. While SGLT2 inhibitors are already in the market; selective SGLT1inhibitors and dual SGLT1/SGLT2 inhibitors, which can target glucose and galactose absorption in the small intestine are being worked upon.
Sotagliflozin (LX4211), a dual SGLT1/SGLT2 inhibitor is undergoing phase 3 for Type 1 diabetes mellitus (T1D) and has completed two successful phase 2 trials in T2DM patients; and the company is planning for phase 3 trials in T2DM patients [52]. Data from the phase 2b study in T2DM patients showed that Sotagliflozin comfortably achieved primary and multiple secondary endpoints; also, a significant reduction in body weight and blood pressure was reported. Sotagliflozin was well tolerated with the overall incidence of adverse events being similar to placebo [52]. No selective GLT1 inhibitor has reached this far till date.
Tetrahydrotriazin Containing Oral Antidiabetic (Glimins)
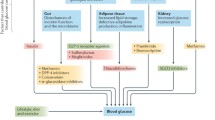
Imeglimin, an oral antidiabetic agent and the first in its class, is defined as a mitochondrial bioenergetics enhancer. Imeglimin simultaneously targets the liver, muscles, and pancreas, thus alleviating the two major defects of diabetes: insulin secretion and insulin sensitivity (Fig. 1).

Action spectrum of Tetrahydrotriazin containing oral antidiabetic (Glimins)
In several phase 2 studies, Imeglimin achieved primary and secondary glycemic endpoints including a statistically significant reduction in HbA1c and was shown to have excellent safety and tolerability similar to placebo. Its developer is aggressively preparing for Phase 3 trials in the US and Europe [53].
Anti-inflammatory Agents
Adipocytes are in a state of chronic inflammation in T2DM and secrete excessive amounts of proinflammatory cytokines/chemokines [tumor necrosis factor (TNF α), interleukin (IL)-1β, IL-6, monocyte chemotactic protein (MCP)]; thus activating proteins including toll-like receptor (TLR)1, TLR4, and activator protein (AP)1 and NFκβ [13, 54]. Several agents that target these inflammatory mediators are under development (Fig. 2).

Role of inflammation in T2DM pathophhysiology. Mechanism of action of anti-inflammatory agents
Diacerein/AC-201 is an orally available, twice-daily, small-molecule agent that inhibits production and activity of IL-1β and down-regulates IL-1R. It is currently in phase3 efficacy study to evaluate glycemic control and liver fat in uncontrolled T2DM subjects (NCT02242149).
Canakinumab, an IL-1 modulator (already approved for cryopyrin-associated periodic syndromes, gouty arthritis, and juvenile rheumatoid arthritis) reached the phase 3 trials but was terminated because of the overall lowering of HbA1c, in combination with Metformin, was inadequate to continue the study into Period IV (NCT00900146).
In a phase 3 randomized trial (NCT00799643) with 286 T2DM patients Salsalate, a Non-Steroidal Anti-Inflammatory Drug (NSAID), lowered HbA1C by 0.33% (P < 0.01 vs. baseline and placebo) [55] through its inhibitory effect on IKKβ/NF-κβ (inhibitor of NF-κB kinase subunit β/nuclear-factor κβ) pathway [56]. Although promising, the result was not sufficient to justify the continuation of the study.
The Newer hypothesis suggests concurrent inhibition of more than one inflammatory pathway to be more promising.
Drug Classes with Newer Mechanism of Action (Molecules in Initial Phases of Development)
Glucokinase Activators
Glucokinase is the member of hexokinase enzyme family with glucose as its preferred substrate under physiological conditions; hence the name glucokinase. Glucokinase is expressed in glucose sensing organs i.e. pancreas, liver, brain and gastrointestinal tract [57]. In pancreatic β-cells glucokinase plays a major role in regulating insulin secretion, while in hepatocytes, it regulates hepatic glucose utilization.
Animal studies showed that glucokinase overexpression accelerates hepatic glucose metabolism, resulting in lower plasma glucose concentration [58]. Although the researchers are continuously working to develop a glucokinase activator (GKA) molecule which can be used in T2DM treatment; but till date no GKA has crossed phase 2 in clinical development.
MK-0941 could not yield sustained glycemic control and was associated with an increased incidence of hypoglycemia and hypertriglyceridemia [59]. In phase 2 studies, similar agents i.e. AZD1656 and Piragliatin respectively showed a lack of sustained glycemic control and increased incidence of hypoglycaemia [60]. Molecules TTP 399, Sinogliatin (HMS-5552) and YH-GKA are still in early phase of development under this class.
G-Protein Coupled Receptor (GPCR/GPR) Agonists
GPCRs in the gut–brain–pancreatic axis is key players in the postprandial control of metabolism and food intake. Cell-specific expression of these receptors causes selective activation of target tissues, including pancreatic β cells, gut endocrine cells and neuronal populations involved in the suppression of appetite [61, 62].
Potential GPCRs targets in pancreatic β-cells include incretin receptors (GLP1R, GIPR), GPR119, FFAR1 (GPR40) and FFAR4 (GPR120); the activation of which stimulates insulin secretion in a glucose-dependent fashion [63].
For GPR40, molecule TAK-875 was the farthest to go but its development was discontinued in phase 3 due to potential liver toxicity [64]. At present, only two GPCR agonists JTT851 and DS-8500 are under phase 2 trials.
Hybrid Peptide Agonists
Intestinal derived peptides, predominantly glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP); secondarily cholecystokinin (CCK), oxyntomodulin (OXM) and peptide YY (PYY) are responsible for the various endocrine functions of GIT. These peptides play role in postprandial insulin release (“incretin effect”) and energy regulation [65,66,67].
Weight loss and improved glycemic control can be achieved by OXM, through activation of glucagon receptors and GLP-1 receptors respectively [67]. Hybrid peptides, like oxyntomodulin, are capable of modulating more than one receptor pathway. Various types of hybrids are GIP/GLP-1chimeric peptides, GLP-1/glucagon co-agonists (TT401/LY2944876-phase 2), GLP-1-gastrin co-agonists and GLP-1-PYY co-agonists. Hybrid peptides are being developed for the treatment of obesity-related diabetes.
Apical Sodium-Dependent Bile Acid Transporter (ASBT) Inhibitors
Bile salts have been found to act as signals on the intracellular nuclear farnesoid X receptor (FXR) [68] and the cell surface Tgr5 G protein-coupled receptor [69]. These receptors are involved in lipid and glucose metabolism. Bile acids, through these receptors, are capable of enhancing gastrointestinal peptides secretion [70,71,72].
Most of the conjugated bile acids secreted in the duodenum, as a response to a meal, are reabsorbed into enterocytes via the ASBT. By decreasing the bile acids uptake, ASBT inhibitors not only stimulate conversion of hepatic cholesterol to bile acids but also enhance gastrointestinal peptides release.
The only molecule under development in this class is GSK672 (GSK2330672). Under phase 2 studies conducted in T2DM subjects maintained on Metformin, GSK672 improved glycemic parameters and lipid profile. But these patients experienced a high incidence of gastrointestinal adverse events [73].
Glucagon Receptor (GCGR) Antagonists
Glucagon and Insulin are physiological antagonists and their interplay helps in maintaining euglycemia. Dysregulation of glucagon secretion, in T2DM patients, results in abnormally increased fasting and postprandial hepatic glucose production [74, 75].
Many animal and human studies have shown GCGr antagonists as a potential treatment option via reduction of hepatic glucose production. Though the concept came in 1980s [76] but still no molecule has reached phase 3 of the development.
Mitochondrial Target of Thiazolidinediones (mTOT Agonists)
A mitochondrial protein complex (mTOT) coordinates between carbohydrates, lipid, and amino acid metabolism according to cell need [77]. It does so by controlling mitochondrial pyruvate transport [78, 79]. Discovery of mTOT supports the possibility of developing insulin-sensitizing drugs that lack TZDs like adverse effects.
Two molecules (MSDC 0160 and MSDC 0602) reached phase 2 trials but none is active now for T2DM, instead, are being worked upon as a treatment for Alzheimer’s and Parkinsonism.
Fructose-1, 6-Bisphosphatase (FBPase) Inhibitors
The enzyme FBPase catalyzes the bidirectional reaction of fructose-1, 6-bisphosphate to fructose-6-phosphate (glycolysis) and back to fructose-1, 6-bisphosphate (gluconeogenesis). Inhibition of FBPase in animal models of T2DM effectively lowers HGP without causing hypoglycemia [80]. An FBPase inhibitor, MB07803 is currently in phase 2 of development.
Diacylglycerolacyl Transferase (DGAT)-1 Inhibitors
Of the two isoenzymes of DGAT, DGAT-1 catalyzes the final step of triglyceride synthesis. Inhibition of DGAT-1 in the GI tract has shown a reduction in postprandial plasma triglyceride excursion, insulin sensitization, and weight loss in preclinical studies. However, marked GI side effects, mainly diarrhea, occurred at doses that inhibited triglyceride excursions by ≥250% [81]. Only molecule P7435 was found to be in phase 1 study, but no recent updates are available.
11β-Hydroxysteroid Dehydrogenase Type 1 (11β-HSD1) Inhibitors
11β-HSD1 catalyzes inert cortisone into physiologically active cortisol. 11β-HSD1 overexpression in adipose tissue of rodents has resulted in metabolic syndrome-like phenotype, while inhibition of 11β-HSD1 has been proposed to be beneficial in T2DM patients [82]. 11β-HSD1 inhibitors were well tolerated in clinical studies and showed improved glycemic control, lipid profile, blood pressure, and induced modest weight loss.
In comparison to other agents, effects of 11β-HSD1 inhibitors are mild and discouraging for further development [83]. Activation of Hypothalamic–pituitary axis is a major concern associated with these molecules.
Other Potential Targets Under Evaluation for T2DM Drug Development
Several candidates under various other classes have also reached phase 2 of clinical development and include, cannabinoid receptor antagonists (GWP42004), glucocorticoid receptor antagonist (IONIS-GCCRRx), HSP (heat shock protein) inducer (BGP-15), and Protein tyrosine phosphatise 1B (PTP1B) inhibitors (IONIS-PTP1BRx). Extent of development and current status of the above mentioned newer potential drug classes/molecules have been summarised in Table 2.
An array of promising agents, for type 2 diabetes, are in early development like hepatic carnitine palmitoyltransferase1 (CPT1) inhibitors, Melanocortin-4 receptor (MC4R) agonist, Fibroblast growth factor (FGF) 21 agonists, modulators of the gut microbiota (Microbiome modulators), Sirtuin1 (SIRT1) activators, activators of glycogen synthase and inhibitors of glycogen phosphorylase.
Proton pump inhibitors (PPIs) also have antihyperglycemic effect, as indicated by several animal and humans studies. A randomized-controlled clinical study demonstrated increased fasting plasma gastrin and insulin, when Pantoprazole was used as add-on therapy in T2DM patients [84]. The actual role of PPIs in diabetes needs to be further explored and investigated.
Future Perspectives
The progressive β-cell failure seen in diabetic patients irrespective of their treatment profile needs to be addressed. Certain lifestyle modifications and drugs have a beneficial effect on β-cell function and attenuate its rate of failure. Genome-wide association studies (GWAS) and preclinical studies have delineated several potential molecular targets and small molecular agonists as an answer to the problem [85]. It is expected to gain few molecules, in developmental pipeline, which may produce a sizeable increase in β-cell mass by modulating pathways governing β-cell survival, function, and proliferation.
The diabetic patients belong to diverse ethnic groups, age groups, and physical characteristics. Currently, the diabetes standard care guidelines, derived from the concept of evidence-based medicine, are based on the above mentioned phenotypic characteristics. But even among diabetic individuals with similar phenotypic profile pathophysiology process may vary, leading to multiple permutations of insulin level and insulin resistance. This, in turn, leads to a variable response to same medication/s and different rates of complication development. The family of antidiabetic agents is growing at a commendable rate but treatment failure still haunts many patients. All these variations, hint towards underlying individual molecular characteristics (omics) deciding the pathophysiology progress, clinical phenotypes and success rate of treatment.
The need of the hour is precision therapy, where molecular characteristics and clinical presentation jointly decides the optimum medication for any patient. Application of bioinformatics derived tools to genomics, transcriptomics, metabolomics and proteomics is being considered as the solution. Several promising findings in this area includes increased sulphonylurea sensitivity in HNF1A MODY patients; [86] increased sulphonylurea efficacy and enhanced hypoglycemia risk in patients with reduced-function polymorphisms in the gene encoding CYP2C9 enzyme [87, 88] and increased GI intolerance to Metformin in patients with reduced function organic cation transporter 1 (OCT1) variants [89]. In a GWAS on diabetic patients, locus on chromosome 11 was discovered to be associated with Metformin action [90].
The concept of personalized medicine has been widely explored in antineoplastic therapies but, insufficient practical evidence exists in the field of antidiabetic drugs. Also, similar to any pathology, newer treatment modalities for T2DM are expected to achieve optimum risk–benefit ratio. This expectation further strengthens the need of molecules, which may target specific molecular characteristics (omics) and breaks the age old “one-drug suits all” approach.
References
IDF diabetes atlas. International Diabetes Federation. 2015. http://www.diabetesatlas.org. Accessed 03 Aug 2016.
DeFronzo RA. Pathogenesis of type 2 diabetes: metabolic and molecular implications for identifying diabetes genes. Diabetes Res. 1997;5:177–269.
Grant RW, Devita NG, Singer DE, Meigs JB. Polypharmacy and medication adherence in patients with type 2 diabetes. Diabetes Care. 2003;26(5):1408–12.
Jayant D, Lawrence B, Richard G. Factor influencing patient acceptability of diabetes treatment regimens. Clin Diabetes. 2000;18(2):61–7.
Ho PM, Rumsfeld JS, Masoudi FA, McClure DL, Plomondon ME, Steiner JF, et al. Effect of medication nonadherence on hospitalization and mortality among patients with diabetes mellitus. Arch Intern Med. 2006;166:1836–41.
Currie CJ, Peyrot M, Morgan CL, Poole CD, Jenkins-Jones S, Rubin RR, et al. The impact of treatment noncompliance on mortality in people with type 2 diabetes. Diabetes Care. 2012;35:1279–84.
Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51.
Bolen S, Feldman L, Vassy J, Wilson L, Yeh HC, Marinopoulos S, et al. Systematic review: comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med. 2007;147:386–99.
Egan AG, Blind E, Dunder K, Graeff PA, Hummer BT, Bourcier T, et al. Pancreatic safety of incretin based drugs—FDA and EMA assessment. N Engl J Med. 2014;370:794–7.
Steiner S, Hompesch M, Pohl R, Simms P, Flacke F, Mohr T, et al. A novel insulin formulation with a more rapid onset of action. Diabetologia. 2008;51:1602–6.
Novo Nordisk files for regulatory approval of faster-acting insulin aspart in the US for the treatment of type 1 and 2 diabetes. Novo Nordisk. 2015. http://www.novonordisk.com/media/news-details.1972083.html. Accessed 04 Aug 2016.
Novo Nordisk files for regulatory approval of faster-acting insulin aspart in the EU for the treatment of type 1 and 2 diabetes. Novo Nordisk. 2015. http://www.novonordisk.com/media/news-details.1971141.html. Accessed 04 Aug 2016.
Mittermayer F, Caveney E, De Oliveira C, Gourgiotis L, Puri M, Tai L-J, et al. Addressing unmet medical needs in type 2 diabetes: a narrative review of drugs under development. Curr Diabetes Rev. 2015;11(1):17–31.
Bergenstal RM, Rosenstock J, Arakaki RF, Prince MJ, Qu Y, Sinha VP, et al. A randomized, controlled study of once-daily LY2605541, a novel long-acting basal insulin, versus insulin glargine in basal insulin-treated patients with type 2 diabetes. Diabetes Care. 2012;35:2140–7.
Rosenstock J, Bergenstal RM, Blevins TC, Morrow LA, Prince MJ, Qu Y, et al. Better glycemic control and weight loss with the novel long-acting basal insulin LY2605541 compared with insulin glargine in type 1 diabetes: a randomized, crossover study. Diabetes Care. 2013;36:522–8.
2015 Integrated report. Eli Lilly and company. 2015. https://www.lilly.com/_assets/pdf/integrated-report.pdf. Accessed 03 Aug 2016.
Taylor R, Magnusson I, Rothman DL, Cline GW, Caumo A, Cobelli C, et al. Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J Clin Invest. 1996;97:126–32.
Singhal P, Caumo A, Carey PE, Cobelli C, Taylor R. Regulation of endogenous glucose production after a mixed meal in type 2 diabetes. Am J Physiol Endocrinol Metab. 2002;283:E275–83.
Zijlstra E, Heinemann L, Plum-Mörschel L. Oral insulin reloaded: a structured approach. J Diabetes Sci Technol. 2014;8(3):458–65.
Santos Cavaiola T, Edelman S. Inhaled insulin: a breath of fresh air? A review of inhaled insulin. Clin Ther. 2014;36:1275–89.
Zisser H, Jovanovic L, Markova K, Petrucci R, Boss A, Richardson P, et al. Technosphere insulin effectively controls postprandial glycemia in patients with type 2 diabetes mellitus. Diabetes Technol Ther. 2012;14:997–1001.
Ceglia L, Lau J, Pittas AG. Meta-analysis: efficacy and safety of inhaled insulin therapy in adults with diabetes mellitus. Ann Intern Med. 2006;145:665–75.
New drug application approval of zafatek® tablets for the treatment of type 2 diabetes in japan. Takeda Pharmaceutical Company Limited. 2015. https://www.takeda.com/news/2015/20150326_6953.html. Accessed 04 Aug 2016.
McKeage K. Trelagliptin: first global approval. Drugs. 2015;75:1161–4.
Inagaki N, Onouchi H, Sano H, Funao N, Kuroda S, Kaku K. SYR-472, a novel once weekly dipeptidyl peptidase-4 (DPP-4) inhibitor, in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2:125–32.
Inagaki N, Onouchi H, Maezawa H, Kuroda S, Kaku K. Once-weekly trelagliptin versus daily alogliptin in Japanese patients with type 2 diabetes: a randomised, double-blind, phase 3, non-inferiority study. Lancet Diabetes Endocrinol. 2015;3:191–7.
Evans PM, Bain SC. Omarigliptin for the treatment of type 2 diabetes mellitus. Expert Opin Pharmacother. 2016;17(14):1947–52.
Merck provides update on filing plans for omarigliptin, an investigational DPP-4 inhibitor for type 2 diabetes. Merck & company. 2016. http://www.mercknewsroom.com/news/company-statements/merck-provides-update-filing-plans-omarigliptin-investigational-dpp-4-inhibi. Accessed 05 Aug 2016.
Evidence review of marizev, a pipeline DPP-4 treatment for type 2 diabetes. Advera health analytics. 2016. http://info.adverahealth.com/marizev-evidence-review. Accessed 06 Aug 2016.
Bunck MC, Cornér A, Eliasson B, Heine RJ, Shaginian RM, Taskinen MR, et al. Effects of Exenatide on measures of β-cell function after 3 years in Metformin treated patients with type 2 diabetes. Diabetes Care. 2011;34:2041–7.
Drucker DJ. Deciphering metabolic messages from the gut drives therapeutic innovation: the 2014 Banting Lecture. Diabetes. 2015;64:317–26.
Novo Nordisk announces positive results for phase 2 trial with oral semaglutide in people with type 2diabetes. Novo Nordisk. 2015. http://www.novonordisk.com/media/news-details.1896081.html. Accessed 07 Aug 2016.
Novo Nordisk to initiate phase 3a development of oral semaglutide, a once-daily oral GLP-1 analogue. Novo Nordisk. 2015. http://www.novonordisk.com/media/news-details.1947638.html. Accessed 07 Aug 2016.
Novo Nordisk successfully completes fifth phase 3a trial with semaglutide in people with type 2 diabetes. Novo Nordisk. 2016. http://www.novonordisk.com/media/news-details.1988465.html. Accessed 11 Sept 2016.
Rohloff CM, Alessi TR, Yang B, Dahms J, Carr JP, Lautenbach SD. DUROS® technology delivers peptides and proteins at consistent rate continuously for 3 to 12 months. J Diabetes Sci Technol. 2008;2(3):461–7.
Henry RR, Rosenstock J, Logan D, Alessi T, Luskey K, Baron MA. Continuous subcutaneous delivery of exenatide via ITCA 650 leads to sustained glycemic control and weight loss for 48 weeks in metformin-treated subjects with type 2 diabetes. J Diabetes Complic. 2014;28(3):393–8.
Intarcia announces new top-line phase 3 results for investigational therapy ITCA 650 in type 2 diabetes: freedom-2 comparative trial demonstrates superior and sustained glucose control and weight reduction vs januvia over 52 weeks. Intarcia Therapeutics, Inc. 2016. http://www.intarcia.com/media/press-releases/2015-aug-18-freedom2.html. Accessed 15 Aug 2016.
ITCA 650. Intarcia Therapeutics, Inc. http://www.intarcia.com/pipeline-technology/itca-650.html. Accessed 07 Aug 2016.
Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–35.
Henriksen K, Byrjalsen I, Qvist P, Beck-Nielsen H, Hansen G, Riis BJ, et al. Efficacy and safety of the PPARγ partial agonist balaglitazone compared with pioglitazone and placebo: a phase III, randomized, parallel-group study in patients with type 2 diabetes on stable insulin therapy. Diabetes Metab Res Rev. 2011;27(4):392–401.
Kim SG, Kim DM, Woo J-T, Jang HK, Chung CH, Ko KS, et al. Efficacy and safety of lobeglitazone monotherapy in patients with type 2 diabetes mellitus over 24-weeks: a multicenter, randomized, double-blind, parallel-group, placebo controlled trial. PLoS ONE. 2014;. doi:10.1371/journal.pone.00928439.
Jin SM, Park CY, Cho YM, Ku BJ, Ahn CW, Cha BS, et al. Lobeglitazone and pioglitazone as add-ons to metformin for patients with type 2 diabetes: a 24-week, multicentre, randomized, double-blind, parallel-group, active-controlled, phase III clinical trial with a 28-week extension. Diabetes Obes Metab. 2015;17(6):599–602.
Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294(20):2581–6.
AstraZeneca annual report 2006. AstraZeneca. 2007. https://www.astrazeneca.com/content/dam/az/PDF/2006_Annual_Report.pdf. Accessed 11 Aug 2016.
Roche halts investigation of aleglitazar following regular safety review of phase III trial. Roche. 2013. http://www.roche.com/media/store/releases/med-cor-2013-07-10.htm. Accessed 15 Aug 2016.
He BK, Ning ZQ, Li ZB, Shan S, Pan DS, Ko BCB, et al. In vitro and in vivo characterizations of chiglitazar, a newly identified ppar pan-agonist. PPAR Res. 2012;. doi:10.1155/2012/546548.
Li P-P, Shan S, Chen Y-T, Ning Z-Q, Sun S-J, Liu Q, et al. The PPAR α/γ dual agonist chiglitazar improves insulin resistance and dyslipidemia in MSG obese rats. Br J Pharmacol. 2006;148(5):610–8.
Phase IIa clinical trial of chiglitazar completed. Chipscreen biosciences. 2007. http://www.chipscreen.com/News/201309140810501322442224.html. Accessed 15 Aug 2016.
Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–42.
Hediger MA, Rhoads DB. Molecular physiology of sodium–glucose cotransporters. Physiol Rev. 1994;74:993–1026.
Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–94.
Clinical trials: sotagliflozin (LX4211). Lexicon pharmaceuticals. 2016. http://www.lexpharma.com/pipeline/lx4211.html. Accessed 23 Aug 2016.
Vuylsteke V, Chastain LM, Maggu GA, Brown C. Imeglimin: a potential new multi-target drug for type 2 diabetes. Drugs R D. 2015;15(3):227–32.
Bays H, Mandarino L, DeFronzo RA. Role of the adipocytes, FFA, and ectopic fat in the pathogenesis of type 2 diabetes mellitus: PPAR agonists provide a rational therapeutic approach. J Clin Endocrinol Metab. 2004;89:463–78.
Goldfine AB, Fonseca V, Jablonski KA, Chen YD, Tipton L, Staten MA, et al. Targeting inflammation using salsalate in type 2 diabetes study team: salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med. 2013;159:1–12.
Esser N, Paquot N, Scheen AJ. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Exp Opin Investig Drugs. 2015;24:283–307.
Matschinsky FM, Magnuson MA, Zelent D, Jetton TL, Doliba N, Han Y, et al. The network of glucokinase- expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes. 2006;55:1–12.
Niswender KD, Shiota M, Postic C, Cherrington AD, Magnuson MA. Effects of increased glucokinase gene copy number on glucose homeostasis and hepatic glucose metabolism. J Biol Chem. 1997;272:22570–5.
Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2560–6.
Wilding JP, Leonsson-Zachrisson M, Wessman C, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes Metab. 2013;15:750–9.
Tan T, Bloom S. Gut hormones as therapeutic agents in treatment of diabetes and obesity. Curr Opin Pharmacol. 2013;13:996–1001.
Mancini AD, Poitout V. GPR40 agonists for the treatment of type 2 diabetes: life after ‘TAKing’ a hit. Diabetes Obes Metab. 2015;17:622–9.
Bharate SB, Nemmani KV, Vishwakarma RA. Progress in the discovery and development of small-molecule modulators of G-protein-coupled receptor 40 (GPR40/FFA1/FFAR1): an emerging target for type 2 diabetes. Expert Opin Ther Pat. 2009;19:237–64.
Oh DY, Olefsky JM. G protein-coupled receptors as targets for anti-diabetic therapeutics. Nat Rev Drug Discov. 2016;15:161–72.
Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic sujbjects. J Clin Investig. 1967;46:1954–62.
Dockray GJ. Cholecystokinin. Curr Opin Endocrinol Diabetes Obes. 2012;19:8–12.
Pocai A. Unraveling oxyntomodulin, GLP1’s enigmatic brother. J Endocrinol. 2012;215:335–46.
Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284(5418):1362–5.
Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–40.
Plaisancié P, Dumoulin V, Chayvialle JA, Cuber JC. Luminal glucagon-like peptide-1(7–36) amide-releasing factors in the isolated vascularly perfused rat colon. J Endocrinol. 1995;145:521–6.
Dumoulin V, Moro F, Barcelo A, Dakka T, Cuber JC. Peptide YY, glucagon-like peptide-1, and neurotensin responses to luminal factors in the isolated vascularly perfused rat ileum. Endocrinology. 1998;139:3780–6.
Adrian TE, Ballantyne GH, Longo WE, Bilchik AJ, Graham S, Basson MD, et al. Deoxycholate is an important releaser of peptide YY and enteroglucagon from the human colon. Gut. 1993;34(9):1219–24.
Nunez DJ, Yao X, Lin J, Walker A, Zuo P, Webster L, et al. Glucose and lipid effects of the ileal apical sodium-dependent bile acid transporter inhibitor GSK2330672: double-blind randomized trials with type 2 diabetes subjects taking metformin. Diabetes Obes Metab. 2016;18(7):654–62.
Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev. 2007;28(3):253–83.
Bagger JI, Knop FK, Holst JJ, Vilsboll T. Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes Metab. 2011;13:965–71.
Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D. Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist. Science. 1982;215(4536):1115–6.
Rohatgi N, Aly H, Marshall CA, McDonald WG, Kletzien RF, Colca JR, et al. Novel insulin sensitizer modulates nutrient sensing pathways and maintains β-cell phenotype in human islets. PLoS ONE. 2013. doi:10.1371/journal.pone.0062012.
Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100.
Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337(6090):93–6.
Van Poelje PD, Potter SC, Chandramouli VC, Landau BR, Dang Q, Erion MD. Inhibition of fructose 1, 6-biphosphatase reduces excessive endogenous glucose production and attenuates hyperglycemia in Zucker diabetic fatty rats. Diabetes. 2006;55:1747–54.
Denison H, Nilsson C, Kujacic M, Lofgren L, Karlsson C, Knutsson M, et al. Proof of mechanism for the DGAT1 inhibitor AZD7687: results from a first-time-in-human single-dose study. Diabetes Obes Metab. 2013;15:136–43.
Hollis G, Huber R. 11β-Hydroxysteroid dehydrogenase type 1 inhibition in type 2 diabetes mellitus. Diabetes Obes Metab. 2011;13(1):1–6.
Anderson A, Walker BR. 11β-HSD1 inhibitors for the treatment of type 2 diabetes and cardiovascular disease. Drugs. 2013;73(13):1385–93.
Singh PK, Hota D, Dutta P, Sachdeva N, Chakrabarti A, Srinivasan A, et al. Pantoprazole improves glycemic control in type 2 diabetes: a randomized, double-blind, placebo-controlled trial. J Clin Endocrinol Metab. 2012;97(11):2105–8.
Vetere A, Choudhary A, Burns SM, Wagner BK. Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov. 2014;13:278–89.
Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362:1275–81.
Zhou K, Donnelly L, Burch L, Tavendale R, Doney AS, Leese G, et al. Loss-of-function CYP2C9 variants improve therapeutic response to sulfonylureas in type 2 diabetes: a Go-DARTS study. Clin Pharmacol Ther. 2010;87:52–6.
Ragia G, Petridis I, Tavridou A, Christakidis D, Manolopoulos VG. Presence of CYP2C9*3 allele increases risk for hypoglycemia in type 2 diabetic patients treated with sulfonylureas. Pharmacogenomics. 2009;10:1781–7.
Dujic T, Zhou K, Donnelly LA, Tavendale R, Palmer CN, Pearson ER. Association of organic cation transporter 1 with intolerance to metformin in type 2 diabetes: a GoDARTS study. Diabetes. 2015;64(5):1786–93.
Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43(2):117–20.
Acknowledgements
All authors have contributed significantly to a collection of information and designing and formatting of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Chikara, G., Sharma, P.K., Dwivedi, P. et al. A Narrative Review of Potential Future Antidiabetic Drugs: Should We Expect More?. Ind J Clin Biochem 33, 121–131 (2018). https://doi.org/10.1007/s12291-017-0668-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12291-017-0668-z