
Abstract
Leukemia is one of the most aggressive hematological malignancies. Leukemia stem cells account for the poor prognosis and relapse of the disease. Decades of investigations have been performed to figure out how to eradicate the leukemia stem cells. It has also been known that cancer cells especially solid cancer cells use energy differently than most of the cell types. The same thing happens to leukemia. Since there are metabolic differences between the hematopoietic stem cells and their immediate descendants, we aim at manipulating the energy sources with which that could have an effect on leukemia stem cells while sparing the normal blood cells. In this review we summarize the metabolic characteristics of distinct leukemias such as acute myeloid leukemia, chronic myeloid leukemia, T cell lymphoblastic leukemia, B-cell lymphoblastic leukemia, chronic lymphocytic leukemia and other leukemia associated hematological malignancies such as multiple myeloma and myelodysplastic syndrome. A better understanding of the metabolic profiles in distinct leukemias might provide novel perspectives and shed light on novel metabolic targeting strategies towards the clinical treatment of leukemias.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Leukemia is a kind of hematological malignancy that often initiates in the bone marrow and leads to abnormal and immature white blood cells which are also named blasts or leukemia cells. In a contemporary view, LSCs is responsible for the initiation and development of the disease and can explain most of the poor prognosis in clinical trials. Numerous studies have shed light on eliminating LSCs based on their distinct genetic and epigenetic characteristics compared with the normal blood stem cells [1, 2]. In recent decades, awareness that the metabolic phenotype of leukemia cells is heterogeneous and distinct from that of their normal counterparts—is increasing. How the leukemia cells survive and propagate in adverse environment such as low glucose, hypoxia and oxidative stress remains poorly understood [3, 4].
Glucose, free fatty acids, glutamine, amino acids are basic and important materials for the catabolism pathways that support the growth and survival of the cancer cells. These catabolites are either assimilated from the blood circulation or synthesized within cancer cells. Glucose is the among the most available nutrients in blood and a metabolic substrate generally utilized by cancer cells [5]. It is well known that anaerobic glycolysis and aerobic mediated oxidative phosphorylation (OXPHOS) are the two major pathways in glucose metabolism. Thus, various drugs that interfere with glycolysis and OXPHOS are being investigated and designed as anti-cancer agents. Fatty acids are also essential substrates for catabolic pathways in cancers. The pentose phosphate pathway (PPP) mediated glucose catabolism is known to be an important initiating point for the production of NADPH and maintenance of a redox balance in normal cells [6]. However, fatty acid oxidation (FAO) take the place of PPP in cancer cells and contributes substantially to these processes under metabolic stress due to the low glucose levels [7]. Contrary to the FAO catabolism, lipids and steroids are de novo synthesized to generate the new phospholipid bilayers, which are essential for the fast proliferation and division of the cancer cells [8]. Glutamine is another indispensable nutrient for cancer cell growth since the amido nitrogen is an essential substrate for hexosamine and nucleotide synthesis [9], and is also getting involved in TCA cycle to produce energy for cancer cells. Other critical metabolic processes include asparagine metabolism, folate metabolism and oxidative stress metabolism. Like solid cancer cells, leukemia cells are not metabolically homogenous and distinct types of leukemia cells utilize special metabolic raw material preferentially. Thus, targeting the metabolic differences between leukemia cells and normal blood cells provides novel anti-leukemia strategy. In this review, we discuss the metabolic profiles of four major leukemias as well as other hematological malignancies. In particular, we underscore potential metabolic vulnerabilities for each type of leukemia.
Glucose Metabolism
The well established Warburg effect has demonstrated that solid cancer cells mainly use glycolysis to meet their energy requirement [10]. The Warburg effect had long been believed to be an adaptation to low-oxygen environment in cancer cells. Since most of the raw materials and energy required for cell proliferation are from glycolysis, cancer cells need to activate glycolysis pathways although they mostly reside in a condition that has enough oxygen. Targeting the glucose metabolism of leukemia cells has already becoming an emerging trend for leukemia treatment.
Role in AML
There are still some controversies concerning whether AML cells use glycolytic metabolism or OXPHOS metabolism although the glucose metabolism in AML has been implicated in many studies. An interesting study by Wang et al. had demonstrated that AML cells are so unique in the way they sense the glucose availability and utilization while normal cells do not get disrupted because they have other glucose metabolism in place in case of an emergency. They suggested that by precise control of the level of glycolysis would be a novel therapeutical strategy for treating AML without affecting the normal function of the HSCs [11]. Besides, Saito et al. indicated that AMPK deletion substantially delays leukemogenesis and eradicates LSCs in MLL-AF9 induced AML by downregulating the expression of glucose transporter 1 (Glut1), enhancing oxidative stress and inducing DNA damage. This study powerfully supports that the Warburg effect does exist in leukemia [12]. Moreover, Chen et al. [13] showed that elevated glycolysis decrease the vulnerability to anti-leukemia agent arabinofuranosyl cytidine (Ara-C) while inhibition of glycolysis suppresses AML cell growth and enhances cytotoxicity of Ara-C in killing leukemic cells. Also, Larrue et al. demonstrated that the glycolytic inhibitor 2-deoxy-d-glucose (2-DG) exhibited great anti-leukemia activity through modulating the expression of receptor tyrosine kinases (RTKs) such as FLT-ITD and KIT. Modulating glycolysis by using 2-DG provides a therapeutic approach in some subset of AML harboring mutated RTKs [14]. One of the traditional ways of treating AML is by chemotherapy but sometimes it is not quite effective because of chemoresistance. A recent research by Song et al. revealed that increased glycolysis and decreased OXPHOS may result in the drug resistance of AML cells. Targeting anaerobic glycolysis metabolism is an effective strategy for manipulating chemoresistance in AML [15]. Despite the numerous evidence proofing that AML mainly uses glycolysis as their glucose utilization pathways, which is consistant with the solid cancer cells, an intriguing investigation by Lagadinou et al. reported that OXPHOS is the unique fuel source for LSCs of AML. They discovered that LSCs in AML reside in a low levels of reactive oxygen species (termed “ROS-low”) condition and exhibit a slower rate of energy metabolism compared with the normal blood cells. Interestingly, they found that the anti-apoptotic molecule BCL-2 is aberrantly overexpressed in ROS-low LSCs. Targeting the BCL-2 dependent OXPHOS could selectively kill LSCs while spare the normal blood stem cells [16] (Table 1).
Role in ALL and CLL
Intriguingly, T-ALL leukemia cells might also prefer to reprogram glycolysis to OXPHOS to survive under the metabolic stress. For example, a study by Kishton et al. [17] revealed that AMPK is essential to maintain mitochondrial metabolism which alleviates metabolic stress and prevents T-ALL cells from apoptosis. Moreover, Liu et al. implied that specifically targeting Glut1 impaired the glycolysis and anabolic metabolism of B-ALL cells harboring BCR-ABL fusion protein [18]. CLL leukemic cells also tend to use glycolysis. Synergistically using a tyrosine kinase inhibitor dasatinib and inhibiting two master metabolic regulators (mTORC1 and AMPK) could target the CLL lymphocytes by reducing lactate production, thus decreasing aerobic glycolysis [19].
Role in CML
Glycolytic dependencies is also reported in CML. For instance, Gottschalk et al. showed that imatinib (STI571), which is a classic inhibitor of the BCR-ABL oncoprotein, induces cell apoptosis in human leukemia BCR-ABL positive cells by altering the glucose metabolism from glycolysis to mitochondrial metabolism [20].
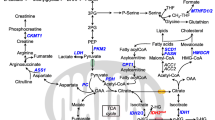
To sum up, leukemia cells may not always depend on anaerobic respiration as their energy source. Moderated aerobic glycolysis, along with alternative metabolic pathways like OXPHOS, may also be necessary to promote the growth and survival of leukemia cells especially when they are in an adverse environment in the long run. The heterogeneity of glucose metabolism in distinct leukemias remains to be studied and more evidence is needed to get a comprehensive map of leukemic glucose metabolism for treatment in the future (Fig. 1).

Basic metabolic pathways in leukemia cells
Lipogenesis Metabolism
The process of lipogenesis, also named lipid synthesis, plays an important role in the pathogenesis of solid cancer. Several enzymes are getting involved in the lipid synthesis pathways, including fatty acid synthase, acetyl-CoA carboxylase, ATP citrate lyase and choline kinase [21,22,23,24,25]. Choline is the raw material in regenerating the membrane phospholipid phosphatidylcholine of cancer cells. The expression and activity of choline kinase are increased in various solid human cancers because of growth factor stimulation and activation of Ras signaling pathway [23]. Many studies support that lipid synthesis favors leukemogenesis (Table 2).
Role in AML
A recent finding by Fraineau et al. [29] revealed that mesenchymal stromal cells (MSCs) derived from bone marrow niche create a favorable microenvironment that sustains the initiation and progression of AML. This discovery also indicates that novel therapeutic treatment could target the leukemia niche which favors more for the survival of cancer cells than the normal blood cells. However, there are evidence showing that adipogenesis was inhibited in some subtype of AML. For example, Kim et al. suggested that adipogenic associated transcription factors are negatively regulated in the promyelocytic leukemia (PML). This contributes to fat accumulation by blocking the differentiation of preadipocytes into adipocytes [27]. In addition, Yasugi et al. [28] showed that peroxisome proliferator-activated receptor gamma ligands (PPARγ) promote the lipogenesis as well as myeloid differentiation in human NB4 cells. This finding seemed to indicate the association of the AML differentiation with lipogenesis but it did not clarify a definite cause and effect relationship between them. It remains to be explored whether lipogenesis contributes to the myeloid differentiation or vice versa.
Role in Other Hematological Malignancies
A study by Medina et al. demonstrate that adiponectin, cytokine generated by adipocytes could significantly suppress the survival of MM cells by activating PKA/AMPK pathway and inactivating the ACC1 activity, thus alleviating lipogenesis [26]. These results not only clarify the positive association between obesity and the trend to develop into MM but also provide a therapeutic strategy by targeting the lipogenesis of MM cancer cells.
To summarize, whether lipogenesis promote the development of leukemia or not is still elusive. Leukemic dependency on lipogenesis may be determined by the genetic and epigenetic characteristics of distinct leukemias.
FAO Metabolism
The fatty acid oxidation (FAO) is considered to be the reverse lipid metabolism in terms of lipogenesis. It is noteworthy that FAO has been associated with chemoresistance and mitochondrial uncoupling. Warburg’s discovery about the glycolysis in cancer cells can be explained as a result of the preferential oxidation of fatty acids in the mitochondrial of cancer cells. In fact, targeting FAO or other anaplerotic pathways that compensate FAO has been demonstrated to provide novel therapeutic strategies in treating distinct leukemias [30].
Role in AML and CML
Carnitine O-palmitoyltransferase I (CPT1) is a critical rate-limiting enzyme, which catalyzes the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine in FAO. Inhibition of CPT1 has been implicated its anti-leukemia effect in vivo and intro. For example, Ricciard et al. [31] demonstrated that a novel CPT1a inhibitor ST1326 exhibits high anti-leukemia activity by inducing cell cycle arrest, mitochondrial damage, and cell apoptosis in several leukemia cell lines including AML, ALL and CLL, with the best efficacy for AML cells. Despite the catalytic enzymes involved in FAO in cytoplasm, the cell surface fatty acid transporters are also novel therapeutic targets for AML. Recently, Ye et al. showed that fatty acid transporter CD36 positive LSCs, which were substantially enriched in gonadal adipose tissue (GAT), have unique metabolic properties and could evade from chemotherapy by the GAT niche. Targeting the CPT1 mediated FAO pathway upsteam of CD36 provides a unique strategy of selective targeting the quiescent LSCs both in human AML and CML treatment [32]. Since free fatty acids are not able to cross the mitochondrial membranes without help, carnitine-acylcarnitine translocase, also named carnitine transporter, is responsible for transporting both carnitine-fatty acid complexes and free carnitine across the inner mitochondrial membrane. A study by Wu et al. revealed that targeting the carnitine transporter 2 (CT2, SLC22A16) which is overexpressed in AML shows the great efficacy in eradicating AML cells [33]. Since FAO is carrying out in mitochondrial, a study by Lee et al. [34] revealed that avocatin B could inhibit the mitochondrial function, which provides a novel strategy for selectively eradicating leukemia cells while sparing their normal counterparts. Moreover, Velez et al. [35] implied that metformin and phenformin inhibit the FAO and sensitize leukemia cells to the BCL-2 inhibitor ABT-737 induced cell-intrinsic apoptosis. Interestingly, Shinohara et al. [37] showed that by synergistically using the anti-cancer fatty acid derivative AIC-47 and imatinib could strengthen the attack on CML cells by inhibiting CPT1c expression and FAO metabolism.
Role in ALL and CLL
Messmer et al. revealed that one antagonist of PPARα called NXT629 efficiently eliminates the CLL cells by suppressing peroxisome proliferator-activated receptor (PPAR)-α, which is a main transcriptional modulator of FAO. This finding strongly supported that the survival and growth of CLL cells also depend on the fatty acid metabolism [36]. In addition, Hermanova et al. suggested that pharmacological inhibition of FAO greatly sensitizes the ALL cells to L-ASP, which is a key agent in treating T-ALL.
To sum up, targeting the leukemia’s “fatty tooth” has exerted a great potential and efficacy in treating distinct kinds of leukemias. Targeting FAO as well as other metabolic pathways simultaneously, with moderate inhibition by combination therapy, provide a great value in leukemia treatment.
Glutaminolysis Metabolism
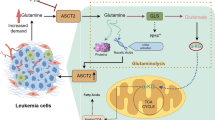
Glutamine is the most abundant amino acid in amino acid metabolism. Glutaminolysis, a process in which glutamine is discomposed to glutamate, aspartate, CO2, pyruvate, lactate, alanine and citric acid, is an extra energy resource in cancer cells particularly when glycolysis is relatively low due to the accumulation of a dimeric form of PKM2. The rate-limiting step in intracellular glutamine catabolism is catalyzed by glutaminase (GA) which converts glutamine to glutamate. The genes that encode GA are called GLS1 and GLS2. They are alternatively spliced and distinct GA isoforms are generated.
Role in AML and CML
Glutaminolysis plays an important role in maintaining the growth and survival of AML cells as is evidenced by the indication that glutaminolysis inhibition through CB-839 concomitant with using the BCL-2 inhibitor ABT-199 induced GLS1 inhibition contributes to the leukemic cell cycle arrest and apoptosis without affecting their normal human CD34+ progenitors [38]. Moreover, an in vitro assay by Goto et al. [39] suggested that glutamine deprivation reduces the intracellular glutathione content and increases reactive oxygen species (ROS) most significantly in four AML cell lines especially in HL-60. The incidence of mutations in isocitrate dehydrogenase 1 and 2 (IDH1/2) is frequently found in AML. These mutations contribute to the glutamine dependency as the main source for α-ketoglutarate which could be converted to the oncoprotein 2-hydroxyglutarate in cancer cells. Targeting the glutamine dependency of the leukemia cells would be mostly beneficial to a specific subset of AML with IDH mutations. For example, Emadi et al. [40] demonstrate that a small molecule of glutaminase inhibitor called Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide BPTES, exclusively suppresses the survival of human primary AML cells expressing mutated IDH compared with those expressing the normal IDH. The impact of glycosylation of ASCT2 which was ever underestimated in metabolic reprogramming could be a therapeutic target in leukemia treatment. For example, Polet et al. [43] showed that the defect in glycosylation of glutamine transporters could result in glycolytic inhibition in AML as well as in CML. Drug resistance resulting from the metabolic reprogramming accounts for the relapse of leukemia treatment. For instance, Stäubert et al. [44] demonstrated that resistant leukemia cells alter their metabolism characterized by reduced dependency on glutamine metabolism at the cost of a higher requirement for glucose and enhanced FAO with decreased pantothenic acid uptake ability. Moreover, Polet et al. implied that phosphoglycerate dehydrogenas (PHGDH) is upregulated after glutamine deprivation. Silencing PHGDH in vitro and the use of diet without serine in vivo suppresses AML. Besides, the glutamine dependency could also be observed in BCR-ABL positive CML. Sontakke et al. indicated that the expression of glutamine importer SLC1A5 is increased in BCR-ABL transduced human cord blood CD34+ cells and these cancer cells are more susceptible to the glutaminase inhibitor BPTES, which implies that the glutamine metabolism is elevated in CML to maintain OXPHOS level [45].
Roles in ALL
Glutaminolysis has been also highly implicated to be a critical process in other forms of leukemia such as T-ALL. For example, Herranz et al. [41] revealed that glutaminolysis is an important pathway for mutated NOTCH1 induced T-ALL growth and a critical determinant of sensitivity in anti-NOTCH1 leukemia treatment in vivo. ASCT2, also named SLC1A5, which is known as a major glutamine transporter, has been reported to participate in the growth of solid cancer cells [42].
To sum up, targeting glutaminolysis have shown a great efficacy in treating AML as well as ALL. More research is needed to determine whether distinct types of leukemia cells glutamine metabolism as a complemental source.
Aspartate Metabolism
l-Asparaginase (I-ASP, erwinia l-asparaginase), which converts l-asparagine to ammonia and aspartic acid, is an anticancer agent exhibiting both asparaginase and glutaminase activity.
Role in AML
I-ASP shows great efficacy in treating AML disease. For example, Parmentier et al. [47] showed that asparaginase activity of I-ASP alone may not be enough for the cytotoxicity to the leukemia cells in T-ALL, and that glutaminase activity may be essential for its full anti-leukemia activity. Moreover, Willems et al. [48] reveal that l-ASP increases the expression of the glutamine synthase (GS) and knockdown of GS contributes to l-ASP induced autophagic process in AML cells.
Role in ALL and CLL
I-ASP has also been demonstrated to be a efficacious amidohydrolase in treating T-ALL and natural killer (NK) cell lymphoma by inhibiting the protein synthesis, inducing the cell cycle arrest and enhancing the ROS level to the mitochondrial permeabilization and subsequent cell apoptosis in cancer cells [46]. However, an study done by Chan et al. showed that the glutaminase activity of L-ASP is not always indispensable for anticancer effect in solid tumors. They implied that the L-ASP’s glutaminase activity is essential for inhibiting the asparagine synthetase (ASNS) positive cells but not for ASNS negative cells. The therapeutic significance of this observation is that glutaminase negative variants of L-ASP instead of wild-type L-ASP should be explored for better treatment of ASNS negative cancers [49]. Although different L-ASP based chemotherapy are highly efficient, disease relapse could happen frequently. For example, Chien et al. [50] reported that cleavage of anti-apoptotic molecule BCL-2A1 contribute to the sensitivity of malignant natural killer (NK) cell lines and B-ALL cells.
In summary, l-ASP associated therapeutics towards distinct types of leukemia provide one-carbon metabolism targeting strategy in leukemia treatment.
Folate Metabolism
Folate metabolism is considered one of another important one-carbon metabolism besides the aspartate metabolism. Antifolates are the first effective chemotherapy drugs which suppress the growth and survival of cancer cells by inhibiting DNA and RNA synthesis.
Role in AML
A study by Lynn et al. discovered that the expression of folate receptor β (FRβ) is elevated when treating AML cells with all-trans retinoic acid (ATRA), which improves immune therapy by using CAR-T cell therapy. This observation provides novel therapeutic treatment by targeting the folate metabolism [51].
Role in ALL
In addition, Methotrexate (MTX) is well known for its role in treating various hematological malignancies by inhibiting dihydrofolate reductase (DHFR). For example, Uchiyama et al. [52] demonstrated that a cyclin-dependent kinase inhibitor called SU9516, which downregulates the expression of both DHFR mRNA and protein, leads to enhanced sensitivity to MTX in human T-cell leukemia Jurkat cell line. However, the polymorphisms of DHFR are concerned with worse ALL outcome, which is associated with higher DHFR expression [53]. Combined effects should be carefully examined on other genes involved in folate metabolism. Drug combination shows very high efficacy as it is evidenced by Teachey et al. [54] showed that mTOR inhibitors (MTIs) substantially increase the sensitivity of ALL cells to MTX by downregulation of both DHFR and cell cycle promotor cyclin D1. Drug resistance also gives rise to major obstacles in applying MTX to treat T-ALL [55]. Thus, individual and personalized treatment based on molecular diagnosis should be provided to the patients who are possibly resistant to MTX. In general, although most of the current investigations of folate metabolism focus on T-ALL treatment, future directions would be extended to other forms of hematological malignancies to advance the understanding of folate metabolism in leukemia.
Oxidative Stress Metabolism
Role in AML and CML
Oxidative stress and OXPHOS are interconnected: OXPHOS occurs in mitochondrial, induces oxidative stress [56], which in turn reduces OXPHOS flux [57]. Reactive oxygen species (ROS) is generated during oxidative stress and plays an important role in regulating cell growth, division, differentiation and apoptosis. A moderate level of is essential to maintain the survival of the cancer cells. However, the LSCs might be vulnerable to high ROS condition, and upregulation of ROS level might even induce the differentiation of leukemia cells. For example, Zhou et al. showed that 3-Deazaneplanocin A (DZNep), a histone methyltransferase inhibitor, downregulates polycomb-repressive complex 2 (PRC2) and induces cell apoptosis by enhancing the oxidative stress in primary AML samples [58]. Recently, a study from Doshi et al. [59] indicated that by jointed using pan-Pim kinase inhibitor AZD1208 and topoisomerase 2 inhibitors significantly contribute to cell apoptosis by inducing increased DNA damage and enhanced ROS level in AML with FLT3-ITD translocation. In addition, Liu et al. revealed that bone marrow niche induces oxidative adaptation and protects leukemia cells from apoptosis in ALL, thus providing cues that targeting redox balance by suppressing antioxidant generation and anti-apoptosis pathways could overcome drug resistance [60]. Moreover, the therapeutic approach by oxidative stress enhancement to kill leukemia cells could be also applied to CML. For example, a recent study by Singh et al. [61] suggested that high endogenous ROS level is able to weaken the nitric oxide (NO) generation in neutrophils (PMN) of CML. To briefly conclude, targeting the oxidative condition represents a general strategy in treating distinct types of leukemias.
Conclusions
Though sharing many similarities, leukemia is genetically and epigenetically different from solid tumor. Besides, the emerging trend of using metabolic strategies to treat leukemia is more or less enlightened by the metabolic treatment in solid tumor. Since leukemia originates from a small subset of cell population called LSCs, it is critical to figure out the metabolic differences between LSCs and HSCs and further develop strategies by specifically targeting LSCs. Similar to their normal counterparts, LSCs are orchestrated in a high hierarchy which is from stem cells, progenitor cells to more differentiated cell lineages. The metabolic profiles might depend on diverse cell lineages that determine the LSC origin in distinct leukemias. Therefore it is not difficult to understand why different type of leukemia would prefer different ways of utilizing energy resources. Moreover, microenvironment could also exert an influence on the leukemia cells and is constantly reshaping their “metabolic behavior”. Due to the complicated niche where the leukemia cells reside in, it is essential to dig into the composition of the niche for each type of leukemia, thus figure out more clearly how the niche changes the “metabolic behavior” in distinct leukemia cells. In other words, it is critical to take the context-dependent determinants into consideration in attempts to study metabolisms in diverse leukemias. To summarize, a better understanding of the connection between the cell origin and their metabolic status, and the function of each leukemia niche would better map the metabolic profiles in distinct hematological malignancies.
With regards to specific targeting strategies based on the metabolic profiles in distinct hematological malignancies, high throughout sequencing and metabolomics would show their power in discovering more potential targets. For example, by comparing different metabolic profiles in the normal human CD34+ cord blood samples and the primary AML patient samples, one can do an analytic selection to target the most potential molecule or molecule combination. Drug inhibition of the metabolic signaling pathways and molecules has been implicated in clinical treatment of leukemias but it is also noteworthy that the normal blood cells should be spared when designing small inhibitors for the potential targets. And sometimes combinatorial use of drugs would potentiate the efficiency in eliminating the leukemia cells and even the LSCs. Moreover, drug deprivation contributes to metabolic compensation, which is recognized as a metabolic adaption and reprogramming. Combined modulation of related metabolic signaling pathways is indispensable because leukemia cells would bypass or utilize other pathways in order to better proliferate and survive. Finally, figuring out novel functional sites of metabolic targets would be helpful of targeting genetic mutation induced drug resistance. Finally mapping a whole and comprehensive metabolic profiles of distinct hematological malignancies is the first and most critical step to address all of the issues that are constantly bewildering us.
Abbreviations
- AML:
-
Acute myeloid leukemia
- CML:
-
Chronic myeloid leukemia
- T-ALL:
-
T-cell lymphoblastic leukemia
- B-ALL:
-
B-cell lymphoblastic leukemia
- CLL:
-
Chronic lymphocytic leukemia
- MM:
-
Multiple myeloma
- MDS:
-
Myelodysplastic syndrome
- LSCs:
-
Leukemia stem cells
- OXPHOS:
-
Oxidative phosphorylation
- FAO:
-
Fatty acid oxidation
- PPP:
-
Pentose phosphate pathway
- Glut1:
-
Glucose transporter 1
- Ara-C:
-
Arabinofuranosyl cytidine
- 2-DG:
-
2-Deoxy-d-glucose
- RTKs:
-
Receptor tyrosine kinases
- ROS:
-
Reactive oxygen species
- CSCs:
-
Cancer stem cells
- PPARγ:
-
Proliferator-activated receptor gamma ligands
- CPT1:
-
Carnitine O-palmitoyltransferase I
- GAT:
-
Gonadal adipose tissue
- GA:
-
Glutaminase
- IDH1/2:
-
Isocitrate dehydrogenase 1 and 2
- PHGDH:
-
Phosphoglycerate dehydrogenas
- I-ASP:
-
l-Asparaginase
- GS:
-
Glutamine synthase
- ASNS:
-
Asparagine synthetase
- PRC2:
-
Polycomb repressive complex 2
References
Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y (2008) PML targeting eradicates quiescent leukaemia-initiating cells. Nature 453(7198):1072–1078
Holtz M, Forman SJ, Bhatia R (2007) Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Can Res 67(3):1113–1120
Cairns RA, Harris IS, Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11(2):85–95
Schulze A, Harris AL (2012) How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491(7424):364–373
Boroughs LK, DeBerardinis RJ (2015) Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17(4):351–359
Kruiswijk F, Labuschagne CF, Vousden KH (2015) p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16(7):393–405
Jeon SM, Chandel NS, Hay N (2012) AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485(7400):661–665
Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP (2017) Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14(2):113
DeBerardinis RJ, Cheng T (2010) Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29(3):313–324
Kumazaki M, Shinohara H, Taniguchi K, Takai T, Kuranaga Y, Sugito N et al (2016) Perturbation of the Warburg effect increases the sensitivity of cancer cells to TRAIL-induced cell death. Exp Cell Res 347(1):133–142
Wang Y-H, Israelsen William J, Lee D, Yu Vionnie WC, Jeanson Nathaniel T, Clish Clary B et al (2014) Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158(6):1309–1323
Saito Y, Chapple RH, Lin A, Kitano A, Nakada D (2015) AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell 17(5):585–596
Chen WL, Wang JH, Zhao AH, Xu X, Wang YH, Chen TL et al (2014) A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 124(10):1645–1654
Larrue C, Saland E, Vergez F, Serhan N, Delabesse E, Mansat-De Mas V et al (2015) Antileukemic activity of 2-deoxy-d-glucose through inhibition of N-linked glycosylation in acute myeloid leukemia with FLT3-ITD or c-KIT mutations. Mol Cancer Ther 14(10):2364–2373
Song K, Li M, Xu X, Xuan LI, Huang G, Liu Q (2016) Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol Lett 12(1):334–342
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M et al (2013) BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12(3):329–341
Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ et al (2016) AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-all cell stress and survival. Cell Metab 23(4):649–662
Liu T, Kishton RJ, Macintyre AN, Gerriets VA, Xiang H, Liu X et al (2014) Glucose transporter 1-mediated glucose uptake is limiting for B-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell Death Dis 5(10):e1470
Martinez Marignac VL, Smith S, Toban N, Bazile M, Aloyz R (2013) Resistance to dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget 4(12):2550–2566
Gottschalk S, Anderson N, Hainz C, Eckhardt SG, Serkova NJ (2004) Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin Cancer Res 10(19):6661–6668
Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24(41):6314–6322
Chajes V, Cambot M, Moreau K, Lenoir GM, Joulin V (2006) Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Can Res 66(10):5287–5294
Clem BF, Clem AL, Yalcin A, Goswami U, Arumugam S, Telang S et al (2011) A novel small molecule antagonist of choline kinase-alpha that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene 30(30):3370–3380
Flavin R, Peluso S, Nguyen PL, Loda M (2010) Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol 6(4):551–562
Mulvihill MM, Nomura DK (2013) Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci 92(8–9):492–497
Medina EA, Oberheu K, Polusani SR, Ortega V, Velagaleti GV, Oyajobi BO (2014) PKA/AMPK signaling in relation to adiponectin’s antiproliferative effect on multiple myeloma cells. Leukemia 28(10):2080–2089
Kim MK, Yang S, Lee KH, Um JH, Liu M, Kang H et al (2011) Promyelocytic leukemia inhibits adipogenesis, and loss of promyelocytic leukemia results in fat accumulation in mice. Am J Physiol Endocrinol Metab 301(6):E1130–E1142
Yasugi E, Horiuchi A, Uemura I, Okuma E, Nakatsu M, Saeki K et al (2006) Peroxisome proliferator-activated receptor gamma ligands stimulate myeloid differentiation and lipogenensis in human leukemia NB4 cells. Dev Growth Differ 48(3):177–188
Le Y, Fraineau S, Chandran P, Sabloff M, Brand M, Lavoie JR et al (2016) Adipogenic mesenchymal stromal cells from bone marrow and their hematopoietic supportive role: towards understanding the permissive marrow microenvironment in acute myeloid leukemia. Stem Cell Rev 12(2):235–244
Samudio I, Fiegl M, Andreeff M (2009) Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Can Res 69(6):2163–2166
Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR et al (2015) Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood 126(16):1925–1929
Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M et al (2016) Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 19(1):23–37
Wu Y, Hurren R, MacLean N, Gronda M, Jitkova Y, Sukhai MA et al (2015) Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis Int J Program Cell Death 20(8):1099–1108
Lee EA, Angka L, Rota SG, Hanlon T, Mitchell A, Hurren R et al (2015) Targeting mitochondria with avocatin B induces selective leukemia cell death. Can Res 75(12):2478–2488
Velez J, Pan R, Lee JT, Enciso L, Suarez M, Duque JE et al (2016) Biguanides sensitize leukemia cells to ABT-737-induced apoptosis by inhibiting mitochondrial electron transport. Oncotarget 7(32):51435–51449
Messmer D, Lorrain K, Stebbins K, Bravo Y, Stock N, Cabrera G et al (2015) A selective novel peroxisome proliferator-activated receptor (PPAR)-alpha antagonist induces apoptosis and inhibits proliferation of CLL cells in vitro and in vivo. Mol Med 21:410–419
Shinohara H, Kumazaki M, Minami Y, Ito Y, Sugito N, Kuranaga Y et al (2016) Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells. Cancer Lett 371(1):1–11
Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L et al (2015) Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 126(11):1346–1356
Goto M, Miwa H, Shikami M, Tsunekawa-Imai N, Suganuma K, Mizuno S et al (2014) Importance of glutamine metabolism in leukemia cells by energy production through TCA cycle and by redox homeostasis. Cancer Investig 32(6):241–247
Emadi A, Jun SA, Tsukamoto T, Fathi AT, Minden MD, Dang CV (2014) Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp Hematol 42(4):247–251
Herranz D, Ambesi-Impiombato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V et al (2015) Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med 21(10):1182–1189
van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D et al (2016) ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 35(24):3201–3208
Polet F, Martherus R, Corbet C, Pinto A, Feron O (2016) Inhibition of glucose metabolism prevents glycosylation of the glutamine transporter ASCT2 and promotes compensatory LAT1 upregulation in leukemia cells. Oncotarget 7(29):46371–46383
Sontakke P, Koczula KM, Jaques J, Wierenga AT, Brouwers-Vos AZ, Pruis M et al (2016) Hypoxia-like signatures induced by BCR-ABL potentially alter the glutamine uptake for maintaining oxidative phosphorylation. PLoS ONE 11(4):e0153226
Polet F, Corbet C, Pinto A, Rubio LI, Martherus R, Bol V et al (2016) Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget 7(2):1765–1776
Alachkar H, Fulton N, Sanford B, Malnassy G, Mutonga M, Larson RA et al (2017) Expression and polymorphism (rs4880) of mitochondrial superoxide dismutase (SOD2) and asparaginase induced hepatotoxicity in adult patients with acute lymphoblastic leukemia. Pharmacogenomics J 17(3):274–279
Parmentier JH, Maggi M, Tarasco E, Scotti C, Avramis VI, Mittelman SD (2015) Glutaminase activity determines cytotoxicity of L-asparaginases on most leukemia cell lines. Leuk Res 39(7):757–762
Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M et al (2013) Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 122(20):3521–3532
Chan WK, Lorenzi PL, Anishkin A, Purwaha P, Rogers DM, Sukharev S et al (2014) The glutaminase activity of L-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood 123(23):3596–3606
Chien WW, Le Beux C, Rachinel N, Julien M, Lacroix CE, Allas S et al (2015) Differential mechanisms of asparaginase resistance in B-type acute lymphoblastic leukemia and malignant natural killer cell lines. Sci Rep 5:8068
Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS et al (2015) Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood 125(22):3466–3476
Uchiyama H, Sowa Y, Wakada M, Yogosawa M, Nakanishi R, Horinaka M et al (2010) Cyclin-dependent kinase inhibitor SU9516 enhances sensitivity to methotrexate in human T-cell leukemia Jurkat cells. Cancer Sci 101(3):728–734
Dulucq S, St-Onge G, Gagne V, Ansari M, Sinnett D, Labuda D et al (2008) DNA variants in the dihydrofolate reductase gene and outcome in childhood ALL. Blood 111(7):3692–3700
Teachey DT, Sheen C, Hall J, Ryan T, Brown VI, Fish J et al (2008) mTOR inhibitors are synergistic with methotrexate: an effective combination to treat acute lymphoblastic leukemia. Blood 112(5):2020–2023
Wojtuszkiewicz A, Peters GJ, van Woerden NL, Dubbelman B, Escherich G, Schmiegelow K et al (2015) Methotrexate resistance in relation to treatment outcome in childhood acute lymphoblastic leukemia. J Hematol Oncol 8:61
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M et al (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 107(19):8788–8793
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48(2):158–167
Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG et al (2011) The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 118(10):2830–2839
Doshi KA, Trotta R, Natarajan K, Rassool FV, Tron AE, Huszar D et al (2016) Pim kinase inhibition sensitizes FLT3-ITD acute myeloid leukemia cells to topoisomerase 2 inhibitors through increased DNA damage and oxidative stress. Oncotarget 7(30):48280–48295
Liu J, Masurekar A, Johnson S, Chakraborty S, Griffiths J, Smith D et al (2015) Stromal cell-mediated mitochondrial redox adaptation regulates drug resistance in childhood acute lymphoblastic leukemia. Oncotarget 6(40):43048–43064
Singh AK, Awasthi D, Dubey M, Nagarkoti S, Kumar A, Chandra T et al (2016) High oxidative stress adversely affects NFkappaB mediated induction of inducible nitric oxide synthase in human neutrophils: implications in chronic myeloid leukemia. Nitric Oxide 58:28–41
Acknowledgements
This study was supported by grants from Project of Shanghai Municipal Health Bureau (Surface Program, SHXH201402).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. BL designed the manuscript and TL and XP wrote the review.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
All authors are compliant with ethical standards.
Consent for publication
All authors approve the manuscript for publication.
Availability of data and materials
Data and materials related to this work are available upon request.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, T., Peng, XC. & Li, B. The Metabolic Profiles in Hematological Malignancies. Indian J Hematol Blood Transfus 35, 625–634 (2019). https://doi.org/10.1007/s12288-019-01107-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12288-019-01107-8