
Abstract
Cardiovascular diseases (CVD) have overtaken infectious diseases and are currently the world’s top killer. A quite strong linkage between this type of ailments and elevated plasma levels of triglycerides (TG) has been always noticed. Notably, this risk factor is mired in deep confusion, since its role in atherosclerosis is uncertain. One of the explanations that aim to decipher this persistent enigma was provided by apolipoprotein C-III (apoC-III), a small protein historically recognized as an important regulator of TG metabolism. Preeminently, hundreds of studies have been carried out in order to explore the APOC3 genetic background, as well as to establish a correlation between its variants and dyslipidemia-related disorders, pointing to an earnest predictive power for future outcomes. Among several polymorphisms reported within the APOC3, the SstI site in its 3′-untranslated region (3′-UTR) was the most consistently and robustly associated with an increased CVD risk. As more genetic data supporting its importance in cardiovascular events aggregate, it was declared, correspondingly, that apoC-III exerts various atherogenic effects, either by intervening in the function and catabolism of many lipoproteins, or by inducing endothelial inflammation and smooth muscle cells (SMC) proliferation. This review was designed to shed the light on the structural and functional aspects of the APOC3 gene, the existing association between its SstI polymorphism and CVD, and the specific molecular mechanisms that underlie apoC-III pathological implications. In addition, the translation of all these gathered knowledges into preventive and therapeutic benefits will be detailed too.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
At the beginning of the 21th century, CVD have become a significant issue on the global health agenda. As early as 2001, these diseases have been documented as the largest single cause of mortalities on the planet [1]. Today, despite the huge growth of biological knowledge and advances in medication, CVD remain at the world’s top rank of all human death reasons [2]. The data leave no room for doubt: Annually, around 17.5 million people die from CVD [3], and this number is predicted to increase dramatically in the next years. By 2030, more than 23.3 million individuals will succumb to this growing burden each year [4], and hence, this pandemic will continue to dominate the mortality trends in the future.
“Cardiovascular diseases” is an umbrella term that encompasses a broad spectrum of cardiologic diagnoses, affecting heart and circulatory system [5]. Disorders under this term primarily comprise coronary heart diseases, cerebrovascular accidents and peripheral vascular diseases [6]. The major underlying cause of CVD appears to be atherosclerosis [7], also known as the hardening of arteries [8]. It is defined as an immunoinflammatory fibroproliferative disease, in which fatty deposits called atheromatous plaque develops, over many decades, inside the inner layers of the arterial wall, and over time, it narrows the artery depriving the vascularized tissue of oxygen [8]. This process has been recognized as a human pathology for more than 3500 years: It occurred in ancient Egyptian mummies, showing the same salient features observed in the modern time [9].
Historically, a broad variety of factors have been linked to the etiology of atherosclerosis and CVD. Its incidence is ascribable to polygenic inheritance coupled with environmental exposures that add to the complexity of the disease [10]. Interestingly, most of the classic risk factors which can intensify and provoke a cardiovascular event are the result of people’s lifestyles and behaviors; therefore, they are modifiable and avoidable [11]. These variables include unhealthy blood lipid profile, hypertension, smoking, type 2 diabetes, obesity and physical inactivity [8]. Other determinants cannot be changed such as increased age, gender, race and genetic background [8]. In order to improve the prognosis of CVD and provide better preventive strategies, it is critical to develop a greater understanding of these factors, especially, the genetic predisposition to these diseases.
Pathogenesis of atherosclerosis
Although our current understanding of atheroma formation is fragmentary, a straightforward illustration of atherogenesis steps was outlined. It was established that the root cause of this pathogenesis is the deposition of lipids, or more precisely the apolipoprotein B-containing lipoproteins, inside the arterial wall, sparking an inflammatory response and promoting atherosclerosis [12]. Circulating low-density lipoprotein (LDL) particles, that are the most abundant atherogenic lipoproteins in the bloodstream, can penetrate via transcytosis across the endothelial cells (EC) monolayer into the sub-endothelial space [13], where they are retained and oxidized [12]. The EC become activated by atherogenic stimuli, and as a result, their expression of adhesion molecules is upregulated, and monocytes are recruited [12]. Within the intima, the monocytes differentiate into macrophages and internalize the oxidized LDL, resulting in the formation of lipid-loaded macrophages called foam cells [12]. With a containing supply of modified lipoproteins, these foam cells eat until they die [12]. This progressively destroys the normal architecture of the intimal layer, and the accumulated cells, lipids and debris, coalesce into enlarging pools, forming a soft and destabilizing core in the center of the plaque [12]. Concomitantly, SMC migrate from the adventitia to the intima, and secrete collagen and elastin adding a thick fibrous cap over the core, directly at the blood interface [8] (Fig. 1). At this point, the rupture of the raised patch favors the platelets to stick to the surface of injury, leading to the formation of a blood clot (thrombosis), and subsequently, the vessel becomes completely obstructed [8].

Schematic of atherogenesis. The atherosclerotic plaque consists of a fibrous cap rich in collagen, surrounding a lipid-rich core comprising lipids, foam cells and debris. Atheroma plaque formation involves a series of steps initiated by the (1) lipid infiltration into the sub-endothelial space, followed by (2) EC dysfunction and activation with increased expression of adhesion molecules on their surface, promoting the attachment and penetration of inflammatory cells into the intima. (3) Monocytes differentiate into macrophages, engulf modified lipids forming foam cells. (4) Meanwhile, SMC proliferate, migrate and synthesize matrix proteins to form a fibrous cap that encloses the growing core
Hypertriglyceridemia and CVD: a renewed interest
Despite several decades of research, the implication of hypertriglyceridemia (HTG) in CVD, either as a direct promoter or merely as a biomarker, remains a topic of debate [14]. For a long time, elevated TG levels have been considered as an important risk factor for CVD [15], but randomized controlled trials aiming to reduce it failed to show any significant medical benefit [16, 17], rendering it a non-persuasive biomarker in cardiovascular pathologies [18]. This long-standing controversy stems from the heterogeneity of TG-rich lipoproteins (TRL) [19], as well as the complexity of TG metabolism described below.
Due to its hydrophobic nature, TG must combine with proteins forming lipoprotein particles that allow its transport through the circulation [20]. In accord with this concept, plasma TG are mostly found in TRL [21], including very low-density lipoproteins (VLDL) which transport it from the liver, and chylomicrons which transport dietary fat from the intestine [22]. Along the luminal surface of capillaries, they are hydrolyzed by the action of lipoprotein lipase (LPL), generating free fatty acids that can be taken up by muscles and adipose tissues [23]. During this phenomenon, an enormous fraction of apolipoproteins and phospholipids moves from TRL into the high-density lipoproteins (HDL), converting VLDL and chylomicrons into remnant particles [23]. Ultimately, these later should be rapidly removed from the plasma, through a specific receptor mediated mechanism in the liver [24]. Under pathological conditions, HTG results from the accumulation of TRL in the bloodstream [25], either by excessive production, or by decreased catabolism [26] due to an LPL dysfunctionality, or to a grave alteration of another LPL-independent pathways, such as the hepatic removal of remnants. In this regard, it is difficult to separate TG from the complicated network of confounding variables in the experimental systems based on cellular mechanisms, clinical trials or epidemiological data [21]. However, renewed interest in this factor is brought by recent observations suggesting that the loss of function mutations in the gene encoding apolipoprotein C-III (APOC3 gene; apoC-III protein), an important regulator of TG homeostasis, is beneficially associated with decreased CVD risk [27, 28]. Based on these findings, the APOC3 gene and its genetic variants have been a great focus of research in the last years.
Spotlight on the APOC3 gene
The APOC3 gene was discovered and characterized nearly 50 years ago [29], but firstly, it did not attract sufficient attention [24]. This gene resides within the APOA5/APOA4/APOC3/APOA1 multigene cluster on the long arm of the human chromosome 11q23 [30]. It comprises 4 exons and 3 introns [31], and encoding a 99 amino acid glycoprotein called apoC-III [32]. This apolipoprotein is mostly expressed in hepatocytes and enterocytes, where it undergoes an intracellular cleavage, yielding the mature 79 amino acid protein [32]. Furthermore, it undergoes a post-translational modification leading to the formation of three distinct isoforms containing zero (apoC-III0), one (apoC-III1) or two (apoC-III2) sialic acid residues, and importantly, all these isoforms exhibit the same plasma half-life and catabolic mechanisms, suggesting similar physiological implications [33]. However, important modifications in their normal distributions were detected in different diseased cases, but it remains unknown if these variations have a pathological consequence [23]. At the transcriptional level, the APOC3 gene expression is tightly regulated by several proposed pathways. A series of in vivo and in vitro studies have demonstrated that its expression is downregulated by insulin [34], peroxisome proliferator-activated receptor α [35], Rev-erb [36] and farnesoid X receptor [37]. Conversely, the positive responsiveness of the APOC3 promoter to glucose was reported. This factor stimulates the gene expression by the activation of the carbohydrate-responsive element binding protein, as well as the hepatocyte nuclear factor-4α [38]. Hence, the opposite interplay between insulin and glucose on modulating APOC3 transcriptional activity may induce an enhanced apoC-III secretion under an insulin-resistant condition associated with hyperglycemia (as in type 2 diabetes) [38]. Also, the total apoC-III levels can be significantly modulated in hyperlipidemic individuals by the dietary intake of low saturated fat and high amounts of monosaturated and omega-3 polyunsaturated fatty acids [39].
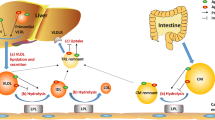
Once secreted, apoC-III is released into the plasma as a major component of circulating TRL and HDL [40]. Today, we know that it is a multifaced apolipoprotein, with a major physiological concern [24]. The prevailing is that apoC-III impairs the LPL-mediated lipolysis of chylomicrons and VLDL, by displacing the lipase from TRL, as well as by antagonizing apolipoprotein C-II, the main protein responsible for the enzyme activation [41]. In addition, apoC-III affects TG catabolism by other LPL-independent pathways. It delays the clearance of remnants, by interfering with their specific binding to hepatic receptors [33]. Besides, it has been proposed to directly influence plasma VLDL levels, by stimulating their intrahepatic secretion [42]. A further illustration of this point comes from the increased VLDL synthesis in genetically modified mice overexpressing apoC-III [43]. However, the reduction of apoC-III levels using antisense oligonucleotides (ASO) in mice did not affect VLDL production [31]. Thus, the biological implication of this protein in VLDL assembly and secretion is still obscure [24], but overall, the combination of all its actions renders it an essential actor in the regulation of TRL metabolism [23] (Fig. 2). Therefore, the overexpression of apoC-III causes an unwanted accumulation of TG inside the vessels, resulting in HTG [44].

The functions of apolipoprotein C-III in TRL metabolism and their impacts on atherogenesis. The red arrows depict the inhibition of LPL activity and TRL remnants hepatic uptake. The suggested intrahepatic effect of apoC-III as an enhancer of VLDL assembly and secretion is indicated with a red plus sign. The dashed blue arrow depicts the resulting accumulation of atherogenic TRL in the atherosclerotic artery
APOC3 genetic variants and CVD: is there a link?
Although interest in apoC-III has continued, it appears theoretically plausible that the deficiency of this protein could be a cardioprotective phenotype, given the relationship that exists between unfavorable TG profiles and cardiovascular events. Correspondingly, two parallel large-scale epidemiological studies have revealed that a striking decrease in ischemic CVD and coronary heart disease risk can be influenced by a series of typical APOC3 mutations (Table 1) [27, 28]. In the first study, four mutations were identified in an Exome Sequencing Project, including a missense mutation (A43T), and three loss-of-function mutations: The null mutation R19X, as well as the two splicing site mutations IVS2 + 1G→A and IVS3 + 1G→T [27]. Even if only one in every 150 persons carried some of these alleles, their existence was coupled with a 39% decrease in TG levels, corresponding to a 40% reduction of CVD risk, as compared with non-carriers [27]. Likewise, the second cohort study (n = 75.725) has shown similar results [28]. Loss-of-function mutations in the APOC3 gene have a conferred 41% decrease in the risk of ischemic vascular disease, and a 36% reduction in ischemic heart disease [28].
On the other side, the important biological functions of apoC-III, together with the aggregated data indicating that a null allele in its gene can lead to a healthy life, deem it necessary to explore the full genetic background of DNA polymorphisms at the APOC3 locus, and how they may correlate to the susceptibility of dyslipidemia and other related disorders, such as CVD. Remarkably, several biallelic polymorphisms in this gene have been investigated as possible markers of HTG [45]. In particular, special attention has been paid to the SstI site, which represents the most extensively studied polymorphism in this gene [45]. Here, we focused specifically on this variant, since it was consistently linked to many pathological traits in the literature.
APOC3 SstI polymorphism (rs5128)
The SstI polymorphism, also recognized as SacI, 3238C > G or 3175C > G [40], was the first polymorphic site discovered within the APOC3 gene [46]. Arising from a Cytosine (C) to Guanine (G) transversion in the 3′ UTR [40], this variant distinguishes between 2 alleles: S1 (frequent allele) and S2 (rare allele) [47]. It involves the formation of a restriction site for the endonuclease SstI [48] that facilitated its exploration. Consequently, the genotype can be easily detected using the polymerase chain reaction-restriction fragment length polymorphism technique.
The correlation of APOC3 SstI polymorphism with several phenotypic variables has been the subject of numerous association studies that have covered many ethnic groups. Even though the published data provided some conflicting results, a recent meta-analysis confirmed that carriers of the rare allele exhibit high plasma concentrations of apoC-III and TG [40]. This polymorphism was also associated with CVD risk, through a large-scale genome wide association study [49]. The discrepancies reported in few populations are not explained, but they strongly postulate a gene-environment interaction [50].
Possible effects of APOC3 SstI polymorphism on the transcriptional activity
Because of its localization in the 3′UTR, the SstI polymorphism was unexpected to have a functional significance [47]. However, elevated APOC3 mRNA levels were detected in 3/5 of hepatic biopsies collected from heterozygotes carriers of the S2 allele [51]. Although the specific molecular mechanism explaining this overexpression is poorly understood [40], different potential processes have been hypothesized.
Firstly, it was believed that the SstI site is located in a sequence that is involved in the regulation of gene expression or mRNA stability [52], on account of the well-known mechanisms of the 3′UTR region [53]. Additionally, it is conceivable that the S2 allele could be acting as a marker for other functional variants, within the APOC3 or in neighboring genes [52]. Accordingly, an indirect functionality was proposed through the strong linkage disequilibrium of this allele with two APOC3 promoter polymorphisms, the − 455 T > C (rs2854117) and the − 482C > T (rs2854116) [54]. These two substitutions are located within an insulin response element [52], and their presence leads to an attenuated suppression by insulin, and subsequently, an increased production of apoC-III [55]. Nevertheless, the analyses of haplotypes combination within the APOA4/APOC3/APOA1 cluster provided evidence for a dominant effect of the SstI variant on lipid abnormalities [56].
Recently, the literature has suggested a possible post-transcriptional regulation of APOC3 mRNA, involving the rare allele of the SstI polymorphic site [54]. In silico assays predicted potential binding losses for 5 miRNAs (has-miR-138, has-miR-185, has-miR-564, has-miR-1207-5p, has-miR-1270), all expressed in human hepatic and intestinal tissues [54]. MiRNAs are a group of small non-coding RNA, that post-transcriptionally down-regulate gene expression, usually by binding target 3′-UTR mRNA sequences [57]. Unfortunately, the in vitro trials aiming to validate this hypothesis have failed [54]. However, it is not ruled out that the S2 allele could suppress the binding sites corresponding to other miRNAs, that are not detected by the bioinformatic analysis, nor expressed in the cell models used [54].
More recently, a new hypothesis was provided explaining the functionality of the SstI polymorphism. It was declared that the SstI site lies within 40 nucleotides from the 3′ poly-A tail [58], a long chain of adenine bases involved in mRNA stability, nucleocytoplasmic transport and translation [59]. Generally, the shortening of the poly-A tail is one of the key steps initiating the decay of the messenger body, when it is no longer being used for translation [60]. In this respect, the inhibition of the binding of the specific proteins that remove the poly-A tail, in the presence of the S2 allele, was hypothesized to explain a possible prolonged half-life of the messenger [58]. Nevertheless, this suggestion requires empirical confirmation. Altogether, the unclear transcriptional regulatory effect of the SstI variant remains to be fully elucidated.
ApoC-III and cardiovascular pathophysiology: more than just a hypothesis
A growing evidence from epidemiological data supported a strong correlation of apoC-III with CVD risk [61]. In addition, the current body of literature contains numerous methodologically sound investigations, that can provide together a consistent proof for the deep implication of this protein in cardiovascular complications. It is now well known that the variation in plasma apoC-III levels is directly related to the atherogenicity of lipoprotein particles in which it resides, and to an alarming increase in cardiovascular risk [62]. This connection was confirmed in an important meta-analysis conducted by Wyler et al., based on the overall published data of 12 eligible studies [63]. Furthermore, apoC-III concentration was also reported, in a 15-year follow-up study, as a potent predictor of cardiovascular mortalities [64]. Accordingly, several clinical reports validated the usefulness of this protein in the prediction of future CVD outcomes, especially coronary artery events [33]. This relationship appears to be extremely complex, taking into consideration the pleiotropic impacts of this apolipoprotein on the cardiovascular system. As discussed previously, apoC-III keeps bad company: It inhibits the clearance of TRL remnants, augmenting their plasma levels and prolonging their residence time in the blood. Consequently, the causal association of apoC-III with CVD has been firstly attributed to its effect on TRL catabolism [41]. While the previous theory is undoubtedly true in part, the apoC-III finds itself, recently, at the scene of the crime. An increasing body of researches reflected that it may be the primary mediator of disease onset and progression, exerting several atherogenic properties [62]. Accordingly, it has been identified as an independent risk factor for CVD [46].
ApoC-III links dyslipidemia with CVD
Circulating lipoproteins normally flux into and out of the arterial wall by transcytosis, a transport system by which these particles can be transported, via specialized clathrin-coated vesicles, across the endothelium [65]. Furthermore, the transport vesicles are approximately 100 nm in diameter, making the transcytotic system restricted to lipoproteins smaller than 70 nm, thereby excluding large VLDL and chylomicrons [66]. However, their remnants are small enough to exceed the size limitation and penetrate to the arterial intima [67] (Fig. 2). Despite being atherogenic, it is not their TG content that participates in the evolution of atheroma [68]. Besides TG, TRL remnants also contain enriched amounts of cholesterol, 40 times more than an LDL particle [69]. Thus, they directly mediate cholesterol deposition within the intima, and facilitate the activation of various inflammatory pathways [67]. Additionally, these particles do not need to be modified/oxidized to become atherogenic [70]. Hence, they can be taken up, in an unregulated fashion, by the arterial macrophages [67], resulting in accelerated foam cells generation. According to these mechanisms, TRL remnants may have a stronger atherogenic effect than LDL [67], and may further increase the risk. As apoC-III is elevated in CVD, it was reasonable to postulate that it might enhance atherosclerosis by enriching the plasma components with these plaque-promoting particles. Nonetheless, it was suggested that apoC-III can facilitate the sub-endothelial accumulation of apolipoprotein B-containing lipoproteins, by influencing their affinity for the artery wall proteoglycans [71]. This mechanism was firstly elusive since apoC-III itself is not able to bind the proteoglycans, but a possible explanation could be that it seems to provoke a modification in the lipid composition of lipoproteins, allowing the apolipoprotein B to adopt a specific conformation, which is more favorable for the binding to the artery wall proteoglycans [71].
ApoC-III is directly involved in atherosclerosis
The plethora of observational studies has provided a mean for testing whether apoC-III is the real disease culprit or an innocent bystander in the manifestation of CVD. Biologists have struggled with this issue, however, due to the rapid evolution of molecular genetics technologies, the apoC-III pathogenicity is uncovered [61]. Today, it is becoming clear that apoC-III has an atherogenic potency, beyond its ability to raise TG levels [62]. It was detected in the site of atherosclerotic lesions of major arteries, such as the coronary artery, the carotid artery and the aorta [72], adding more weight to its direct implication in CVD. Also, it was found that patients with elevated levels of apoC-III-containing lipoproteins have an increased risk of both fatal and nonfatal myocardial infarction [73]. In the light of all these facts, arduous efforts have been made to explore the exact molecular mechanisms, by which this protein can induce numerous atherogenic conditions.
CVD are not only a complication of a disordered lipid profile, but they are also considered as inflammatory diseases. As aforementioned, the development of atheroma requires, in addition to the lipoproteins accumulation, a concomitant inflammation in the arterial wall. Interestingly, several inflammatory effects have been ascribed to apoC-III [74], opening entirely new prospects on its participation in disease progression. This protein was found to be involved in crucial events of the early pathological stage, including endothelial dysfunction, recruitment of blood monocytes to the site of injury, and their subsequent extravasation through the endothelium into the sub-endothelial area [75]. Indeed, a series of interactions between immune and endothelial cells can be promoted by the action of apoC-III, alone or as a lipoprotein component [74]. It is deemed to activate the protein kinase C-β (PKC-β), and posteriorly the nuclear factor kappa B (NFκB) in the vascular EC, pushing them to express two types of adhesion molecules, the vascular cell adhesion molecule 1 and the intracellular adhesion molecule 1, on their surface [76]. Remarkably, a specific antibody targeting apoC-III was able to abolish this effect [46]. Moreover, because apoC-III-containing particles hold other apolipoproteins, many assays were designed in order to validate that the increased adhesiveness of EC is attributed to the apoC-III itself [77].
In addition to exerting effects on the EC layer, apoC-III can enhance the attachment of circulating monocytes to the vascular wall, by acting directly on these immune cells. It was shown to increase their surface expression of the β1-integrin [18], a member of the receptors family that mediates cell–cell and cell-extracellular matrix contact [78]. Eminently, the contribution of Kawakami et al. has delved into the intricacies of the induced signal transduction axis [79]. They have demonstrated that this process is fulfilled through the activation of the pertussis toxin-sensitive G protein pathway in monocytic cells [79]. Particularly, it was revealed that apoC-III stimulates the phosphatidylcholine-specific phospholipase C (PC) activity, and this later, in turn, catalyzes the PC to generate phosphatidylcholine and diacylglycerol (DAG) [79]. Then, DAG triggers the activation of PKC-α, resulting in the translocation of NFκB into the nuclei, where it increases the transcription of the gene encoding the β1-integrin [79]. Also, Ras homolog family member A may be partially involved [76]. Eventually, the net result of this long cascade is an enhanced crawling of monocytes on the endothelial surface [18]. Although these findings seem to have illustrated a complete and compelling figure concerning the inflammatory role of apoC-III, they leave some unanswered questions. For example, the specific signaling pathway by which apoC-III can activate the PKC-β in EC, as well as the possible involvement of other mediators in PKC-α activation in monocytes, present an unexplained conundrum that provides fertile ground for future research.
Simultaneously, laboratory assays have continued to unravel additive mechanistic information about the pathophysiology of apoC-III. From an attractive standpoint, the study of its impact on SMC has yielded a new elucidation of its atherogenic capacity. Various transgenic and knocked-out mouse models have been created, and they were used to demonstrate that the apoC-III can trigger the proliferation of SMC, participating in the worsening of atherosclerosis [80]. ApoC-III takes part in this process via the stimulation of the Akt signaling pathway in these cells [80]. Nonetheless, it was examined whether the apoC-III-poor particles could elicit similar effects, since this apolipoprotein resides typically on the surface of TRL. It was indicated that apoC-III-enriched TRL induce aortic SMC proliferation and arterial stenosis, while apoC-III-poor TRL don’t [80]. In summary, all these advances have robustly deepened our understanding of apoC-III atherogenicity.
ApoC-III alters HDL functionality
To more complicate this issue, the direct contribution of the apoC-III in atherosclerosis was not limited to the previously mentioned scenarios. Looking beyond inflammation and fibroproliferation, apoC-III emerged as an actor in the profound modification of the functionality of HDL. This lipoprotein is an intriguing particle that has attracted the attention of the scientific community for many years, mostly by its atheroprotective potential [81]. Besides its considerable anti-inflammatory and anti-apoptotic properties, it can promote classically the reverse efflux of cholesterol from the lipid-loaded macrophages in the plaque, to the liver [82]. Unluckily, all of these characteristics can be altered in many pathological conditions, including CVD [18]. As a matter of fact, a strong correlation of this dysfunctionality with the presence of apoC-III has been well established [18]. Based on proteomics analyses with different methodologies, HDL particles isolated from CVD patients were remarkably enriched by this atherogenic apolipoprotein [83, 84]. Further, apoC-III in these lipoproteins was found to be responsible for the activation of the p38-mitogen activated protein kinase signaling pathway in EC, followed by an excessive expression of the pro-apoptotic protein truncated-BID, therefore, converting HDL from cytoprotective into pro-apoptotic molecules [83]. The significant value of this result comes from the usual EC turn over and death in the atherosclerotic injury-prone regions within the vasculature [84]. Overall, these newly discovered functions of the apoC-III have confirmed that this protein is a causal factor for CVD. Importantly, they have also highlighted a new target for therapeutic interventions, allowing the development of apoC-III based treatments with respect to HTG and atherosclerosis.
Lesson learned from genomics: why the APOC3 fingerprint should be pursued?
While the picture is less hazy in the era of “Mendelian” diseases that typically arise from a single gene rare mutation, the heritable component of complex conditions is attributable to a constellation of several variants, spreading across different genomic loci [85]. Altogether, they can disrupt a key physiological mechanism, in a clear illustration of the so-called “polygenic inheritance” pattern [86]. In other words, a genetic variation, credibly correlated to a common disease, is not necessarily the straight cause of illness [10], but it can account for only a minuscule fraction of the trait in question [85]. However, it can be used as a marker that will help in the prediction of a future pathological event [10].
The past few years witnessed substantial progress in the genomic field, especially in the horizon of high throughput analyses of single nucleotide polymorphisms, leading to the identification of a large number of CVD-associated genetic loci [87], and providing an unprecedented opportunity to assess these widespread disorders, that clinicians encounter daily. Owing to the small effect of each polymorphism, the theory of the “genetic risk score” has been conceived [88]. Fundamentally, aggregating information about diverse sets of risk-conferring alleles into a single algorithm, has become a useful tool for examining the cumulative predictive ability of these variants on subclinical CVD-related phenotypes [88].
Generally speaking, the determination of the personal genetic makeup in candidate genes may improve the overall cardiovascular outcomes, by identifying relative at-risk patients before that the symptoms manifest, motivating them to change their health behaviors [5]. Moreover, this genotype-guided approach can lower the burden of CVD by bringing the attention of clinicians to medical follow-up for these subjects, and helping them to make better treatment decisions [5], under a personalized medicine mode. In this context, the harnessing of the available APOC3 genetic information, considering its serious contribution in the onset, as well as the aggravation of atherosclerosis, may yield further advances in risk stratification. The “cardiogenomic profiling”, or the “Heart health tests”, are methods used to evaluate single nucleotide polymorphisms in genes that modulate lipid balance, blood pressure, inflammation and oxidative stress, and they continue to be offered in the health care market [5]. They are already advertised on the Internet, and it can be ordered online, without the direct involvement of a physician [5]. Interestingly, the APOC3 is included in some of these panels, besides other relevant genes [5, 89]. Additionally, Palomaki et al. have summarized the CVD-associated variants observed in the white population [5]. In this study, the SstI polymorphism was mentioned with moderate credibility [5], making it an important high-risk predictor, that may provide discriminatory power in the assessment of cardiovascular disorders.
ApoC-III as a therapeutic target
While multiple lipid-lowering drugs, such as atorvastatin [90], fibrates [35] and others, decrease moderately the plasma apoC-III levels by 10–30% [23], there is still no effective compound that can suppress robustly this protein overexpression [91]. The apoC-III emerging roles have been the catalyst for a remarkable endeavor in the therapeutic field. Currently, a promising strategy in the treatment of hyperlipidemia, based on the ASO technology, is under development [91].
In general, the ASO application is a newest type of pharmacological approaches pertaining to nucleic acid-based gene silencing [92]. Briefly, a heavily modified single-stranded deoxyribonucleotide can serve as a selective sequence that pairs to specific regions of a given mRNA, and regulates its translation into a functional protein [92]. The ASO can bind complementarily to the targeted mRNA by Watson–Crick hybridization and, in consequence, block the expression of this genetic material via many distinct mechanisms [92]. When the synthetic antisense sequence contains stretches of > 5 DNA nucleotides, it can elicit the activation of the ribonuclease H1 [92]. This enzyme is an endogenous endonuclease that degrades selectively the cognate RNA strand of an RNA–DNA heteroduplex [93] (Fig. 3), thus decreasing the amount of translated protein.

Mechanism of action of ASO. The ASO have been proposed to traverse cell and nuclear membrane, bind to cognate mRNA sequences, and activate RNase-H1. Upon its activation, this enzyme cleaves the mRNA to prevent translation of the targeted protein
Fortunately, a chemically modified-second generation anti-apoC-III ASO (5′-AGCTTCTTGTCCAGCTTTAT-3′), formally called ISIS 304801 or ISIS-ApoCIIIRx and now referred to as “Volanesorsen” [91], can be powerful in the management of HTG and future CVD. It has been shown to selectively reduce the biosynthesis of apoC-III and TG in all preclinical tested rodents and nonhuman primates [31]. Following these results, the drug was examined in healthy volunteers in a double-blind, placebo-controlled, dose-ranging trial, during the initial phase of clinical applications, revealing a dose-dependent reduction of apoC-III and TG concentrations [31].
Based on the previous proof-of-concept published data, the phase II trials were rapidly initiated. Gaudet et al. decided to test the effect of this agent in three hypertriglyceridemic patients with a familial chylomicronemia syndrome (FCS), and after treatment, these participants have benefited from more than 70% reduction in plasma apoC-III levels [94]. FCS is a rare autosomal recessive disorder, usually caused by loss of function mutations in the genes encoding LPL or LPL-modulator proteins, and it is characterized by high plasma concentrations of chylomicrons, severe hypertriglyceridemia, and consequently, an elevated risk for recurrent episodes of abdominal pain and pancreatitis [95]. The efficacy of the anti-apoC-III ASO in this case supported the physiological concept that apoC-III is involved in the regulation of TRL metabolism by LPL-independent pathways, besides its effect on LPL activity. In another larger trial, “Volanesorsen” was administered by patients with HTG, either as a monotherapy, or in combination with a stable fibrate-based therapy [96]. This cohort confirmed a dose-dependent reduction in both plasma apoC-III and TG levels. In addition, the compound was examined in a randomized double-blind study including 15 participants with type 2 diabetes and HTG [97], revealing that dyslipidemia, as well as the whole-body insulin sensitivity, were improved after this treatment [97]. Furthermore, it has been advantageous that “Volanesorsen” was generally safe and well-tolerated in phases I and II of investigations, and only mild local cutaneous reactions at the injection site were reported at a low rate [91]. In the third phase of clinical trials, two important randomized, double-blind, placebo-controlled studies were designed. The first (APPROACH trial) included 66 FCS patients [98], and the second (COMPASS study) recruited 113 subjects with severe HTG [91]. About 70–80% reduction of apoC-III and TG concentrations were reported in the released data of these studies.
Even though there are not many treatment options for FCS patients, “Volanesorsen” was not approved by the FDA in August 2018 as a medication for this illness. This negative decision was only based on safety considerations, since the APPROACH study indicated that an unexplained grade 4 thrombocytopenia (low blood platelet counts) occurred in three enrolled patients, and it ended when they stopped the drug administration. Otherwise, the COMPASS study did not report a similar observation, therefore, the current thinking is that the underlying mechanism could be linked to the methodology of drug delivery, and not to the apoC-III itself. Recently, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted an important positive opinion concerning “Volanesorsen” in March 2019, with respect to the treatment of genetically confirmed adult FCS patients, who are at a high pancreatitis risk and their response to diet interventions and lipid-lowering drugs was inadequate [24]. Till now, ongoing randomized trials are underway, and the prospect looks very hopeful.
Conclusion
Modern research has greatly expanded our knowledge on the sizable effect of apoC-III in TG homeostasis, inflammation and atherosclerosis. In addition, the emerging genetic results and association studies have shed light on the usefulness of specific APOC3 variants in pursuing future CVD. Particularly, the S2 allele of SstI polymorphism was identified as a good predictor for the HTG susceptibility, implying notoriously the susceptibility to cardiovascular disorders. Moreover, the novel understanding of the apoC-III atherogenicity renders it an attractive target for treatment strategies, and by finishing the inaugurated phase III of clinical trials, an anti-apoC-III ASO could offer an intriguing therapy for cardiovascular protection.
Abbreviations
- ApoC-III:
-
Apolipoprotein C-III
- ASO:
-
Antisense oligonucleotides
- bp:
-
Base pairs
- C:
-
Cytosine
- CVD:
-
Cardiovascular diseases
- DAG:
-
Diacylglycerol
- DNA:
-
Deoxyribonucleic acid
- EC:
-
Endothelial cells
- FCS:
-
Familial chylomicronemia syndrome
- G:
-
Guanine
- has-miR:
-
Homo Sapiens-micro ribonucleic acid
- HDL:
-
High-density lipoproteins
- HTG:
-
Hypertriglyceridemia
- LDL:
-
Low-density lipoproteins
- LPL:
-
Lipoprotein lipase
- miRNA:
-
Micro ribonucleic acid
- mRNA:
-
Messenger ribonucleic acid
- NFκB:
-
Nuclear factor kappa B
- PC:
-
Phospholipase C
- PKC:
-
Protein kinase C
- RNA:
-
Ribonucleic acid
- SMC:
-
Smooth muscle cells
- TG:
-
Triglycerides
- TRL:
-
Triglyceride rich lipoproteins
- UTR:
-
Untranslated region
- VLDL:
-
Very low-density lipoproteins
References
Castellano JM, Narula J, Castillo J et al (2014) Promoting cardiovascular health worldwide: strategies, challenges, and opportunities. Rev Esp Cardiol (Engl Ed) 67(9):724–730
Mc Namara K, Alzubaidi H, Jackson JK (2019) Cardiovascular disease as a leading cause of death: how are pharmacists getting involved? Integr Pharm Res Pract 4(8):1–11
Hajar R (2016) Framingham contribution to cardiovascular disease. Heart Views 17(2):78–81
Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3(11):e442
Palomaki GE, Melillo S, Neveux L et al (2010) Use of genomic profiling to assess risk for cardiovascular disease and identify individualized prevention strategies–a targeted evidence-based review. Genet Med 12(12):772–784
Glaudemans AW, Slart RH, Bozzao A et al (2010) Molecular imaging in atherosclerosis. Eur J Nucl Med Mol Imaging 37(12):2381–2397
Frostegård J (2013) Immunity, atherosclerosis and cardiovascular disease. BMC Med 11:117
Barr AJ (2018) The biochemical basis of disease. Essays Biochem 62(5):619–642
Allam AH, Thompson RC, Wann LS et al (2011) Atherosclerosis in ancient Egyptian mummies: the Horus study. JACC Cardiovasc Imaging 4(4):315–327
Gibbons GH, Liew CC, Goodarzi MO et al (2004) Genetic markers: progress and potential for cardiovascular disease. Circulation 109(25 Suppl 1):IV47–IV58
Buttar HS, Li T, Ravi N (2005) Prevention of cardiovascular diseases: role of exercise, dietary interventions, obesity and smoking cessation. Exp Clin Cardiol 10(4):229–249
Linton MF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, Doran AC, Vickers KC (2019) The role of lipids and lipoproteins in atherosclerosis. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland HJ, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Purnell J, Singer F, Stratakis CA, Trence DL, Wilson DP (eds) Endotext [Internet]. MDText.com, Inc., South Dartmouth (MA), p 2000
Huang L, Chambliss KL, Gao X et al (2019) SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 569(7757):565–569
Singh AK, Singh R (2016) Triglyceride and cardiovascular risk: a critical appraisal. Indian J Endocrinol Metab 20(4):418–428
Hokanson JE, Austin MA (1996) Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk 3(2):213–219
Bosch J, Gerstein HC, Dagenais GR et al (2012) n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med 367:309–318
Ginsberg HN, Elam MB, Lovato LC et al (2010) Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 362:1563–1574
Luo M, Peng D (2016) The emerging role of apolipoprotein C-III: beyond effects on triglyceride metabolism. Lipids Health Dis 15(1):184
Hodis HN (1999) Triglyceride-rich lipoprotein remnant particles and risk of atherosclerosis. Circulation 99(22):2852–2854
Do R, Willer CJ, Schmidt EM et al (2013) Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet 45(11):1345–1352
Lewis GF, Xiao C, Hegele RA (2015) Hypertriglyceridemia in the genomic era: a new paradigm. Endocr Rev 36(1):131–147
Navar AM (2019) The evolving story of triglycerides and coronary heart disease risk. JAMA 321(4):347–349
Norata GD, Tsimikas S, Pirillo A, Catapano AL (2015) Apolipoprotein C-III: from pathophysiology to pharmacology. Trends Pharmacol Sci 36(10):675–687
Taskinen MR, Packard CJ, Borén J (2019) Emerging evidence that ApoC-III inhibitors provide novel options to reduce the residual CVD. Curr Atheroscler Rep 21(8):27
Gordts PL, Nock R, Son NH et al (2016) ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest 126(8):2855–2866
Miller M, Stone NJ, Ballantyne C et al (2011) Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 123(20):2292–2333
Crosby J, Peloso GM et al (2014) Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 371(1):22–31
Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A (2014) Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 371(1):32–41
Brown WV, Levy RI, Fredrickson DS (1969) Studies of the proteins in human plasma very low density lipoproteins. J Biol Chem 244(20):5687–5694
Huff MW, Hegele RA (2013) Apolipoprotein C-III: going back to the future for a lipid drug target. Circ Res 112(11):1405–1408
Graham MJ, Lee RG, Bell TA III et al (2013) Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res 112(11):1479–1490
Bolanos-Garcia VM, Miguel RN (2003) On the structure and function of apolipoproteins: more than a family of lipid-binding proteins. Prog Biophys Mol Biol 83(1):47–68
Ooi EM, Barrett PH, Chan DC, Watts GF (2008) Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci (Lond) 114(10):611–624
Altomonte J, Cong L, Harbaran S et al (2004) Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J Clin Invest 114(10):1493–1503
Hertz R, Bishara-Shieban J, Bar-Tana J (1995) Mode of action of peroxisome proliferators as hypolipidemic drugs. Suppression of apolipoprotein C-III. J Biol Chem 270(22):13470–13475
Coste H, Rodríguez JC (2002) Orphan nuclear hormone receptor Rev-erbalpha regulates the human apolipoprotein CIII promoter. J Biol Chem 277(30):27120–27129
Claudel T, Inoue Y, Barbier O et al (2003) Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology 125(2):544–555
Caron S, Verrijken A, Mertens I et al (2011) Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler Thromb Vasc Biol 31(3):513–519
Pieke B, von Eckardstein A, Gülbahçe E et al (2000) Treatment of hypertriglyceridemia by two diets rich either in unsaturated fatty acids or in carbohydrates: effects on lipoprotein subclasses, lipolytic enzymes, lipid transfer proteins, insulin and leptin. Int J Obes Relat Metab Disord 24(10):1286–1296
Song Y, Zhu L, Richa M, Li P, Yang Y, Li S (2015) Associations of the APOC3 rs5128 polymorphism with plasma APOC3 and lipid levels: a meta-analysis. Lipids Health Dis 14:32
Zheng C (2014) Updates on apolipoprotein CIII: fulfilling promise as a therapeutic target for hypertriglyceridemia and cardiovascular disease. Curr Opin Lipidol 25(1):35–39
Sundaram M, Zhong S, Bou Khalil M et al (2010) Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J Lipid Res 51(1):150–161
Yao Z (2012) Human apolipoprotein C-III - a new intrahepatic protein factor promoting assembly and secretion of very low-density lipoproteins. Cardiovasc Hematol Disord Drug Targets 12(2):133–140
Ito Y, Azrolan N, O’Connell A, Walsh A, Breslow JL (1990) Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science 249(4970):790–793
Talmud PJ, Humphries SE (1997) Apolipoprotein C-III gene variation and dyslipidaemia. Curr Opin Lipidol 8(3):154–158
Jin JL, Guo YL, Li JJ (2016) Apoprotein C-III: a review of its clinical implications. Clin Chim Acta 460:50–54
Li Y, Li C, Gao J (2016) Apolipoprotein C3 gene variants and the risk of coronary heart disease: a meta-analysis. Meta Gene 9:104–109
Salas J, Jansen S, López-Miranda J et al (1998) The SstI polymorphism of the apolipoprotein C-III gene determines the insulin response to an oral-glucose-tolerance test after consumption of a diet rich in saturated fats. Am J Clin Nutr 68(2):396–401
Adams JN, Raffield LM, Freedman BI et al (2014) Analysis of common and coding variants with cardiovascular disease in the Diabetes Heart Study. Cardiovasc Diabetol 13:77
López-Miranda J, Jansen S, Ordovas JM et al (1997) Influence of the SstI polymorphism at the apolipoprotein C-III gene locus on the plasma low-density-lipoprotein-cholesterol response to dietary monounsaturated fat. Am J Clin Nutr 66(1):97–103
Esterbauer H, Hell E, Krempler F, Patsch W (1999) Allele-specific differences in apolipoprotein C-III mRNA expression in human liver. Clin Chem 45(3):331–339
Hoffer MJ, Sijbrands EJ, De Man FH, Havekes LM, Smelt AH, Frants RR (1998) Increased risk for endogenous hypertriglyceridaemia is associated with an apolipoprotein C3 haplotype specified by the SstI polymorphism. Eur J Clin Invest 28(10):807–812
Vislovukh A, Vargas TR, Polesskaya A, Groisman I (2014) Role of 3’-untranslated region translational control in cancer development, diagnostics and treatment. World J Biol Chem 5(1):40–57
Dancer M, Caussy C, Di Filippo M, Moulin P, Marçais C, Charrière S (2016) Lack of evidence for a liver or intestinal miRNA regulation involved in the hypertriglyceridemic effect of APOC3 3’UTR variant SstI. Atherosclerosis 255:6–10
Li WW, Dammerman MM, Smith JD, Metzger S, Breslow JL, Leff T (1995) Common genetic variation in the promoter of the human apo CIII gene abolishes regulation by insulin and may contribute to hypertriglyceridemia. J Clin Invest 96(6):2601–2605
Groenendijk M, Cantor RM, Blom NH, Rotter JI, de Bruin TW, Dallinga-Thie GM (1999) Association of plasma lipids and apolipoproteins with the insulin response element in the apoC-III promoter region in familial combined hyperlipidemia. J Lipid Res 40(6):1036–1044
Felekkis K, Touvana E, Stefanou Ch, Deltas C (2010) microRNAs: a newly described class of encoded molecules that play a role in health and disease. Hippokratia 14(4):236–240
Galton DJ (2017) Clarifying complex inheritance: apolipoprotein C3 and atherosclerosis. Curr Opin Lipidol 28(4):308–312
Searfoss AM, Wickner RB (2000) 3’ poly(A) is dispensable for translation. Proc Natl Acad Sci USA 97(16):9133–9137
Chen CY, Shyu AB (2011) Mechanisms of deadenylation-dependent decay. Wiley Interdiscip Rev RNA 2(2):167–183
Taskinen MR, Borén J (2016) Why is apolipoprotein CIII emerging as a novel therapeutic target to reduce the burden of cardiovascular disease? Curr Atheroscler Rep 18(10):59
Kohan AB (2015) Apolipoprotein C-III: a potent modulator of hypertriglyceridemia and cardiovascular disease. Curr Opin Endocrinol Diabetes Obes 22(2):119–125
Wyler von Ballmoos MC, Haring B, Sacks FM (2015) The risk of cardiovascular events with increased apolipoprotein CIII: a systematic review and meta-analysis. J Clin Lipidol 9(4):498–510
Scheffer PG, Teerlink T, Dekker JM et al (2008) Increased plasma apolipoprotein C-III concentration independently predicts cardiovascular mortality: the Hoorn Study. Clin Chem 54(8):1325–1330
Fogelstrand P, Borén J (2012) Retention of atherogenic lipoproteins in the artery wall and its role in atherogenesis. Nutr Metab Cardiovasc Dis 22(1):1–7
Borén J, Matikainen N, Adiels M, Taskinen MR (2014) Postprandial hypertriglyceridemia as a coronary risk factor. Clin Chim Acta 431:131–142
Peng J, Luo F, Ruan G, Peng R, Li X (2017) Hypertriglyceridemia and atherosclerosis. Lipids Health Dis 16(1):233
Nordestgaard BG (2016) Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res 118(4):547–563
Borén J, Taskinen MR, Olofsson SO, Levin M (2013) Ectopic lipid storage and insulin resistance: a harmful relationship. J Intern Med 274(1):25–40
Nordestgaard BG, Varbo A (2014) Triglycerides and cardiovascular disease. Lancet 384(9943):626–635
Hiukka A, Ståhlman M, Pettersson C et al (2009) ApoCIII-enriched LDL in type 2 diabetes displays altered lipid composition, increased susceptibility for sphingomyelinase, and increased binding to biglycan. Diabetes 58(9):2018–2026
Hoff HF, Heideman CL, Jackson RL, Bayardo RJ, Kim HS, Gotto AM Jr (1975) Localization patterns of plasma apolipoproteins in human atherosclerotic lesions. Circ Res 37(1):72–79
Sacks FM, Alaupovic P, Moye LA, Cole TG, Sussex B, Stampfer MJ, Pfeffer MA, Braunwald E (2000) VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the cholesterol and recurrent Events (CARE) trial. Circulation 102(16):1886–1892
Kawakami A, Yoshida M (2009) Apolipoprotein CIII links dyslipidemia with atherosclerosis. J Atheroscler Thromb 16(1):6–11
Tao Y, Xiong Y, Wang H et al (2016) APOC3 induces endothelial dysfunction through TNF-α and JAM-1. Lipids Health Dis 15(1):153
Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM (2006) Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation 114(7):681–687
Kawakami A, Aikawa M, Libby P, Alcaide P, Luscinskas FW, Sacks FM (2006) Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation 113(5):691–700
Galkina E, Ley K (2007) Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol 27(11):2292–2301
Kawakami A, Aikawa M, Nitta N, Yoshida M, Libby P, Sacks FM (2007) Apolipoprotein CIII-induced THP-1 cell adhesion to endothelial cells involves pertussis toxin-sensitive G protein- and protein kinase C alpha-mediated nuclear factor-kappaB activation. Arterioscler Thromb Vasc Biol 27(1):219–225
Li H, Han Y, Qi R et al (2015) Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: the effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc Res 107(4):579–589
Constantinou C, Karavia EA, Xepapadaki E et al (2016) Advances in high-density lipoprotein physiology: surprises, overturns, and promises. Am J Physiol Endocrinol Metab 310(1):E1–E14
Rosenson RS, Brewer HB Jr, Ansell B et al (2013) Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation 128(11):1256–1267
Riwanto M, Rohrer L, Roschitzki B et al (2013) Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation 127(8):891–904
Xiong X, Liu H, Hua L, Zhao H, Wang D, Li Y (2015) The association of HDL-apoCIII with coronary heart disease and the effect of statin treatment on it. Lipids Health Dis 14:127
Dainis AM, Ashley EA (2018) Cardiovascular precision medicine in the genomics era. JACC Basic Transl Sci 3(2):313–326
Mitchell KJ (2012) What is complex about complex disorders? Genome Biol 13(1):237
Giudicessi JR, Kullo IJ, Ackerman MJ (2017) Precision cardiovascular medicine: state of genetic testing. Mayo Clin Proc 92(4):642–662
Smith JA, Ware EB, Middha P, Beacher L, Kardia SL (2015) Current applications of genetic risk scores to cardiovascular outcomes and subclinical phenotypes. Curr Epidemiol Rep 2(3):180–190
Humphries SE, Ridker PM, Talmud PJ (2004) Genetic testing for cardiovascular disease susceptibility: a useful clinical management tool or possible misinformation? Arterioscler Thromb Vasc Biol 24(4):628–636
Dallinga-Thie GM, Berk-Planken II, Bootsma AH, Jansen H (2004) Diabetes Atorvastatin Lipid intervention (DALI). Study Group Atorvastatin decreases apolipoprotein C-III in apolipoprotein B-containing lipoprotein and HDL in type 2 diabetes: a potential mechanism to lower plasma triglycerides. Diabetes Care 27(6):1358–1364
Gouni-Berthold I (2017) The role of antisense oligonucleotide therapy against apolipoprotein-CIII in hypertriglyceridemia. Atheroscler Suppl 30:19–27
Miller CM, Harris EN (2016) Antisense oligonucleotides: treatment strategies and cellular internalization. RNA Dis 3(4):e1393
Aboul-Fadl T (2005) Antisense oligonucleotides: the state of the art. Curr Med Chem 12(19):2193–2214
Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, Geary RS, Baker BF, Graham MJ, Crooke RM, Witztum JL (2014) Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 371(23):2200–2206
Brahm AJ, Hegele RA (2015) Chylomicronaemia–current diagnosis and future therapies. Nat Rev Endocrinol 11(6):352–362
Gaudet D, Alexander VJ, Baker BF et al (2015) Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med 373(5):438–447
Digenio A, Dunbar RL, Alexander VJ et al (2016) Antisense-mediated lowering of plasma apolipoprotein C-III by volanesorsen improves dyslipidemia and insulin sensitivity in type 2 diabetes. Diabetes Care 39(8):1408–1415
Blom DJ, O’Dea L, Digenio A et al (2018) Characterizing familial chylomicronemia syndrome: baseline data of the APPROACH study. J Clin Lipidol 12(5):1234-1243.e5
Khetarpal SA, Zeng X, Millar JS et al (2017) A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med 23(9):1086–1094
Reyes-Soffer G, Sztalryd C, Horenstein RB et al (2019) Effects of APOC3 heterozygous deficiency on plasma lipid and lipoprotein metabolism. Arterioscler Thromb Vasc Biol 39(1):63–72
Acknowledgments
All authors are acknowledged in the authorship
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors had the idea for the article. ID conducted the literature search and did writing. HN and AK conceptualized the ideas and contributed to writing and preparing the final version of the manuscript. ID prepared the figures. HN, AK, RC and YM critically revised the work. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Research involving human and animal participants
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dib, I., Khalil, A., Chouaib, R. et al. Apolipoprotein C-III and cardiovascular diseases: when genetics meet molecular pathologies. Mol Biol Rep 48, 875–886 (2021). https://doi.org/10.1007/s11033-020-06071-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-06071-5