
Abstract
The current epidemic affecting Indians is coronary artery disease (CAD), and is currently one of the most common causes of mortality and morbidity in developed and developing countries. The higher rate of CAD in Indians, as compared to people of other ethnic origin, may indicate a possible genetic susceptibility. Hence, Lp(a), an independent genetic risk marker for atherosclerosis and cardiovascular disease assumes great importance. Lp(a), an atherogenic lipoprotein, contains a cholesterol rich LDL particle, one molecule of apolipoprotein B-100 and a unique protein, apolipoprotein (a) which distinguishes it from LDL. Apo(a) is highly polymorphic and an inverse relationship between Lp(a) concentration and apo(a) isoform size has been observed. This is genetically controlled suggesting a functional diversity among the apo(a) isoforms. The LPA gene codes for apo(a) whose genetic heterogeneity is due to variations in its number of kringles. The exact pathogenic mechanism of Lp(a) is still not completely elucidated, but the structural homology of Lp(a) with LDL and plasmin is possibly responsible for its acting as a link between atherosclerosis and thrombosis. Upper limits of normal Lp(a) levels have not been defined for the Indian population. A cut off limit of 20 mg/dL has been suggested while for the Caucasian population it is 30 mg/dL. Though a variety of assays are available for its measurement, standardization of the analytical method is highly complicated as a majority of the methods are affected by the heterogeneity in apo(a) size. No therapeutic drug selectively targets Lp(a) but recently, new modifiers of apo(a) synthesis are being considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The current epidemic affecting Indians is coronary artery disease (CAD), and is currently one of the most common causes of mortality and morbidity in developed and developing countries. [1]. By 2015, this is expected to rise by 103 % in men and by 90 % in women [2]. It is predicted that in the next 15 years India would have more than half of the world’s cases of coronary vascular disease, which includes both CAD and stroke [3]. More importantly, CAD is affecting Indians 5–10 years earlier than other communities [4] and its incidence is 12–16 % as compared to 5 % in the West [5, 6]. This premature form of CAD is more diffuse and follows a malignant course. There is a clustering of risk factors among ethnic Indians, so the effect is a magnified risk, which is multiplicative rather than additive. The higher rate of CAD in Indians, as compared to people of other ethnic origin may indicate a possible genetic susceptibility. Since the affected population is in the working age-group, this has far reaching economic consequences for a developing economy like India.
Lp(a) is an LDL like particle discovered by Berg in 1963 and is an important independent risk factor for CAD. Its indeterminate pathophysiological mechanism, as well as the considerable variations found in its blood levels in different racial/ethnic groups, have consistently intrigued researchers. Though still not completely elucidated, Lp(a) may constitute a link between the processes of atherosclerosis and thrombosis. Also, new facts about the interactions between Lp(a) and other established risk factors like LDL, HDL and homocysteine have emerged in recent studies [7, 8]. The study of genetics has played an important role in establishing Lp(a) as an important risk biomarker for CAD and currently research is focusing on the LPA gene in this regard [9, 10].
Lp(a) is a plasma lipoprotein synthesized by the liver, containing a cholesterol rich LDL particle, one molecule of apolipoprotein B-100 and another protein, apolipoprotein (a) attached via a disulfide bond (Fig. 1). The blood levels, to a large extent, are genetically determined via variation in the apolipoprotein (a) gene [11]. Depending on the size of apo (a), 34 different isoforms of Lp(a) have been observed and it has been estimated that more than 90 % of the interindividual variation in plasma Lp(a) has been attributed to the apo(a) gene while 70 % is related to the size of apo(a) isoforms [12]. Life long levels of Lp(a) are attained by the age of two and it is highly heritable. The high consistency of Lp(a) levels over many years in an individual, stresses the fact that Lp(a) does not have any substantial correlation with either lifestyle modifications or any of the established risk factors. This again emphasizes the importance of the LPA locus on Lp(a) levels and, unlike other markers, confers the advantage of assessing CAD risk by a single measurement [13] (Box 1).

Structure of Lp(a)
Reports of other factors affecting Lp(a) levels in the blood are few. However, an effect of interleukin-6 (IL-6) on Lp(a) expression has been described [14] and there is some suggestion that estrogen may effect its blood level [15]. Also, Lp(a) has neither consistently nor convincingly been associated or correlated to other risk factors in plasma, such as cholesterol, LDL, HDL, triglycerides or C-reactive protein (CRP) [13] or even smoking. These findings further support the theory that the locus harboring the LPA gene plays an important role in determining the concentration of Lp(a) in blood.
Coronary Risk Association
Various studies on the relationship between Lp(a) and CAD have shown discordant results but majority have found the association to be positive between the two. Some prospective studies have suggested that this association may be more marked in subjects with a raised LDL level [16, 17]. A study by Albers et al. in [18] showed that elevated Lp(a) levels were a significant risk factor in patients with myocardial infarction, especially in the younger age group [18]. In another study involving Swedish male patients who were monitored for 6 years, the Lp(a) levels in the fatal or non fatal CAD group were higher than the control group [19]. In yet another study by Ornek et al., it was concluded that serum Lp(a) was higher in unstable angina pectoris and MI patients in comparison to stable angina patients and the levels also correlated positively with the severity of CAD which was independent of other lipid parameters and CAD risk factors [20].
With CAD rising in the Indian population due to affluence, urbanization and lifestyle changes, a number of studies conducted on the Indian population have observed higher Lp(a) levels in CAD patients than in controls [21–26]. Vasisht et al. [23], examined the association of apo(a) polymorphism with Lp(a) levels in CAD in North Indians, and observed a strong association between raised Lp(a) and CAD. In another study, Lp(a) levels of CAD patients undergoing coronary angiography were compared with controls. The CAD patients showed higher levels of Lp(a), however those who were alcohol drinkers had lower levels [27]. In a few studies involving racial and ethnic groups, higher Lp(a) levels have been reported in Asian Indians as compared to Chinese [28, 29]. Other studies have also reported higher Lp(a) levels in Asian Indians as compared to non Hispanic whites but the results are conflicting [30, 31]. D Rajashekher et al., observed a positive correlation of Lp(a) with CHD at >25 mg/dL in a South Indian population, which is a lower cut off than for other populations, with Lp(a) also showing a positive association with angiographic severity [32].
A study by Pedreno et al. has shown no gender difference in Lp(a) levels in both patients and controls [33]. However Rajashekhar et al. [32] showed higher Lp(a) levels in females as compared to males which was in agreement with another report by Wong et al. [34]. Higher levels are also found in post- menopausal women. Though influence of sex on Lp(a) is not yet established in literature but the lowering effect of testosterone could be the cause of lower Lp(a) levels in males [35].
In the Indian population, upper limits of normal Lp (a) levels have not been defined. A cut off limit of 30 mg/dL has been followed in the Caucasian population whereas for the Indian population, Enas et al. suggested a level of 20 mg/dL as upper limits of normal [36]. In an another study, among Asian Indians worldwide, mean Lp(a) has been defined as 18–20 mg/dL [37]. Higher Lp(a) levels have also been observed in individuals with strong family history of CAD [38] and in patients with familial and non familial hypercholesterolemia [39]. Moreover, in Asian Indians diagnosed with NIDDM, Lp(a) has been demonstrated to be an independent risk factor for Coronary Heart Disease (CHD) [40].
Pathophysiology of Lp(a)
Lp(a) is an LDL like particle composed of a lipid core and two disulphide linked subunits : apolipoprotein B 100 and apolipoprotein, apo (a). The lipid core and apo B 100 are similar to that in LDL. The essential difference between the structure of Lp(a) and LDL is the presence of the glycoprotein apo(a), which is structurally similar to plasminogen, a precursor of plasmin, the fibrinolytic enzyme. This allows Lp(a) to bind to fibrin and to the membrane proteins of endothelial cells and monocytes. Since Lp(a) resembles both LDL and plasminogen, it could possibly act as a link between atherosclerosis and thrombosis [41]. The accumulation of Lp(a) on the surface of fibrin and cell membranes as well as the inhibition of plasmin generation favors the deposition of fibrin and cholesterol at sites of vascular injury. After transfer into the arterial intima from the plasma, Lp(a) gets retained much more than LDL as it binds to the extracellular matrix through apo (a) as well as the apolipoprotein B component [42], thus contributing to the atherosclerotic plaque.
Recent studies have shown that Lp(a) inhibits the generation of transforming growth factor-β (TGF-β) [43] leading to migration and proliferation of smooth muscle cells into the intima, thus further enhancing the formation of atheroma plaque. However this hypothesis needs further confirmation. Hence, though the exact mechanism of the pathophysiology of Lp(a) in atherosclerosis is still not clearly elucidated, the current evidence suggests that apo(a) inhibits plasmin formation and the LDL component favors cholesterol accumulation.
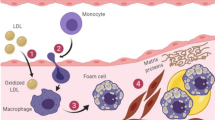
Other athero-thrombogenic mechanisms of Lp(a) include: (a) stimulation of Plasminogen Activator Inhibitor-1(PAI-1) and inhibition of tissue Plasminogen Activator (t-PA), thus enhancing Lp(a) dependent hypofibrinolyis (b) binding of Lp(a) to extra cellular matrix components like proteoglycans or glycosaminoglycans leading to accumulation of Lp(a) in the vascular wall and (c) Lp(a) and LDL are sensitive to exudation and phagocytosis of these oxidized particles results in formation of foam cells [42] (Fig. 2; Box 2).

Schematic diagram representing different modes of action of Lp(a) in the vessel wall
Metabolism
The primary site of synthesis of Lp(a) is the hepatocyte which also synthesizes apo B100. Apo(a), on secretion, is then assembled with plasma LDL to form Lp(a) by the formation of a disulfide bond between apoB100 in LDL and kringle IV in apo(a) [44]. The apolipoprotein (a) genotype alone accounts for 90 % of the concentration in blood since it determines both the rate of synthesis as well as the size of the apo(a) moiety [45, 46]. Catabolism of Lp(a) occurs primarily by hepatic and renal pathways though they do not govern the plasma levels. Metabolism of Lp(a) is independent of other lipoproteins and variations in plasma levels are due to synthesis rather than due to degradation [44] (Box 3).
Genetic Polymorphism and Functional Heterogeneity of Lp(a)
Genetics has played an important role in further enhancing the study of Lp(a) and the role of LPA gene in developing the risk of CHD [9, 10]. It is now a well known fact that apo(a) gene regulates the circulating concentration of Lp(a). According to amino acid sequencing & cDNA cloning, apo(a) contains a variable number of kringle domains (i.e. 80–90 amino acid sequences arranged in triple loop tertiary structure stabilized by disulphide bridges) that are similar to kringle IV of plasmingen [47, 48]. Kringle IV copies of plasminogen in apo(a) are similar but not identical and have been classified into 10 different types [49]. Variability in this type of kringle in apo(a) has lead to the identification of 34 isoforms of Lp(a) [50, 51]. The LPA gene which resides on chromosome 6q26–27, codes for apo(a) whose genetic heterogeneity is due to variations in its number of kringles. Solving the puzzle of kringle variability led to further research on the effect of LPA gene on cardiovascular disease. Kraft et al. showed the effect of kringle number variation on CHD risk [52], while Holmer et al. [53], tested the association of polymorphisms in LPA gene with the risk of atherosclerosis and its consequences. From these studies we can conclude that there is suggestive evidence that LPA gene confers risk of CAD though this risk relationship needs further detailing.
The smaller isoforms (i.e. relatively low number of kringle IV repeats of the Lp(a) polypeptide chains) are associated with a higher Lp(a) concentration [54, 55]. So the general consensus is that smaller this hyper variable region, and therefore smaller the size of the apo(a) isoform, higher the plasma concentration of Lp(a). This has been corroborated by studies [56–58] which have observed that low molecular mass isoforms were found more frequently in patients with high Lp(a) concentrations & a history of myocardial infarction. Short apo(a) alleles increased Lp(a) levels, thus favoring atherosclerosis. Also, since the inverse relationship between apo(a) size & Lp(a) is not linear, this suggests a functional diversity among the apo(a) isoforms [41]. Another study by Hervio et al. [59] suggested that the variable number of kringles in apo(a) influences its ability to bind to fibrin. Thus, low molecular mass isoforms of apo(a) with the highest affinity for fibrin are the best competitors for plasminogen & so are related to higher cardiovascular risk [58–60].
Since it is almost certain that the main structural difference between the isoforms is due to the variable number of kringles in apo(a), further investigations are required to confirm whether these variations are due to the presence or absence of any specific kringle sequences. Also, due to the inverse relationship between isoform size and affinity for fibrin, isoforms of low molecular size show highest affinity for fibrin. So according to this hypothesis, some Lp(a) phenotypes may not actually be related to CHD. This then introduces a novel concept of “functional heterogeneity” of Lp(a), adding a new dimension that not all individuals with elevated Lp(a) would be at risk for cardiovascular disease [41].
Measurement of Lp(a)
A variety of assays are available for the measurement of Lp(a). They include immunoturbidity, nephelometry, enzyme linked immunosorbent assays, and fluorescence assays. The standardization of the analytical method for Lp(a) is highly complicated as the majority of the methods determining its serum concentration are affected by the heterogeneity in apo(a) size. Also, in most of the clinical studies, Lp(a) has been estimated by methods affected by apo(a) size heterogeneity. So for these assays to be considered valid, we have to assume that the distribution of apo(a) isoforms is similar between cases and controls to minimize the potential of method dependent over or under estimation of Lp(a). However, to achieve assay standardization, each assay must be evaluated for its sensitivity to apo(a) size polymorphism (SM Marcovine in his NHLBI conference presentation) [45] (Box 4).
Lp(a) is usually reported as mg/dL representing the entire Lp(a) mass (protein, lipid and carbohydrate). However, these assays must be validated with reference standards because despite using polyclonal antibodies independent of isoform size, apo(a) size dependent bias has been observed with assay calibrators. Since poor agreement between various Lp(a) methods is a major problem, an IFCC Working Group for Standardization of Lp(a) assays had initiated a project to select a suitable secondary reference material for Lp(a). As the major problem facing Lp(a) estimation is lack of accuracy, the IFCC Working Group suggested that the testing should be done only by validated methods that are not affected by apo(a) size heterogeneity. These assays measure Lp(a) as moles of apo(a) protein using specific monoclonal antibodies and are independent of isoform size [61]. It was then decided that the IFCC reference material with an assigned value of 107 should be used as point of reference for assay calibrator with the units as nmol/L. The nmol/L value can be converted to mg/dL by dividing by a factor 2.8.
Hence, manufacturers of Lp(a) testing should focus on producing assays which are minimally impacted by apo(a) size variability, assay imprecision as well as the variable effects of the antibodies used. Also data should be compared from different populations to exclude ethnic and race related differences. Finally, stringent protocols for blood collection and sample storage should be developed to minimize their impact on the measurement.
Treatment
Currently there are no guidelines recommending treatment of elevated Lp(a). It has been suggested that lowering of Lp(a) may prove to be beneficial in at least some subgroups of patients but these subgroups need to be properly defined with respect to Lp(a) levels, apo(a) size, presence or absence of other risk factors etc. [17, 62]. Many studies have shown that using Niacin [63–68] alone or in combination with statins reduces Lp(a) levels by 30–40 % [69]. However, since Niacin also helps in lowering LDL-C, total cholesterol, triglycerides and raising HDL-C, its beneficial effects are not entirely due to the lowering of Lp(a). Nevertheless, these studies have shown clearly that using Niacin for lowering Lp(a) is safe and beneficial. Also, since Lp(a) is relatively refractory to both lifestyle and drug interventions, the use of statins and fibrates have shown limited and variable results [70, 71].
The most effective therapy to reduce plasma Lp(a) concentration significantly is therapeutic apheresis. In young and middle aged patients with progressive CAD and markedly elevated Lp(a), LDL apheresis can be considered which removes Lp(a) quite efficaciously. However, apheresis is laborious and expensive, and impractical for most patients and treatment centers [72].
It is therefore clear that a more detailed study of Lp(a) metabolism is required to develop the therapeutic interventions for lowering Lp(a) levels. Since the synthesis of Lp(a) is largely responsible for its blood levels, it would be appropriate to target the synthesis of apolipoprotein (a) and/or the formation of Lp(a) [46, 73]. Antisense oligonucleotide and thyroid hormone analogue therapies directed at apolipoprotein(a) synthesis may hold therapeutic promise in the future [74, 75].
Conclusion
Lp(a) levels provide additional information on cardiovascular risk and is today an important component of the lipid profile test. An independent and continuous association between Lp(a) and risk of CAD has been firmly established by a number of studies. Lp(a) is highly stable in individuals across many years and is only weakly correlated with known risk factors. Though the exact mechanism of action is still not clear, it is now suggested that due to the presence of both LDL and plasminogen like moiety, Lp(a) may form a link between atherosclerosis and thrombosis. Since Lp(a) levels differ significantly between various racial/ethnic groups, multi-ethnic studies are required in different populations to provide insight into the variability found in its blood levels. As of now, there are no therapeutic drugs that selectively reduce Lp(a) levels, but recently, new modifiers of apo(a) synthesis are being identified. A routine screening of Lp(a) levels in blood along with apo(a) phenotyping may help in identification of individuals with a high genetic predisposition to CAD. Finally, further research is required on the inverse relationship between apo(a) size polymorphism and Lp(a) levels to help understand better, its role as a unique independent risk biomarker for CAD.
References
Sekhri T, Kanwar RS, Wilfred R, Chigh P, Chillar M, Aggarwal R, et al. Prevalence of risk factors for coronary artery disease in an urban Indian population. BMJ Open. 2014;4:e005346. doi:10.1136/bmjopen-2014-005346.
Bulatao RA, Stephens PW. Global estimates and projections of mortality by cause, 1970–2015. Preworking paper 1007. Washington: Population and Nutrition Department, World Bank 1992.
Enas EA, Yusuf S, Mehta J. Meeting of international working group on coronary artery disease in South Asians. Indian Heart J. 1996;48:727–32.
Rissam HS, Kishore S, Trehan N. Coronary artery disease in young Indians—the missing link. J Indian Acad Clin Med. 2001;2(3):128–31.
Negus BH, Williard JE, Glamam DB, Landau C, Snyder RW 2nd, Hillis LD, et al. Coronary anatomy and prognosis of young asymptomatic survivors of myocardial infarction. Am J Med. 1994;96:354–8.
Mammi MVI, Pavithran P, Rahman PA, Pisharody R, Sugathan K. Acute MI in North Kerala. A 20 years hospital based study. Indian Heart J. 1991;43:93–6.
Maher VM, Brown BG, Marcovina SM, Hilger LA, Zhao XQ, Albers JJ. Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein (a). JAMA. 1995;274:1771–4.
Foody JM, Milberg JA, Robinson K, Pearce GL, Jacobson DW, Sprecher DL. Homocysteine and lipoprotein (a) interact to increase CAD risk in young men and women. Arterioscler Thromb Vasc Biol. 2000;20:493–9.
Hobbs HH, White AL. Lipoprotein (a): intrigues and insights. Curr Opin Lipidol. 1999;10:225–36.
Utterman G. Genetic architecture and evolution of the lipoprotein (a) trait. Curr Opin Lipidol. 1999;10:133–41.
Uttermann G. Lipoprotein (a). In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw Hill; 2001. p. 2753–87.
Enas EA, Garg A, Davidson MA, Nair VM, Huet BA, Yusuf S. Coronary heart disease and its risk factors in first-generation immigrant Asian Indians to the United States of America. Indian Heart J. 1996;48:343–53.
Bennet A, Di Angelantonio E, Erqou S, Eiriksdottir G, Sigurdsson G, Woodward M, et al. Lipoprotein (a) levels and risk of future coronary heart disease: large scale prospective data. Arch Intern Med. 2008;168:598–608.
McCormick SP. Lipoprotein (a): biology and clinical importance. Clin Biochem Rev. 2004;25:69–80.
Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein (a), hormone replacement therapy, and the risk of future cardiovascular events. J Am Coll Cardiol. 2008;52:124–31.
Cantin B, Gagnon F, Moorjani S, Despres JP, Lamarche B, Lupien PJ, et al. Is lipoprotein (a) an independent risk factor for ischemic heart disease in men? The Quebec Cardiovascular Study. J Am Coll Cardiol. 1998;31:519–25.
Suk DJ, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular event among initially healthy women. JAMA. 2006;296:1363–70.
Albers JJ, Marcoina SM, Lodge MS. The unique lipoprotein (a): properties and immunochemical measurement. Clin Chem. 1990;36:2019–26.
Rosengren A, Wilhelmsen L, Eriksson E, Risberg B, Wedel H. Lipoprotein (a) and coronary heart disease: a prospective case-control study in a general population sample of middle aged men. BMJ. 1990;301:1248–51.
Ornek E, Murat S, Duran M, Turfan M, Kurtul A, Demircelik MB, et al. The relationship between lipoprotein (a) and coronary artery disease, as well as its variable nature following myocardial infarction. Clin Investig Med. 2011;34(1):E14–20.
Gambhir JK, Kaur H, Gambhir DS, Prabhu KM. Lipoprotein(a) as an independent risk factor for coronary artery disease in patients below 40 years of age. Indian Heart J. 2000;4:411–5.
Luthra K, Vasisht S, Chhabra S, Raju KR, Agarwal DP, Manchanda SC, et al. LP (a) phenotypes and levels in angiographically proven coronary heart disease patients and controls. Indian J Clin Biochem. 1998;13(1):12–9.
Vasisht S, Gulati R, Srivastava LM, Narang R, Chopra V, Srivastava N, et al. Apolipoprotein(a) polymorphism and its association with plasma lipoprotein(a) levels: a North Indian Study. Indian Heart J. 2005;2:407–10.
Mohan V, Deepa R, Rema M. Lipoprotein (a) and coronary artery disease in Indians. JAPI. 1998;46:979–80.
Geethanjali FS, Luthra K, Lingerhel A, Kanagasabapathy AS, Jacob J, Srivastava LM, et al. Analysis of the apo (a) size polymorphism in Asian Indian populations: association with Lp(a) concentration and coronary heart disease. Atherosclerosis. 2003;169:121–30.
Gupta R, Kastia S, Rastogi S, Kaul V, Nagar R, Enas EA. Lipoprotein(a) in coronary heart disease: a case-control study. Indian Heart J. 2002;52:407–10.
Vasisht S, Agarwal DP, Wasir HS, Srivastava LM. Lipoprotein (a) levels in alcohol drinking and alcohol non-drinking coronary artery disease patients. Indian J Clin Biochem. 1996;11:176–9.
Anand SS, Yusuf S, Vuksan V, Devanesen S, Teo KK, Montague PA, et al. Differences in risk factors, atherosclerosis, and cardiovascular disease between ethnic groups in Canada: the Study of Health Assessment and Risk in ethnic groups (SHARE). Lancet. 2002;356(9226):279–84.
Low PS, Heng CK, Saha N, Tay JSH. Racial variation of cord plasma lipoprotein (a) level in relation to coronary risk level: a study in three ethnic groups in Singapore. Pediatr Res. 1996;40(5):718–22.
Anand SS, Enas EA, Pogue J, Haffner S, Pearson T, Yusuf S. Elevated lipoprotein(a) level in South Asians in North America. Metabolism. 1998;47(2):182–4.
Tavridou A, Unwin N, Bhopal R, Laker MF. Predictors of lipoprotein (a) level in a European and South Asian population in the Newcastle Heart Project. Eur J Clin Investig. 2003;33(8):686–92.
Rajasekhar D, Saibaba KSS, Srinivasa Rao PVLN, Latheef SAA, Subramanyam G. Lipoprotein(a): better assessor of coronary heart disease risk in South Indian population. Indian J Clin Biochem. 2004;19(2):53–9.
Pedreno J, Fernandez R, Ballester A, Jornet A, Uson M, Camela J, et al. Lack of association with serum Lp(a) with Type 2 Diabetes Mellitus in patients with angiographically defined coronary artery disease. Int J Cardiol. 2000;79:159–67.
Wong MS, Chew WL, Aw TC. Serum lipoprotein (a) profiles in Singaporean population. Pathology. 1999;31:225–9.
Henrikson P, Angelin P, Berglund L. Hormonal regulation of serum Lp(a) levels: opposite effects after oestrogen treatment and orchidectomy in males with prostatic carcinoma. J Clin Investig. 1992;89:1166–71.
Enas EA, Dhawan J, Petkar S. Coronary Artery Disease in Asian Indians: Lessons learned so far and the role of Lp(a). Ind Heart J. 1997;49:25–34.
Gupta R, Gupta VP. Meta-analysis of coronary heart disease prevalence in India. Indian Heart J. 1996;48:241–5.
Durrington PN, Ishola M, Hunt L, Arrol S, Bhatnagar D. Apolipoprotein(a), A1 and B and parental history in men with early onset ischemic heart disease. Lancet. 1998;1:1070–3.
Schaefer EJ, Lamon-Fava S, Jenner JL, Mcnamara JR, Ordovas JM, Davis CE, et al. Lipoprotein (a) levels and risk of coronary heart disease in men. The Lipid Research Clinics Coronary Primary Prevention Trial. JAMA. 1994;271:999–1003.
Mohan V, Deepa R, Haranath SP, Premalatha G, Rema M, Sastry NG, et al. Lipoprotein (a) is an independent risk factor for coronary artery disease in NIDDM patients in South India. Diabetes Care. 1998;21:1819–23.
Angles-Cano E. Structural basis for the pathophysiology of lipoprotein (a) in the athero-thrombotic process. Braz J Med Biol Res. 1997;30:1271–80.
Neilsen LB. Atherogenicity of lipoprotein (a) and oxidized low density lipoprotein: insight from in vivo studies of arterial wall influx, degradation and efflux. Atherosclerosis. 1999;143:229–43.
Kojima S, Harpel PC, Rifkin D. Lipoprotein (a) inhibits the generation of transforming growth factor β: an endogenous inhibitor of smooth muscle cell migration. J Cell Biol. 1991;113:1439–45.
Luthra K, Misra A, Srivastava LM. Lipoprotein (a): biology and role in atherosclerotic vascular diseases. Curr Sci. 1999;76:1553–60.
Marcovina SM, Koschinsky ML, Albers JJ, Scarlatos S. Report of the national heart, lung and blood institute workshop on lipoprotein (a) and cardiovascular disease: recent advances and future directions. Clin Chem. 2003;49:1785–96.
Koschinsky M, Marcovina SM. Lipoprotein (a). In: Ballantyne C, editor. Clinical lipidology: a companion to Braunwald’s heart disease. Philadelphia: Saunders Elselvier; 2009. p. 130–43.
McLean J, Tomlinson J, Kuang W, Eaton D, Chen E, Fless G, et al. cDna sequence of human apololipoprotein(a) is homologous to plasminogen. Nature. 1978;330:132–7.
Eaton D, Fless GM, Kohr W, McLean J, Xu Q, Miller C, et al. Partial amino acid sequence of apolipoprotein(a) shows that it is homologous to plasminogen. Proc Natl Acad Sci USA. 1987;84:3224–8.
Guevara J, Knapp RD, Honda S, Northup SR, Morrisett JD. A structural assessment of the apo (a) protein of human lipoprotein (a). Proteins. 1992;12:188–99.
Kambon MI, Ferrell RE, Kottke BA. Expressed hypervariable polymorphism of apolipoprotein(a). Am J Hum Genet. 1991;49:1063–74.
Lackner G, Boerwinkle E, Leffert CC, Rahmig T, Hobbs HH. Molecular basis of apolipoprotein(a) isoform size heterogeneity as revealed by pulsed-field gel electrophoresis. J Clin Investig. 1991;87:2153–61.
Kraft HG, Lingenhel A, Kochi S, Hoppichler F, Kronenberg F, Abe A, et al. Apolipoprotein(a) kringle IV repeat number predicts risk for coronary heart disease. Arterioscler Thromb Vacs Biol. 1996;16(6):713–9.
Holmer SR, Hengstenberg C, Kraft HG, Mayer B, Pöll M, Kürzinger S, et al. Association of polymorphisms of the apolipoprotein(a) gene with lipoprotein(a) levels and myocardial infraction. Circulation. 2003;107(5):696–701.
UtermannG G, Kraft HG, Menzel H, Hopferweiser T, Seitz C. Genetics of the quantitative Lp(a) lipoprotein trait I. Relation of Lp(a) glycoprotein phenotypes to Lp(a) lipoprotein concentrations in plasma. Hum Genet. 1998;7:41–6.
Uutermann G, Duba C, Menzel H. Genetics of the quantitative Lp(a) lipoprotein trait II.Inheritance of Lp(a) glycoprotein phenotypes. Hum Genet. 1998;78:47–50.
Molgaard J, Klasusen IC, Lassvik C, Faergeman O, Gerdes LU, Olsson AG. Significant association between low-molecular-weight apolipoprotein (a) isoform and intermittent claudication. Arterioscler Thromb. 1992;12:895–901.
Seed M, Hoppichler F, Reaveley D, McCarthy S, Thompson GR, Boerwinkle E, et al. Relation of serum lipoprotein (a) and apolipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia. N Engl J Med. 1990;322:1494–9.
Sandholzer C, Saha N, Kark JD, Rees A, Jaross W, Dieplinger H, et al. Apo(a) isoform predict risk for coronary heart disease. A study in six populations. Arterioscl Thromb. 1992;12:1214–26.
Hervio L, Chapman MJ, Thilet J, Loyau S, Angles-Cano E. Does apolipoprotein (a) heterogeneity influence lipoprotein (a) effect on fibrinolysis? Blood. 1993;82:392–7.
Rouy D, Grailhe P, Chapman J, Angles-Cano E. Lipoprotein (a) impairs generation of plasmin by fibrin bound tissue-type plasminogen activator. Arterioscl and Thromb. 1991;11:629–38.
Tsimikas S, Hall JL. Lipoprotein (a) as a potential causal genetic risk factor of cardiovascular disease. J Am Coll Cardiol. 2012;60:716–21.
Cantin B, Despres JP, Lamarche B, Moorjani S, Lupien PJ, Bogaty P, et al. Association of fibrinogen and lipoprotein (a) as a coronary heart disease risk factor in men (The Quebec Cardiovascular Study). Am J Cardiol. 2002;89(6):662–6.
Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010;210:353–61.
Carlson LA, Rosenhamer G. Reduction in mortality in the Stockholm Ischemic Heart Disease Secondary Prevention Study by combined treatment with clofibrate and nicotinic acid. Acta Med Scand. 1988;233:405–18.
Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, et al. Fifteen year mortality in Coronary Drug Project patients: long term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–55.
Cashin-Hemphill L, Mack WJ, Pogoda JM, Sanmarco ME, Azen SP, Blankenhorn DH. Beneficial effects of colestipol-niacin on coronary atherosclerosis. A 4-year follow-up. JAMA. 1990;264:3013–7.
Brown G, Albers JJ, Fisher LD, Schaefer SM, Lin JT, Kaplan C, et al. Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N Engl J Med. 1990;323:1289–98.
Taylor AJ, Villines TC, Stanek EJ, Devine PJ, Griffen L, Miller M, et al. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med. 2009;361:2113–22.
Chapman MJ, Redfern JS, McGovern ME, Giral P. Niacin and fibrates in atherogenic dyslipidemia: pharmacotherapy to reduce cardiovascular risk. Pharmacol Ther. 2010;126:314–45.
Tziomalos K, Athyros VG, Wierzbicki AS, Mikhailidis DP. Lipoprotein(a): where are we now? Curr Opin Cardiol. 2009;24:351–7.
Gonbert S, Malinsky S, Sposito AC, Laouenan H, Doucet C, Chapman MJ, et al. Atorvastatin lowers lipoprotein (a) but not apolipoprotein (a) fragment levels in hypercholesterolemic subjects at high cardiovascular risk. Atherosclerosis. 2002;164:305–11.
Thompson GR. Recommendations for the use of LDL apheresis. Atherosclerosis. 2008;198:247–55.
Koschinsky ML, Marcovina SM. Structure—function relationship in apolipoprotein(a): insight into lipoprotein (a) assembly and pathogenicity. Curr Opin Lipidol. 2004;15:167–74.
Merki E, Graham MJ, Mullick AE, Miller ER, Crooke RM, Pitas RE, et al. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein (a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118:743–53.
Ladenson PW, Kristensen JD, Ridgway EC, Oisson AG, Carlsson B, Klein I, et al. Use of the thyroid hormone analogue eprotirome in statin treated dyslipidemia. N Engl J Med. 2010;362:906–16.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Manocha, A., Srivastava, L.M. Lipoprotein (a): a Unique Independent Risk Factor for Coronary Artery Disease. Ind J Clin Biochem 31, 13–20 (2016). https://doi.org/10.1007/s12291-015-0483-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12291-015-0483-3