
Abstract
AML (acute myeloid leukemia) cells have a unique reliance on mitochondrial metabolism and fatty acid oxidation (FAO). Thus, blocking FAO is a potential therapeutic strategy to target these malignant cells. In the current study, we assessed plasma membrane carnitine transporters as novel therapeutic targets for AML. We examined the expression of the known plasma membrane carnitine transporters, OCTN1, OCTN2, and CT2 in AML cell lines and primary AML samples and compared expression to normal hematopoietic cells. Of the three carnitine transporters, CT2 demonstrated the greatest differential expression between AML and normal cells. Using shRNA, we knocked down CT2 and demonstrated that target knockdown impaired the function of the transporter. In addition, knockdown of CT2 reduced the growth and viability of AML cells with high expression of CT2 (OCI-AML2 and HL60), but not low expression. CT2 knockdown reduced basal oxygen consumption without a concomitant increase in glycolysis. Thus, CT2 may be a novel target for a subset of AML.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fatty acid oxidation (FAO) is a biochemical process in which fatty acid molecules are metabolized in the mitochondria to acetyl-CoA. Acetyl CoA enters the citric acid cycle with the resultant production of NADH and FADH2 which serve as electron donors for the respiratory chain. Thus, FAO is a source of ATP and energy production for the cell.
Metabolism of fatty acids requires the co-factor carnitine. Via the enzyme carnitine palmitoyltransferase 1 (CPT1), long chain acyl-CoA is bound to carnitine to form long chain acylcarnitine which is transported from the cytosol into the mitochondrial matrix via carnitine-acylcarnitine translocase (CACT). In the mitochondria, long chain acylcarnitine is converted back to long chain acyl-CoA by carnitine palmitoyltransferase 2 (CPT2) and then undergoes multiple rounds of oxidation to generate acetyl-CoA [1–3].
Carnitine is obtained from the diet or synthesized endogenously from lysine and methionine in the kidney, liver and brain [3, 4]. Other cells acquire carnitine through uptake from the plasma via an active carnitine transport system located on the cell membrane. Three plasma membrane carnitine transporters have been identified to date, OCTN1 (SLC22A4), OCTN2 (SLC22A5) and CT2 (SLC22A16) [5]. In normal tissues, OCTN1 and OCTN2 are more ubiquitously expressed while CT2 is primarily expressed in the testis, kidney, and hematopoietic cells [6–8]. The expression of these transporters on malignant cells is less well understood.
AML cells have increased dependence on FAO compared to normal hematopoietic cells and strategies that block FAO at the level of CPT1 selectively induce death in these malignant cells [9, 10]. Here, we examined the expression of carnitine transporters in AML cell lines and primary AML samples and explored the impact of inhibiting carnitine uptake into AML cells at the level of the plasma membrane by down regulating the carnitine transporter CT2.
Materials and methods
Cell lines and growth conditions
OCI-AML2 and HL60 leukemic cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM) containing 10 % fetal bovine serum (FBS), 100 units/mL streptomycin and 100 μg/mL penicillin. TEX leukemic cells were cultured in IMDM with 20 % FBS, 100 units/mL streptomycin, 100 μg/mL penicillin, 20 ng/mL stem cell factor (SCF), 2 ng/mL IL-3, and 2 mM l-glutamine. All cells were maintained in a 37 °C humidified incubator supplemented with 5 % CO2.
Primary AML and normal hematopoietic cells
Primary human bulk AML cells were isolated from peripheral blood samples from consenting patients with AML (n = 10) or myelofibrosis (n = 1) by Ficoll density centrifugation. Similarly, primary normal hematopoietic cells were obtained from healthy consenting volunteers (n = 4) who received G-CSF in preparation for donating stem cells for allotransplantation. All primary cells were maintained at 37 °C in IMDM with 20 % FBS, penicillin and streptomycin. The collection and use of human tissue for this study were approved by the University Health Network institutional review board.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Quantitative reverse transcription polymerase chain reaction assays were performed as previously described [11, 12]. The cDNAs encoding CT2, OCTN1, OCTN2 and 18 s were amplified using the following forward/reverse primer pairs: CT2 Forward 5′-ACTGGGATCGGTGACTTTTG-3′ and CT2 Reverse 5′-CCCACCACAAGATAGCCACT-3′; OCTN1 Forward 5′-CTGCCCAGGCGTTATATCAT-3′ and OCTN1 Reverse 5′-GGTTGGGTAGAGCTCAGCAG-3′; OCTN2 Forward 5′-CTGGTGGTTCATCCCTGAGT-3′ and OCTN2 Reverse 5′-GGACTGCTGCTTCTTGGAAC-3′; 18 s Forward 5′- AGGAATTGA CGGAAGGGCAC-3′ and 18 s Reverse 5′- GGACATCTAAGGGCATCACA-3′. Transcript level of 18 s gene was used for normalization of the other genes. qRT-PCR reactions were prepared by adding equal amount of cDNA of each sample to a premade master mix (SYBR Green PCR Master Mix; Applied Biosystems, Foster City, CA) followed by processing on an ABI Prism 7900 sequence detection system (Applied Biosystems). The relative abundance of a transcript was represented by the threshold cycle of amplification (CT), which was inversely correlated to the amount of cDNA or RNA in the original sample. The comparative CT method was calculated according to the manufacturer’s protocol. The expression level of CT2 relative to the baseline level was calculated as 2–ΔC (CT2)T , where ΔCT is (average CT2 CT–average 18 s CT) and CT is (average CT-AML sample–average CT-normal sample) or (average CT-knockdown sample–average CT-control sample) based on the experimental conditions. Relative expression of OCTN1 and OCTN2 to baseline was calculated with the same method.
Radiolabeled carnitine uptake assay
To measure carnitine uptake, AML cells were washed with PBS and re-suspended in carnitine uptake buffer (140 mM NaCl, 25 mM Hepes pH7.4, 5 mM KCl, 1 mM MgSO4, 1.2 mM CaCl2, 5 mM glucose). Cells were then seeded in 24-well plates at 2.5 × 105 cells/well. L-[methyl-3H] carnitine hydrochloride (100 nM final concentration, 60–86 Ci/mmol) (PerkinElmer, Waltham, MA) was added to the cells. After 1 h of incubation (37 °C, 5 % CO2), cells were washed three times with cold PBS and lysed in 500 μl of 0.05 M NaOH. Radioactivity was quantified using Beckman LS6000IC liquid scintillation counter (Beckman Coulter, Fullerton, CA) according to the manufacturer’s instructions. Radiolabelled counts were normalized for cell number.
shRNA knockdown of CT2
Construction of hairpin-pLKO.1 puromycin resistant vectors containing short hairpin RNA (shRNA) sequences and production of shRNA viruses have been previously described in detail [13]. The shRNA targeting the CT2 coding sequence (CT2, Accession no. NM_033125) and the control shRNA targeting the GFP sequence (GFP, Accession no. clonetechGfp_587s1c1) were as follows: CT2 5′-CCGGGCTGTCTAGCACCTGAAATATCTCGAGATATTTCAGGTGCTAGACAG-3′; GFP 5′-TGCCCGACAACCACTACCTGA-3′. Lentiviral infections were performed as previously described in Moffat et al [13]. Concisely, 2 mL of virus was added to 5 mL of media containing 5 × 106 cells and protamine sulfate (5 µg/mL). Cells were incubated overnight (37 °C, 5 % CO2) to allow infection by virus. The following day, cells were spun down and re-suspended in 25 mL fresh medium with puromycin and subjected to selection for 3 days. Cells (2 × 106) were then cultured over time in fresh medium without puromycin to measure viability and growth using trypan blue exclusion assay. Cell death was measured by flow cytometry with annexin V fluorescein isothiocyanate (FITC) and propidium iodide (PI) (Biovision Research Products, Mountain View, CA) staining according to the manufacturer’s instructions.
Immunoblotting analysis
Cell lysate preparation and western blot analysis were performed as previously described [14]. Cells (5 × 106) were washed with PBS followed by cell lysis in 1X SDS buffer (60 mM Tris-Cl, pH 6.8, 2 % SDS, 10 % glycerol). Cell lysates were heated at 95 °C for 5 min and protein concentration was measured by DC Protein Assay (Bio-Rad, Hercules, CA). Equal amounts of protein were loaded and fractionated on 10 % SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were probed with anti-SLC22A16 1:400 (Sigma-Aldrich AV44073) or anti-SLC22A16 1:500 (Sigma-Aldrich HPA036902) and secondary anti-rabbit 1:1000 (GE Healthcare NA934 V).
Sensitivity of CT2 knockdown cells to bleomycin
Control cells (1 × 104 cells/well) and CT2 knockdown cells (1.5 × 104 cells/well) were incubated (37 °C, 5 % CO2) with bleomycin (20 μg/mL A2 and A5 mix) for 72 h in 96-well plates. Cell viability was then measured by the Cell Titer-Fluor™ cell viability assay (Promega, Madison, WI) according to manufacturer’s instructions.
Cell cycle analysis
Cells (4 × 105) were washed twice with PBS and suspended in 70 % ethanol in PBS followed by incubation overnight at −20 °C. Cells were then washed twice with PBS and treated with 200 µg/mL RNase A (Hoffmann-La Roche, Mississauga, ON) in PBS, and incubated at 37 °C for 30 min. Cells were washed with PBS and stained with propidium iodine (Sigma-Aldrich, St. Louis, MO). DNA content was analyzed by flow cytometry (BD, Franklin Lakes, NJ). The percentage of cells in each phase of the cell cycle was calculated with FlowJo software (FlowJo LLC, Ashland, OR).
Measurement of basal oxygen consumption and extracellular acidification
Measurement of basal oxygen consumption and extracellular acidification were performed using a Seahorse XF96 Analyzer (Seahorse Bioscience, North Billerica, MA) as per the manufacturer’s instructions. Control and CT2 knockdown cells were spun down and re-suspended in bicarbonate-free alpha MEM medium containing 2 % FBS and penicillin-streptomycin. Cells were seeded in XF96 plates at a density of 1.5 × 105 cells/well and equilibrated at 37 °C in a CO2-free incubator for 45 min prior to analysis. The basal oxygen consumption rate and extracellular acidification rate were measured using the XF96 analyzer.
Combinations of CT2 knockdown with cytotoxic agents
Control and CT2 knockdown cells (1.2 × 104 cells/well) were incubated (37 °C, 5 % CO2) with chemotherapeutic agents (daunorubicin, cytarabine), or metabolic regulators (tigecycline, and etomoxir) at increasing concentrations for 72 h in 96-well plates. Cell viability was then measured by the Cell Titer-Fluor™ cell viability assay (Promega, Madison, WI) according to manufacturer’s instructions.
Statistical analysis
All data were expressed as mean and standard deviation (SD) of representative experiments, unless noted otherwise in figure legends. Statistical analyses were performed by unpaired Student’s t test and one-way ANOVA as indicated in figure legends. Differences were considered statistically significant if p < 0.05.
Results
CT2 is over-expressed in AML cell lines and primary AML patient samples
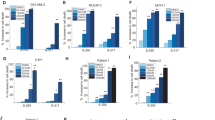
OCTN1, OCTN2, and CT2 are the three major plasma membrane carnitine transporters in human cells. We examined the mRNA expression of these transporters in a panel of AML cell lines as well as primary AML samples (bulk AML blasts) and G-CSF mobilized bulk mononuclear hematopoietic cells. Compared to the mean expression in normal hematopoietic cells, CT2 mRNA was increased in OCI-AML2, THP1, U937, and HL60 leukemic cells (Fig. 1a). OCTN1 and OCTN2 were also over-expressed in a subset of these cell lines, but the difference in expression of these transporters between AML and normal hematopoietic cells was much smaller.

CT2 is over-expressed in AML cell lines and primary patient samples. a CT2, OCTN1, and OCTN2 mRNA expression were measured by qRT-PCR in OCI-AML2, THP-1, U937, TEX, and HL60 leukemic cells as well as normal hematopoietic samples. Expression of each carnitine transporter was normalized to mean expression in the normal cells. Data represent the mean mRNA expression level ±SD. b OCI-AML2 and TEX cells were incubated for 1 h with 100 nM of 3H radiolabeled carnitine with or without 10 μM of unlabeled carnitine. Radioactive counts were recorded. Data represent mean ±SD radioactive counts per 2.5 × 105 cells. One-way ANOVA analysis was performed (p = 0.016) followed by a Student’s t test comparing OCI-AML2 and TEX cells (p = 0.014). (c) CT2, OCTN1, and OCTN2 mRNA expression were measured by qRT-PCR in a panel of primary AML patient samples and normal hematopoietic samples. Expression was normalized to the mean expression in the normal cells. The bar represents the mean mRNA expression level in the samples. * p value < 0.05 and ** p value < 0.0005 determined by Student’s t test
We next compared the uptake of carnitine between OCI-AML2 cells with high expression of carnitine transporters and TEX cells with low expression by incubating cells with radiolabeled 3H carnitine for 1 h. Carnitine uptake was significantly higher in OCI-AML2 cells compared to TEX cells (Fig. 1b). Of note, a sizeable amount of non-specific binding or uptake of 3H carnitine was observed in both cell lines as 3H signal was detectable in both cell lines despite the addition of 100 molar excess of non-labeled carnitine.
We also examined the expression of the carnitine transporters in primary AML cells from 11 patients (AML n = 10 and myelofibrosis with circulating myeloid blasts in the peripheral blood n = 1). The patient characteristics are shown in supplemental Table 1. CT2 expression was increased in primary samples compared to normal hematopoietic cells. In 10 of 11 samples, expression was more than fivefold higher compared to normal hematopoietic cells (Fig. 1c and supplemental Fig. 1). Similar to the AML cell lines, a subset of primary samples had increased expression of the other transporters, but the magnitude of differential expression was much smaller and not statistically significant.
Knockdown of CT2 impairs the function of the transporter and reduces intracellular carnitine
Given the increased expression of CT2 in a subset of AML cells and primary AML patient samples, we used a genetic approach to examine the impact of CT2 inhibition on AML cells. OCI-AML2 cells were infected with shRNA targeting CT2 or control sequence. Target knockdown was confirmed by qRT-PCR (Fig. 2a). We attempted to confirm knockdown by immunoblotting, but despite testing two different antibodies, we were not able to conclusively identify the CT2 protein in lysates of AML cells. Therefore, we used alternate approaches to validate CT2 knockdown in these cells.

Knockdown of CT2 diminishes carnitine uptake and rescues bleomycin toxicity. a OCI-AML2 leukemic cells were infected with CT2 shRNA (shCT2) or control shRNA sequence (control shRNA) in lentiviral vectors, and a population of puromycin-resistant transduced cells was selected. Total mRNA was extracted from the selected cell population and CT2 mRNA expression was quantified by qRT-PCR and normalized to expression in the control cells. Data represent the mean mRNA expression level ±SD. b Control cells and CT2 knockdown cells were incubated for 1 h with 100 nM of 3H radiolabeled carnitine with or without 10 μM of unlabeled carnitine. Radioactive counts were recorded. Data represent mean radioactive counts ±SD per 2.5 × 1 05 cells. One-way ANOVA analysis was performed (p = 0.0002) followed by a Student’s t test comparing the control (group 1) and the CT2 knockdown cells (group 2). c Control cells and CT2 knockdown cells were incubated with bleomycin (20 μg/mL) for 72 h. After incubation, cell growth and viability were assessed by Cell Titer-Fluor viability assay. * p value < 0.05 determined by Student’s t test
To determine whether knockdown of CT2 altered uptake of carnitine by OCI-AML2 cells, we incubated cells after target knockdown with radiolabeled 3H carnitine for 1 h. Knockdown of CT2 reduced carnitine uptake compared to control cells (Fig. 2b). Moreover, knockdown of CT2 reduced carnitine uptake in OCI-AML2 cells to background non-specific levels.
The CT2 transporter mediates the uptake of the chemotherapeutic agent bleomycin [15]. Therefore, as an additional approach to assess the impact of CT2 shRNA knockdown on the function of the transporter, we knocked down CT2 in OCI-AML2 cells and treated them with bleomycin. Compared to control cells, knocking down CT2 rendered OCI-AML2 cells resistant to bleomycin, consistent with a functional effect on the transporter (Fig. 2c). Thus, taken together knockdown of CT2 affected the function of the transporter and was sufficient to reduce intracellular carnitine.
Knockdown of CT2 reduces the growth and viability of AML cells
AML cells are highly dependent on FAO [9, 10]. Therefore, we evaluated the effect of CT2 knockdown on the growth and viability of AML cells. Compared to control shRNA, knockdown of CT2 significantly reduced growth and viability of OCI-AML2 and HL60 cells that expressed higher levels of CT2. In contrast, knockdown of CT2 did not affect the growth of TEX cells that expressed low levels of CT2 (Fig. 3).

Knockdown of CT2 decreases proliferation of AML cells. a AML cells were infected with CT2 shRNA (shCT2) or control shRNA sequence (control shRNA) in lentiviral vectors, and a population of puromycin-resistant transduced cells was selected. Cell growth and viability was determined using the trypan blue exclusion assay at increasing times after selection. Data represent mean cell counts ±SD from representative experiments. b AML cells were infected with CT2 shRNA or control shRNA sequence in lentiviral vectors, and a population of puromycin-resistant transduced cells was selected. Total mRNA was extracted from the selected cell population and CT2 mRNA expression was quantified by qRT-PCR and normalized to expression in the control cells. Data represent the mean mRNA expression level ± SD. Of note, the same OCI-AML2 knockdown cells and qRT-PCR figure were used in Figs. 2a and 3b. * p value < 0.05 determined by Student’s t test
Knockdown of CT2 impairs cell cycle progression in AML cells
To assess the mechanism by which CT2 knockdown impaired the growth of AML, we examined apoptosis in OCI-AML2 cells by annexin V/PI staining. No increase in annexin V staining was observed after CT2 knockdown suggesting that the reduction in cell growth was not due to increased apoptosis (Fig. 4a). Next, we examined changes in the cell cycle after CT2 knockdown. Knockdown of CT2 increased in the population of cells in the G1 phase and decreased in the G2 population, suggesting that impaired growth of AML cells after CT2 knockdown may be partly due to cell cycle arrest (Fig. 4b).

Knockdown of CT2 does not induce apoptosis in AML cells but impairs cell cycle progression. a Apoptosis was examined by annexin V and PI staining of control and CT2 knockdown OCI-AML2 cells at 24 and 72 h after selection. Data represent the mean ± SD percent annexin V positive cells. b Cell cycle analysis with PI staining was performed on control and CT2 knockdown OCI-AML2 cells at 24 and 72 h after selection. Data represent mean ± SD percent of cells in each cell phase. * p value < 0.05 determined by Student’s t test
Knockdown of CT2 decreases basal oxygen consumption
Oxidation of fatty acids generates electrons that flow through the respiratory chain to generate ATP and consume oxygen. Therefore, we examined the impact of CT2 knockdown on basal oxygen consumption. OCI-AML2 cells were infected with shRNA targeting CT2 or control sequences. Basal oxygen consumption was measured using the Seahorse XF96 analyzer at 24, 48 and 72 h after selection. Compared to control cells, knockdown of CT2 decreased basal oxygen consumption at all measured time points, consistent with impairment of FAO (Fig. 5a). Of note, glycolysis did not increase in response to the reduction in oxygen consumption (Fig. 5b), consistent with a recent study by Lagadinou et al., demonstrating that AML cells were unable to upregulate glycolysis upon impairment of oxidative phosphorylation [16]. Thus, knockdown of CT2 impaired mitochondrial metabolism.

Knockdown of CT2 lowers oxidative phosphorylation but not glycolytic activity. a Basal oxygen consumption rate of control and CT2 knockdown OCI-AML2 cells were measured with the Seahorse XF96 analyzer at increasing times after selection. Data represent mean percentage ± SD relative to control cells. b Extracellular acidification rate of control and CT2 knockdown OCI-AML2 cells were measured with the Seahorse XF96 analyzer at increasing times after selection. Data represent mean percentage ± SD relative to control cells. * p value < 0.05 determined by Student’s t test
Knockdown of CT2 shows additive effect with chemotherapeutic agents and metabolic regulators
To explore the potential combinatorial effect of CT2 inhibition with chemotherapeutic agents and metabolic regulators, OCI-AML2 cells with CT2 knockdown were treated with increasing concentrations of the front-line AML chemotherapeutic agents, daunorubicin or cytarabine. Cells were also treated with increasing concentrations of tigecycline, an inhibitor of mitochondrial translation [17] or etomoxir, an inhibitor of CPT1 [9]. Knockdown of CT2 produced primarily additive effects compared to control cells (Fig. 6; Table 1).

Knockdown of CT2 shows primarily additive effect with chemotherapeutic agents and metabolic regulators. Control and CT2 knockdown OCI-AML2 cells were incubated with increasing concentrations of daunorubicin, cytarabine, tigecycline or etomoxir for 72 h. After incubation, cell growth and viability were assessed by Cell Titer-Fluor viability assay. Data represent mean ± SD percent viable cells relative to untreated control cells
Discussion
FAO is an important energy pathway that supports cancer cell growth and survival, and AML cells are highly dependent on this pathway [1, 9, 10]. For example, Skrede et al., demonstrated increased rates of FAO, palmitate uptake and oxygen consumption in a rat model of leukemia [10]. Likewise, Samudio et al. studied human AML cell lines and primary AML samples and demonstrated upregulation of FAO and an increased reliance on this pathway for survival [9]. Carnitine is a critical co-factor for FAO, and we evaluated the expression of carnitine transporters in AML and the impact of inhibiting the CT2 plasma membrane carnitine transporter in AML cells.
CT2 expression was higher in a subset of AML cell lines and patient samples compared to normal cells. In addition, carnitine uptake was higher in AML cells with high transporter expression compared to cells with lower transporter expression. Interestingly, AML cells have high levels of non-specific binding or uptake of carnitine. The non-specific binding of carnitine at the plasma membrane may be necessary to facilitate uptake through the transporters in keeping with a previous study [18 ].
Previous studies have examined the impact of blocking FAO at other points in the pathway. For example, carnitine palmitoyltransferase 1 (CPT1) has been evaluated as a novel therapeutic target for the treatment of malignancies including AML [9, 19, 20]. CPT1 is an enzyme located in the outer mitochondrial membrane and catalyzes the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to L-carnitine, thus facilitating the translocation of fatty acids into the mitochondria. According to the Cancer Cell Line Encyclopedia (http://www.broadinstitute.org/ccle/home), AML cells have the highest expression of CPT1 compared to other cancer cells which further denotes the importance of FAO in AML. Targeting FAO with the CPT1 inhibitor etomoxir effectively reduces proliferation of AML cells and increases their sensitivity to apoptotic inducing agents [9].
Alternatively, other studies have examined fatty acid synthase as a therapeutic target in malignancy. Fatty acid synthase is an enzyme involved in de novo fatty acid synthesis which combines acetyl-CoA with malonyl-CoA to primarily produce palmitic acid [21]. Blocking fatty acid synthase not only inhibits fatty acid synthesis but also results in accumulation of malonyl-CoA which is a potent endogenous inhibitor of FAO. In studies of human breast cancer, C75 and cerulenin, two fatty acid synthase inhibitors, induced apoptosis in breast cancer cells and reduced the growth of breast cancer in vivo without toxicity to normal tissues [22, 23]. Likewise, Pizer et al. demonstrated anti-leukemic effect of inhibiting fatty acid synthase using HL60 leukemic cells [24]. However, more studies are needed to confirm efficacy of the strategy in other leukemia cell lines and primary samples.
Indirect methods of inhibiting FAO by blocking the upstream transcription factor PPAR α have also been studied in malignancies such as chronic lymphocytic leukemia (CLL) [25]. PPAR α is a transcription factor that promotes oxidation of fatty acids when activated [26]. Spaner et al. showed an upregulation of FAO in CLL and the PPAR α antagonist MK886 was cytotoxic to CLL cells [25].
To our knowledge, our study is the first to examine the impact of targeting FAO at the level of plasma membrane carnitine transporters. The three known human carnitine transporters OCTN1, OCTN2 and CT2 are structurally similar with 12 α-helical transmembrane domains (TMD), a cytoplasmic N-terminus, a cytoplasmic C-terminus, and glycosylation and phosphorylation sites within the TMDs [5, 7, 27]. They regulate the transport of L-carnitine and other organic cations across cell membrane. Of the three known plasma membrane carnitine transporters (OCTN1, OCTN2, and CT2), CT2 displayed the greatest differential expression between AML and normal hematopoietic cells. While AML samples derived from peripheral blood had higher CT2 expression than normal hematopoietic cells, additional studies are required to determine the expression of CT2 in AML stem cells and AML samples isolated from patients’ marrows as the expression levels may be different in these populations.
Given the increased expression of CT2 in AML, we focused our studies on this transporter and demonstrated that knockdown of CT2 reduced the viability of AML cells and impaired oxidative phosphorylation. Thus, CT2 may be a novel therapeutic target for AML. Supporting utility of CT2 as a novel therapeutic target for AML, CT2 is primarily expressed in the testis, kidney, and hematopoietic cells [6, 7]. Potentially, this restricted expression could result in less toxicity from CT2 inhibition and might provide a wider therapeutic window than blocking other FAO pathway components such as CPT1, fatty acid synthase, or PPAR α. However, in vivo studies would be necessary to fully understand the toxicity of CT2 inhibition. In addition, understanding the phenotype of CT2 knockout mice could also help define the toxicity of such inhibitors.
In our studies, knockdown of CT2 reduced basal oxygen consumption without a corresponding increase in glycolysis. The lack of compensation by upregulating glycolysis is consistent with a recent study by Lagadinou et al [16]. In this paper, AML cells and stem cells were unable to upregulate glycolysis in response to inhibiting oxidative phosphorylation with oligomycin and FCCP. The molecular basis for why AML cells are unable to upregulate glycolysis in response to inhibition of oxidative phosphorylation is unknown, but may explain the heightened sensitivity of AML cells to strategies that block mitochondrial metabolism.
In summary, we demonstrated that AML cell lines and primary AML patient samples had increased expression of the carnitine transporter CT2 and down regulating this transporter reduced proliferation of these malignant cells. The loss of cell viability was associated with decreased oxidative phosphorylation without a compensatory increase in glycolysis. Thus, our work highlights the potential of CT2 as a new therapeutic target for a subset of AML.
References
Carracedo A, Cantley LC, Pandolfi PP (2013) Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 13:227–232
Eaton S, Bartlett K, Pourfarzam M (1996) Mammalian mitochondrial beta-oxidation. Biochem J 320(Pt 2):345–357
Flanagan JL, Simmons PA, Vehige J, Willcox MD, Garrett Q (2010) Role of carnitine in disease. Nutr Metab 7:30
Vaz FM, Wanders RJ (2002) Carnitine biosynthesis in mammals. Biochem J 361:417–429
Koepsell H, Lips K, Volk C (2007) Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res 24:1227–1251
Gong S, Lu X, Xu Y, Swiderski CF, Jordan CT, Moscow JA (2002) Identification of OCT6 as a novel organic cation transporter preferentially expressed in hematopoietic cells and leukemias. Exp Hematol 30:1162–1169
Koepsell H (2013) The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Aspects Med 34:413–435
Tamai I (2013) Pharmacological and pathophysiological roles of carnitine/organic cation transporters (OCTNs: SLC22A4, SLC22A5 and Slc22a21). Biopharm Drug Dispos 34:29–44
Samudio I, Harmancey R, Fiegl M et al (2010) Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Investig 120:142–156
Skrede S, Iversen PO (1995) Enhanced oxygen consumption and fatty acid metabolism in rat bone marrow with acute promyelocytic leukaemia. Leuk Res 19:463–467
Beheshti Zavareh R, Lau KS, Hurren R et al (2008) Inhibition of the sodium/potassium ATPase impairs N-glycan expression and function. Cancer Res 68:6688–6697
Schimmer AD, Thomas MP, Hurren R et al (2006) Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor-family death receptors. Cancer Res 66:2367–2375
Moffat J, Grueneberg DA, Yang X et al (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124:1283–1298
Xu GW, Ali M, Wood TE et al (2010) The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 115:2251–2259
Aouida M, Poulin R, Ramotar D (2010) The human carnitine transporter SLC22A16 mediates high affinity uptake of the anticancer polyamine analogue bleomycin-A5. J Biol Chem 285:6275–6284
Lagadinou ED, Sach A, Callahan K et al (2013) BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12:329–341
Skrtic M, Sriskanthadevan S, Jhas B et al (2011) Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20:674–688
Gustafson B, Ransnas LA (1997) Regulation of carnitine binding to plasma membranes by an ATP-dependent mechanism. Biochem Biophys Res Commun 231:249–253
Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M (2011) Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta 1807:726–734
Tirado-Velez JM, Joumady I, Saez-Benito A, Cozar-Castellano I, Perdomo G (2012) Inhibition of fatty acid metabolism reduces human myeloma cells proliferation. PLoS ONE 7:e46484
Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr (2013) Cellular fatty acid metabolism and cancer. Cell Metab 18:153–161
Pizer ES, Thupari J, Han WF et al (2000) Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res 60:213–218
Thupari JN, Pinn ML, Kuhajda FP (2001) Fatty acid synthase inhibition in human breast cancer cells leads to malonyl-CoA-induced inhibition of fatty acid oxidation and cytotoxicity. Biochemical and biophysical research communications 285:217–223
Pizer ES, Wood FD, Pasternack GR, Kuhajda FP (1996) Fatty acid synthase (FAS): a target for cytotoxic antimetabolites in HL60 promyelocytic leukemia cells. Cancer Res 56:745–751
Spaner DE, Lee E, Shi Y et al (2013) PPAR-alpha is a therapeutic target for chronic lymphocytic leukemia. Leukemia 27:1090–1099
Ferre P (2004) The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes 53(Suppl 1):S43–S50
Enomoto A, Wempe MF, Tsuchida H et al (2002) Molecular identification of a novel carnitine transporter specific to human testis. Insights into the mechanism of carnitine recognition. J Biol Chem 277:36262–36271
Acknowledgments
We thank Jill Flewelling for administrative assistance. This work was supported by the Canadian Stem Cell Network, the Ontario Ministry of Research and Innovation, the Princess Margaret Cancer Centre Foundation, and the Ministry of Long Term Health and Planning in the Province of Ontario, and the National Institutes of Health.
Conflict of interest
The authors declare that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, Y., Hurren, R., MacLean, N. et al. Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis 20, 1099–1108 (2015). https://doi.org/10.1007/s10495-015-1137-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-015-1137-x