
Abstract
Establishing slash pine plantations is the primary method for restoring sandification land in the Houtian area of South China. However, the microbial variation pattern with increasing stand age remains unclear. In this study, we investigated microbial community structure and function in bare sandy land and four stand age gradients, exploring ecological processes that determine their assembly. We did not observe a significant increase in the absolute abundance of bacteria or fungi with stand age. Bacterial communities were dominated by Chloroflexi, Actinobacteria, Proteobacteria, and Acidobacteria; the relative abundance of Chloroflexi significantly declined while Proteobacteria and Acidobacteria significantly increased with stand age. Fungal communities showed succession at the genus level, with Pisolithus most abundant in soils of younger stands (1- and 6-year-old). Turnover of fungal communities was primarily driven by stochastic processes; both deterministic and stochastic processes influenced the assembly of bacterial communities, with the relative importance of stochastic processes gradually increasing with stand age. Bacterial and fungal communities showed the strongest correlation with the diameter at breast height, followed by soil available phosphorus and water content. Notably, there was a significant increase in the relative abundance of functional groups involved in nitrogen fixation and uptake as stand age increased. Overall, this study highlights the important effects of slash pine stand age on microbial communities in sandy lands and suggests attention to the nitrogen and phosphorus requirements of slash pine plantations in the later stages of sandy management.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
China is among the nations facing severe challenges to its ecological security, including soil erosion, rock and land desertification. By 2022, the province of Jiangxi in southern China has 64,000 hectares of sandy land (http://ly.jiangxi.gov.cn). The sandy land in Jiangxi Province is mainly distributed along the south bank of the Yangtze River, around Poyang Lake, and along five major rivers in the province, including the Ganjiang River, the largest tributary of Poyang Lake. During the 1980s, the Houtian region, which is located downstream of the Ganjiang River, was subject to heavy sandification. However, this was successfully mitigated through the implementation of slash pine (Pinus elliottii) plantations. Presently, areas undergoing significant sandification in Jiangxi Province continue to be effectively managed through the establishment of slash pine plantations.
Slash pine, a species native to the southeastern United States, has been used to restore sandy land in Jiangxi province (Long et al., 2021). However, most studies on slash pine plantations on degraded land have focused on the above-ground components, neglecting soil microorganisms. Soil microorganisms play a crucial role in plant-soil interactions (Trivedi et al., 2020). The growth of plants and their capacity to withstand stresses, both biotic and abiotic, are influenced by changes in soil microbial communities (Allsup et al., 2023; Coban et al., 2022; Pennisi & Cornwall, 2020; Zhao et al., 2020). Therefore, it is imperative to understand the dynamic pattern of the microbial community during plant development to provide guidance for management (Gao et al., 2020; Zhao et al., 2020).
Previous studies on microbial communities in slash pine plantations have either used relatively old methods (Izumi et al., 2008; Wu et al., 2015) or focused on a single stand age (Long et al., 2021; Ning et al., 2019a, 2021; Zhou et al., 2017), and therefore, there is still a limited understanding of the dynamics of microbial communities during the growth stages of slash pine. Recent studies have demonstrated a significant influence of stand age on the structure of microbial communities (Liu et al., 2021b; Wang et al., 2023b). Changes in microbial communities can reflect the “health” of the soil system (Fierer et al., 2021), particularly fungal communities, may indirectly contribute to plantation degradation, and consequently, impede effective management of sandy land. For instance, a decrease in the relative abundance of symbiotrophs, predominantly ectomycorrhizal fungi (EMF), and an increase in the relative abundance of saprotrophs with increasing stand age were found to be associated with degradation of Pinus sylvestris plantations in the Mu Us Desert, Northern China (Zhao et al., 2020). Furthermore, plant growth may be associated with EMF communities as well as with certain EMF exploration types (Anthony et al., 2022). EMF exploration types are classified according to the general morphological characteristics of their colonizing root tips and emanating hyphae as contact, short-distance, medium-distance smooth, medium-distance fringe, and long-distance types (Agerer, 2001), To some extent, the abundance of these types can reflect the ecological niche differentiation of EMF (Jörgensen et al., 2023; Wang et al., 2023a). Previous studies have demonstrated that Rhizopogon (long-distance) is the predominant EMF in slash pine seedlings and 10-year-old stands (Long et al., 2021; Ning et al., 2021). However, Rhizopogon is not common in 30-year-old slash pine plantations and is replaced by EMF belonging to the Thelephoraceae and Russulaceae (Ning et al., 2019a). Thus, the fungal partners of slash pine may undergo succession as the stand ages.
Enhancing our understanding of the mechanisms governing the assembly of microbial communities will enable us to effectively harness the beneficial and indirect impacts of these communities on plants (Gao et al., 2020). Some recent studies on coniferous forests have demonstrated that the assembly of microbial communities is determined by both deterministic and stochastic processes, with stochastic processes being dominant (Wang et al., 2021, 2023b). Other studies on soil microbial communities in subtropical forests of varying stand ages have revealed that community assembly mechanisms differ among different stages, with deterministic processes governing early-stage community assembly and stochastic processes dominating as stand age increases or during later stages of succession (Gao et al., 2015; Liu et al., 2021b). However, the contribution of deterministic or stochastic processes to microbial communities in slash pine plantations with different stand ages growing on sandy land is currently unclear.
We therefore selected the MaAnLing sampling site in the Houtian region, which has a gradient of four stand ages (1, 6, 11, 16-year-old) of slash pine. In addition, because the sampling site is a national ecological benefit forest, it is virtually unaffected by anthropogenic activities such as fertilization or tapping. We collected soil samples from the forest stands, and conducted Illumina Miseq sequencing and quantitative real-time PCR (qPCR) on the 16S rRNA gene and ITS region sequence to investigate the changes in microbial community structure and absolute abundance with the increasing age of slash pine trees on sandy land. In addition, we analyzed soil properties and recorded stand information from slash pine plantations. The aims of this study were: (1) To reveal the succession pattern of microbial community composition and function with increasing age of slash pine plantations; (2) to identify the mechanisms underlying the assembly of soil microbial communities in slash pine plantations on sandy land; and (3) to elucidate the principal factors affecting microbial communities in slash pine plantations on sandy land.
Materials and Methods
Sampling Site Description and Experimental Design
MaAnLing (N28°24′10.79″, E115°47′24.00″) has four age groups that were established in 2003, 2008, 2013, and 2018, respectively. The climate of this region has been described previously (Long et al., 2021). In August 2019, a total of five sampling gradients were established, comprising bare sandy land (BSL), stand age one year (YR01), stand age six years (YR06), stand age eleven years (YR11), and stand age sixteen years (YR16) (Fig. 1). Each sampling gradient was set with three quadrats. For stands older than six years, three 20 m × 20 m quadrats were set for each gradient, with each quadrat more than 50 m apart. Due to the limited planting area, three 10 m × 10 m quadrats were set for YR01, with each quadrat more than 20 m apart. For BSL, three 5 m × 5 m quadrats were set, with each quadrat more than 20 m apart. After removing the litter layer, five bulk soil samples (four samples approximately 1 m from the four corners of the sample square and one sample at the center of the sample square) were collected at a depth of 0–20 cm in each quadrat using a sterile shovel, and five samples in one sample square then mixed into one sample. Finally, a total of fifteen soil samples were collected. Subsamples for the soil chemical and physical and microbiological analysis were processed as previously described (Long et al., 2021). For stand information, the mean diameter at breast height (MD, measured at 1.3 m above ground, and the value of YR01 was substituted by the ground diameter) and mean height (MH) were recorded for at least eight trees within each quadrat (Table 1).

The sampling site, MaAnLing, is situated in the Houtian region of the lower Ganjiang River (A). A total of 15 sample quadrats were established at the site, encompassing bare sandy land (BSL) and four slash pine stand age groups (YR01, YR06, YR11, and YR16) (B)
Soil Physical and Chemical Property Analysis
Soil water content (SWC), pH, total organic carbon (TOC) content, total nitrogen (TN) content, and available phosphorus (AP) content were measured by previous methods (Long et al., 2021; Tai et al., 2013). The total phosphorus (TP) content was measured using the Mo-Sb anti-colorimetric analysis method (Zhao et al., 2020). The soil C/N ratio was calculated as TOC divided by TN.
Illumina Sequencing and Data Processing
The extraction of DNA from each soil sample is subject to the manufacturer's instructions for the E.Z.N.A.® soil DNA Kit (Omega Bio-tek). The bacterial 16S rRNA V3-V4 region, and fungal ITS1 region were amplified with primers 338F/806R (Gao et al., 2022), and ITS1F/ITS2R (Hu et al., 2021). The purified amplicons (PCR amplification system and conditions, and purification process please see Tables S1 and S2) were then run on the Illumina Miseq platform (Shanghai Majorbio Bio-pharm Technology Co., Ltd.). After merging and removing barcodes and primers, the sequencing company provided clean amplicon data for subsequent data processing. The data processing was done using the EasyAmplicon v1.14 pipeline (https://github.com/YongxinLiu/EasyAmplicon) (Liu et al., 2023). In brief, the clean amplicons were denoised into amplicon sequence variants (ASVs) by employing -unosie3 in USEARCH. The ASVs were removed when the total number of sequences present in all the samples was below 20. Representative sequences for each ASV of bacteria and fungi were assigned based on the best matches in the SILVA database (v123, https://www.arb-silva.de/) (Quast et al., 2013) and the UNITE database (v8.3, https://unite.ut.ee/) (Nilsson et al., 2019), respectively.
Bacterial ASVs were functionally annotated using the FAPROTAX database (Louca et al., 2016). The functional annotation of fungal ASVs was performed on the FUNGuild platform (Nguyen et al., 2016). Only ASVs that have confidence rankings of 'highly probable' and 'probable' from FUNGuild were accepted for subsequent analysis. Unassigned ASVs and combined trophic modes were integrated as other fungi, and combined guilds were integrated as other guilds, as previously described (Long et al., 2021; Zhao et al., 2020). For the determination of distance exploratory EMF: long-distance, medium-distance, and short-distance exploration types, we make qualitative decisions based on the previous literature (Table S3) (Agerer, 2001; Peay et al., 2011; Wang et al., 2023a).
Real-Time PCR (qPCR)
The qPCR technique was employed to determine the absolute abundance of bacterial 16S rRNA genes and fungal ITS region sequences, as previously reported (Hu et al., 2021). The primer pairs used in qPCR amplification were identical to those used in Illumina sequencing, i.e., 338F-806R for 16S rRNA and ITS1F-ITS2R for the ITS region, respectively. The standard curves were generated from a clone with 16S rRNA and ITS amplicon inserts by utilizing tenfold serial dilutions of the plasmid. The amplifications were performed using the ABI7300 Real-Time PCR System (Applied Biosystems). Assays were performed in triplicate, each 20 μl reaction contained: 10.0 μl ChamQ SYBR Color qPCR Master Mix (2×, Vazyme Biotech Co., Ltd.), 0.8 μl of each primer (5 μM), 0.4 μl ROX Reference Dye 1 (50×), 2.0 μl template DNA, and 6 μl ddH2O. Thermocycling consisted of an initial denaturation at 95 °C for 3 min, followed by 40 cycles of 95 °C for 5 s, annealing temperature of 58 °C for 16S rRNA and 62 °C for ITS for 30 s, extension at 72 °C for 1 min, and finally 10 °C until terminated.
Statistical Analysis
All statistical analyses were performed using R (version 4.1.3, https://www.r-project.org/). Prior to calculating the soil microbial diversity, we resampled the ASV tables from each sample to obtain an identical number of sequences, namely 25,000 for bacteria and 25,000 for fungi. The alpha diversity metrics, including the Shannon diversity index and Chao1 richness estimator, were calculated for each sample. The significance of differences in soil physicochemical properties, alpha diversity, and functional groups of microbial communities was assessed through a one-way analysis of variance (ANOVA, with ‘Tukey HSD’ correction). The beta diversity was analyzed through the principal-coordinate analysis (PCoA) based on Bray–Curtis distance metrics using the microeco R package (Liu et al., 2021a). The significance of the distinction between different stand ages was tested by the ‘ADONIS’ function with 999 permutations. Hierarchical clustering of different taxonomy levels based on average agglomerative clustering method (Unweighted Pair Group Method with Arithmetic Mean, UPGMA) using the cluster R package (Maechler, 2019).
To estimate the contribution of stochastic processes in bacterial and fungal communities, Sloan’s neutral community model (NCM) was applied (Sloan et al., 2006). The migration rate (m) calculation method and R code for the neutral model were provided by (Gao et al., 2022). Further, null model (NM) was used to assess the stochasticity of microbial community assembly, we computed the beta Nearest Taxon Index (βNTI) and Bray–Curtis-based Raup-Crick Index (RCI) using previous methods (Gao et al., 2020; Stegen et al., 2013; Wang et al., 2023b; Zhou & Ning, 2017).
Prior to conducting the correlation analysis of environmental factors (stand information and soil properties) and microbial communities, the ASV data underwent transformation using the ‘Hellinger’ method, while the microbial diversity indices, absolute abundance, and environmental factors data underwent logarithmic conversion [namely, x = log (e, n + 1), where n is the value of the environmental factor]. The Spearman’s rank correlation analysis was employed to determine the correlations between microbial diversity indices, absolute abundance, functional group, and environmental factors, using the magrittr (https://github.com/tidyverse/magrittr) and corrplot R packages (Friendly, 2002).
We investigated the association between environmental factors (Euclidean distances) and microbial community structure (Bray–Curtis distances) using the Mantel test method with 999 permutations. Further, the distance-based redundancy analysis (db-RDA) was used to analyze the effects of environmental factors on microbial communities (ASV levels), and variation partitioning analysis (VPA) was conducted to clarify the relative contribution of stand information and soil properties on microbial community turnover using the functions ‘capscale’ and ‘varpart’ of the vegan R package (Oksanen et al., 2007), respectively. The db-RDA used variance inflation factor (VIF) analysis to remove colinear factors of environmental factors until the VIF < 10, and before the VPA, environmental factors were subjected to forward selection until the p value was < 0.05 (Gao et al., 2022).
Data Deposit
The clean amplicon data were submitted to the NCBI Sequence Reading Archive (SRA) database with SRA accession numbers PRJNA979784 for 16S rDNA sequences and PRJNA979787 for ITS region sequences.
Results
Stand Information and Soil Physicochemical Properties at Different Stand Ages
The MD and MH increased significantly with increasing stand age (Table 1). The characteristics of 15 soil samples were analyzed regarding pH, SWC, TN, TOC, AP, and TP (Table 1). Only SWC showed significant variation among stand ages, with SWC significantly higher in YR16 than in BSL, YR01, and YR06. With increasing stand age, the trends in TOC, TN, and C/N were increasing, but they were not significant. The decline in soil AP after 11-year plantations was also insignificant.
The Soil Microbial Alpha Diversity Metrics and Community Structure in Different Slash Pine Stand Ages
As the age of slash pine stands increased, the Shannon index and Chao1 index of both bacterial and fungal communities showed an increasing trend (Fig. 2). However, only the fungal Shannon index was significantly different, namely, YR16 was significantly higher than YR06 and YR11 (Fig. 2D).

The alpha diversity of the microbial community in different stand ages. The Chao1 (A) and Shannon index (C) of bacteria, and the Chao1 (B) and Shannon index (D) of fungi. Different lowercase letters represent statistically significant differences between values. (ANOVA, with ‘Tukey HSD’ correction, p < 0.05)
For bacterial communities, Chloroflexi, Actinobacteria, Proteobacteria, and Acidobacteria were the dominant phyla (Fig. 3A). As the age of the stand increased, the relative abundance of Chloroflexi declined significantly at YR11 and YR16 (p < 0.05), whereas the relative abundance of Proteobacteria and Acidobacteria increased significantly at YR11 and YR16 (p < 0.05) (Fig. S1A). At the genus level, the unassigned taxa belonging to phylum Chloroflexi showed dominance (Fig. S1B). Besides, among the genera with a mean relative abundance greater than 1%, Burkholderia, Bradyrhizobium, Variibacter, Candidatus_Solibacter, and Mycobacterium showed significant variations in relative abundance (Fig. S1C). Clustering analysis showed that both bacterial phylum level communities (Fig. 3A) and bacterial genus level communities (Fig. S1B) clustered into two clusters of younger stands (YR01, YR06) and older stands (YR11, YR16) The results of PCoA analysis at the ASV level were similar to those of cluster analysis, with ADONIS R2 = 0.61, p < 0.001 (Fig. 3B).

Hierarchical clustering analysis (UPGMA) of bacterial (A) and fungal (C) communities at the phylum level, and PCoA of bacterial (B) and fungal (D) communities at the ASV level based on Bray–Curtis distance
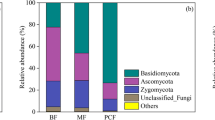
For fungal communities, the Basidiomycota and Ascomycota were the dominant phyla (Fig. 3C). The relative abundance of all phyla did not differ significantly with increasing stand age (Fig. S1D). At the genus level, cluster analysis showed that samples of different stand ages clustered into three clusters, one for BSL, one for younger stands (YR01 and YR06), and one for older stands (11-year stands, 16-year stands) (Fig. S1E). Furthermore, among the genera with a mean relative abundance greater than 1%, Pisolithus and Tomentella showed significant variations in relative abundance. Pisolithus was predominant in YR01 and YR06, and significantly higher in relative abundance than YR11 and YR16 (p < 0.05), while Tomentella had the highest relative abundance in YR16 (p < 0.05) (Fig. S1F). Cluster analysis at the fungal phylum level based on Bray–Curtis distances also showed no clear pattern across forest age samples (Fig. 3C). However, PCoA analysis at the ASV level showed that fungal communities of different forest ages clustered together, with ADONIS R2 = 0.55, p < 0.001 (Fig. 3D).
Assembly Processes of Bacterial and Fungal Communities
The NCM revealed that the assembly of bacterial communities was driven by a combination of deterministic and stochastic processes, with deterministic processes being predominant throughout plantation chronosequence (R2 = 0.2155, m = 0.0923, Fig. 4A). And deterministic processes dominated the bacterial community with succession from BSL to YR16 (R2 = 0.232–0.362, Fig. S2A). In contrast, the fungal communities did not fit the NCM either throughout plantation chronosequence (R2 = − 0.490, m = 0.0005, Fig. 4B) or by each gradient (R2 < 0, Fig. S2B).

Fit of Sloan's neutral community model (NCM) displaying predicted occurrence frequencies versus relative abundance of bacterial (A) and fungal (B) ASVs. The solid blue lines represent the optimal fit to the NCM, while the dashed blue lines indicate 95% confidence intervals surrounding model predictions. ASVs exhibiting greater or lesser frequency than anticipated by the NCM are depicted in varying colors. R2 and m value denote model goodness–of–fit and estimated migration rate, respectively. Negative R2-values can be observed when there is no fit to the model. Distribution of bacterial (C) and fungal (D) beta nearest taxon index (βNTI). The fraction of turnover in the assembly of soil bacterial and fungal communities, as governed primarily by deterministic, homogeneous (HS), variable selection (VS) and stochastic processes (ST)
The NM results suggest that both stochastic (51.4%) and deterministic processes (48.6%) dominate the bacterial community assembly throughout plantation chronosequence (Fig. 4C), while the fungal community assembly was dominated by stochastic processes (84.76%, Fig. 4D). With increasing stand age, the bacterial community assembly shifted from being primarily determined by deterministic processes to being predominantly influenced by stochastic processes (Fig. S3A), while the fungal community assembly was mainly governed by stochastic processes (drift dominated) in all stages (Fig. S3B).
The Soil Functional Groups of Microbial Communities at Different Stand Ages
For bacteria, a total of 52 functional groups were obtained from the FAPROTAX database (Fig. 5A), and nine of these are significantly different (Fig. S2). Among the functional groups with a mean relative abundance greater than 1%, the relative abundance of nitrogen fixation taxa, primarily Bradyrhizobium, exhibited an increase with increasing stand age (Fig. 5B).

The heatmap displays the functional groups of bacteria, with row data z-score normalized. Functional groups with an average abundance greater than 1% are highlighted in red boxes (A). Relative abundance of fungal trophic modes and exploration types. (B). Boxes indicate 25–75 percentiles, vertical lines indicate 10–90 percentiles, horizontal lines inside the boxes are median values. Different lowercase letters represent statistically significant differences between values. (ANOVA, with ‘Tukey HSD’ correction, p < 0.05). (C). Variations of bacterial genera belonging to nitrogen fixation and EMF exploration types vary across different stand ages
For the fungal functional group (Fig. 5C, Fig. S5), the EMF is the most abundant in each slash pine plantation. Moreover, the relative abundance of long-distance EMF decreased with increasing stand age, but the difference was not significant. The relative abundance of medium-distance EMF increased with increasing stand age and was highest at YR16 (p < 0.05), while the relative abundance of short-distance EMF was highest at YR11 (p < 0.05) (Fig. 5B).
Environmental Factors Affecting Microbial Community Diversity, Composition, and Function
The Mantel test results (Fig. 6A, Fig. S6A) indicate that MD, MH, and AP significantly influence the microbial community composition. Additionally, SWC also has a significant impact on bacterial community composition.

A Pairwise comparisons of environmental factors are presented with a color gradient indicating Pearson's correlation coefficient. The taxonomic community composition (after Hellinger transformation) was assessed for its relationship to each environmental factor using Mantel tests. Edge width corresponds to the Mantel’s r statistic for the corresponding distance correlations, and edge color indicates the statistical significance based on 999 permutations. *p < 0.05; **p < 0.01. B Spearman's rank correlation analysis between microbial diversity indices, absolute abundance, nitrogen fixation group, ectomycorrhizal (EMF) exploration types, and environmental factors. MD mean diameter at breast height, MH mean height, SWC soil water content, TN soil total nitrogen, TOC soil total organic carbon, AP soil available phosphorus, TP soil total phosphorus, TK soil total potassium, C/N the TOC content division by TN content
The results of the db-RDA analysis indicated that SWC, AP, and MD were the main factors significantly associated with bacterial and fungal communities (Fig. S6B). VPA analysis revealed that plant (MD) and soil factors (SWC, AP) explained 4.8 and 9.1%, respectively, of bacterial community turnover, while the majority of the variation in community turnover (69.2%) remained unexplained (Fig. S6C). For fungal community turnover, plants (MD) and soils (SWC, AP) explained 7.5 and 4.5%, respectively; however, more variation in fungal community turnover (82.2%) could not be explained compared to bacterial communities.
The results of Spearman's rank correlation analysis (Fig. 6B) also indicated significant correlations between microbial diversity index, absolute abundance, and functional taxa (with a focus on nitrogen-fixing taxa, and EMF) with plant aboveground traits (MD and MH), SWC, and AP.
Discussion
The restoration of degraded land is a long process (Strickland et al., 2017). Indeed, in this study, only SWC exhibited a significant increase with increasing stand age, whereas other soil physicochemical properties did not show statistically significant changes. These findings indicate that the recovery process of sandy land in this study area is slow. Notably, AP decreased as the stand age increased, consistent with the findings of a previous study on slash pine of varying stand ages (Wu et al., 2015). However, our previous research indicated that planting slash pine resulted in an increase in soil AP compared with that in BSL (Long et al., 2021), possibly because our previous study only focused on a single stand age. In both this and our prior research, the changes in AP with increasing stand age did not reach significant levels.
Changes in Microbial Abundance and Diversity
Understanding the dynamic processes of the microbial community with increasing stand age can help us to guide plantation management (Wang et al., 2022a; Zhao et al., 2020). In addition, some microbes can serve as bioindicators (Fierer et al., 2021). For example, the ratio of absolute abundance of bacteria and fungi (B/F) can reflect the “health” of the soil system (Fierer et al., 2021). Changes in the structure of microbial communities can reflect their ecological strategy (Fierer et al., 2007), and be consistent with changes in above-ground plants during different stages of restoration (Bi et al., 2021). In this study, the absolute abundance of bacteria and fungi exhibited a positive correlation with increasing stand age, whereas the B/F ratio displayed a negative trend. Although none of these trends reached the level of statistical significance, a reduced B/F ratio means more sustainable soil systems (Fierer et al., 2021). Similarly, we found that the fungal Shannon index was significantly higher in 16-year-old plantations than in younger ones, suggesting that the ability of the fungal community to withstand external disturbances increases with increasing stand age (Bi et al., 2021).
Bacterial Community Structure and Function
In this study, comparison of relative abundance values showed that Chloroflexi was the dominant bacterial phylum in stands of all ages (Fig. 3A), which verified the results of our earlier study (Long et al., 2021). As monoderms, the Chloroflexi are more drought tolerant than are other phyla, such as the diderms Proteobacteria and Acidobacteria (Xu & Coleman-Derr, 2019). Indeed, with increasing stand age, SWC increased and the relative abundance of Chloroflexi decreased significantly (Fig. S1A). The relative abundance of Actinobacteria did not change significantly with increasing stand age (Fig. S1A), although Actinobacteria are also drought-tolerant monoderms (Xu & Coleman-Derr, 2019). This result suggests that the relative abundance of Actinobacteria does not change in a predictable manner with changes in soil nutrient availability (Fierer et al., 2007). In contrast, as diderm bacteria (Xu & Coleman-Derr, 2019), Proteobacteria and Acidobacteria showed significant increases in their relative abundance with increasing stand age (Fig. S1A). Proteobacteria has been classified as a fast-growing copiotrophic group (Fierer et al., 2007). The accumulation of TOC can stimulate a rapid increase in Proteobacteria, as found in this study and previous studies (Jiang et al., 2021; Wang et al., 2022b).
The FAPROTAX results revealed a significant increase in the functional group of bacteria related to nitrogen fixation with increasing stand age (Fig. S4), and Bradyrhizobium exhibited the highest relative abundance within this group (Fig. 5B). A previous study (Wu et al., 2015) demonstrated a significant decrease in TN with increasing age of slash pine plantations in southern China, suggesting that the nitrogen demand of slash pine on sandy land may increase with stand age, although the changes in TN were not significant in this study (Table 1). Besides, it is noteworthy that the relative abundance of Bradyrhizobium is usually significantly and negatively correlated with soil Olsen P content (a measure of plant-available phosphorus) (Hermans et al., 2017). In this study, the relative abundance of Bradyrhizobium increased with stand age and was significantly and negatively correlated with soil AP content (Fig. 6B). This again suggests that Bradyrhizobium has the potential to serve as a biologically relevant indicator of important soil variables such as AP and the overall effectiveness of land management (Hermans et al., 2017).
Fungal Community Structure and Guilds
For the fungal community, Ascomycota and Basidiomycota were the dominant groups at the phylum level, consistent with the results of other studies on Pinaceae plantations in degraded or desertified lands (Bi et al., 2021, 2022; Liu et al., 2019a, 2019b; Long et al., 2021; Wang et al., 2019b, 2022b; Zhao et al., 2020). However, changes in their abundance did not show a regular trend with increasing stand age (Fig. 3C). At the genus level, the fungal community structure underwent succession as the stand age increased (Fig. S1E), which is a trend that has also been detected in other studies on Pinaceae plantations (Koizumi et al., 2018; Kyaschenko et al., 2017). This succession was mainly reflected by EMF, which showed the highest relative abundance of all stand-age functional guilds (Fig. 5C). The results of EMF explorer analysis indicated a tendency towards an increase in the proportion of short-distance and medium-distance EMF and a decrease in the proportion of long-distance EMF as the stand age increased (Fig. 5B). On the one hand, short-distance and medium-distance smooth EMF have been proposed to promote rapid uptake of mobile nitrogen (Hobbie & Agerer, 2010; Jörgensen et al., 2023). Notably, the relative abundance of Tomentella (Medium-distance smooth) which has been described as nitrophilic (Jörgensen et al., 2023; Kranabetter et al., 2009), increased significantly with stand age in this study (Fig. S1F). This is consistent with the results of previous studies that found that Tomentella was the most abundant EMF in mature slash pine and other Pinus forests (Ning et al., 2019a; Wang et al., 2021) and again, implies an increased demand for nitrogen in the later stages of growth of slash pine plantations.
On the other hand, previous studies have highlighted the significance of Rhizopogon (long-distance EMF) in the initial colonization of pine trees (Koizumi et al., 2018; Long et al., 2021; Ning et al., 2019b; Policelli et al., 2019; Zhao et al., 2020). Rhizopogon was found to be dominant in slash pine seedlings (Ning et al., 2021) and 10-year-old plantations (Long et al., 2021). Similarly, in this study, Rhizopogon was distributed in all stand ages except YR01, and its relative abundance gradually increased with increasing stand age (Fig. S1F). However, Pisolithus (long-distance EMF) dominated the soil samples of younger stands (YR01, YR06, Fig. S1F), highlighting its significance during the early establishment of slash pine in southern China (Hua et al., 1995), especially in sandy land (Sebastiana et al., 2020). This finding is consistent with those of an earlier survey on ectomycorrhizal resources in southern pine forests of China (including slash pine), which concluded Pisolithus tincorius was the most important pioneer mycorrhizal fungus (Hua et al., 1995). A recent study showed that Pisolithus influences the availability of soil phosphorus (Stuart et al., 2022), suggesting that Pisolithus may also be able to help plants access phosphorus.
Assembly of the Microbial Community in Stands of Different Ages
Understanding the mechanism of microbial community assembly is one of the central topics in microbial ecology (Zhou & Ning, 2017). In this study, bacterial community assembly was dominated by both deterministic and stochastic processes, and deterministic processes dominated in the early stage of stand age succession. This is consistent with the results of a study on the early stages of restoration after deforestation (Zhang et al., 2016), and emphasizes the environmental constraints of degraded land (Huo et al., 2023; Wang et al., 2023b; Zhang et al., 2016). In addition, the assembly of bacterial communities shifted from being dominated by deterministic to stochastic processes with increasing forest age (Figs. S2A and S3A), suggesting that the ecological environment of sandy land gradually recovered with increasing forest age.
In contrast, although the fungal community assembly did not fit the NCM, similar to the results of another coniferous forest study (Kang et al., 2022), the extremely low m-values suggest that fungal community assembly was strongly dispersal-limited (Fig. 4B, Fig. S2B), i.e., governed by a stochastic process, consistent with the results of the NM in this study (Fig. S3B). This result is similar to the findings of previous studies on fungal communities in secondary forests and Pinus plantations (Huo et al., 2023; Wang et al., 2021).
Driving Factors of Bacterial and Fungal Community
SWC is an important parameter for predicting bacterial community structure (Delgado-Baquerizo et al., 2018; Fierer, 2017). Accordingly, we found that SWC significantly affected bacterial community composition, diversity, and B/F in this study (Fig. 6 and Fig. S6). These results and those of another study (Xu & Coleman-Derr, 2019) highlight the sensitivity of the soil bacterial community to variations in SWC in sandy land in Southern China. Besides, AP significantly affected both bacterial and fungal communities and functional groups (Fig. 6 and Fig. S6), indicating the important effects of AP on the soil microbial community during the development of pine forest (Bi et al., 2021; Wu et al., 2021). We found that MD was the most important factor predicting variations in microbial communities, suggesting that stand age has a significant effect on microbial communities (Wang et al., 2023b), and MD is a good indicator that is easier to measure.
A previous study detected a significant negative correlation between C/N and the soil microbial diversity of slash pine plantations at different stand ages (Wu et al., 2015), but no such correlation was detected in our study (Fig. 6B), possibly because of the low soil C and N contents in the soil layer of the sandy land studied here. The accumulation of slash pine litter with a high C/N ratio increases the soil C/N ratio with increasing stand age (Wu et al., 2015), consistent with the results of this study. However, a high C/N ratio may lead to nitrogen deficiency (Jiang et al., 2021), and thus exert selective pressure on microorganisms (Wang et al., 2019a). This may also be the reason why the abundance of nitrogen-fixing bacteria increased with increasing stand age in this study (Fig. 5B).
Conclusion
The results of our study show that the sandy land ecosystem gradually improves as the stand age of the slash pine plantation increases, but the process is slow. At each stage, the dominant phylum in the bacterial community was Chloroflexi, while fungal community succession occurred at the genus level, with Pisolithus most abundant in soils of younger stands (YR01 and YR06). This emphasizes the importance of Pisolithus in the early establishment of slash pine in Southern China. In addition, the mechanisms of bacterial and fungal community assembly differed, with bacterial community assembly shifting from being governed by deterministic to stochastic processes with increasing stand age, while fungal communities were always governed by stochastic processes. Notably, the relative abundance of Bradyrhizobium, the main taxon in the nitrogen-fixing functional group, and Tomentella (medium-distance smooth EMF), which potentially promotes mobile nitrogen uptake, increased significantly with stand age, suggesting an increased nitrogen demand in slash pine plantations during the later stages of restoration on sandy lands. Furthermore, given the significant negative correlation between the relative abundance of Bradyrhizobium and soil AP content, we re-emphasize its potential as a bioindicator. The results of Mantel tests and db-RDA analysis showed that both bacterial and fungal communities were significantly correlated with SWC, AP, and MD. Moreover, Spearman’s rank correlation analyses revealed strong correlations between SWC, AP, and MD and microbial diversity, abundance, and functional groups. We concluded that the development of slash pine plantations on sandy land significantly affects the microbial community structure, function, and assembly mechanism, and the increase in stand age may lead to an increase in nitrogen and phosphorus demand. These findings provide a theoretical basis for future management of slash pine plantations on sandy land.
Data Availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files]. Sequences generated for this study have been made available on the NCBI Sequence Reading Archive (SRA) database with SRA accession numbers PRJNA979784 for 16S rDNA sequences and PRJNA979787 for ITS region sequences. Other data will be made available on request.
Code Availability
The software used in this study is R (version 4.1.3) and its open-source packages.
References
Agerer, R. (2001). Exploration types of ectomycorrhizae. Mycorrhiza, 11, 107–114.
Allsup, C. M., George, I., & Lankau, R. A. (2023). Shifting microbial communities can enhance tree tolerance to changing climates. Science, 380, 835–840.
Anthony, M. A., Crowther, T. W., van der Linde, S., Suz, L. M., Bidartondo, M. I., Cox, F., Schaub, M., Rautio, P., Ferretti, M., Vesterdal, L., et al. (2022). Forest tree growth is linked to mycorrhizal fungal composition and function across Europe. The ISME Journal, 16, 1327–1336.
Bi, B., Yuan, Y., Zhang, H., Wu, Z., Wang, Y., & Han, F. (2022). Rhizosphere soil metabolites mediated microbial community changes of Pinus sylvestris var. mongolica across stand ages in the Mu Us Desert. Applied Soil Ecology, 169, 104222.
Bi, B., Zhang, H., Yuan, Y., Wu, Z., Wang, Y., & Han, F. (2021). Dynamic changes of soil microbial community in Pinus sylvestris var. mongolica plantations in the Mu Us Sandy Land. Journal of Environmental Management, 287, 112306.
Coban, O., De Deyn, G. B., & van der Ploeg, M. (2022). Soil microbiota as game-changers in restoration of degraded lands. Science, 375, abe0725.
Delgado-Baquerizo, M., Oliverio, A. M., Brewer, T. E., Benavent-González, A., Eldridge, D. J., Bardgett, R. D., Maestre, F. T., Singh, B. K., et al. (2018). A global atlas of the dominant bacteria found in soil. Science, 359, 320–325.
Fierer, N. (2017). Embracing the unknown: Disentangling the complexities of the soil microbiome. Nature Review Microbiology, 15, 579–590.
Fierer, N., Bradford, M. A., & Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology, 88, 1354–1364.
Fierer, N., Wood, S. A., & Bueno de Mesquita, C. P. (2021). How microbes can, and cannot, be used to assess soil health. Soil Biology and Biochemistry, 153, 108111.
Friendly, M. (2002). Corrgrams: Exploratory displays for correlation matrices. The American Statistician, 56, 316–324.
Gao, C., Montoya, L., Xu, L., Madera, M., Hollingsworth, J., Purdom, E., Singan, V., Vogel, J., Hutmacher, R. B., Dahlberg, J. A., et al. (2020). Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nature Communications, 11, 34.
Gao, C., Zhang, Y., Shi, N. N., Zheng, Y., Chen, L., Wubet, T., Bruelheide, H., Both, S., Buscot, F., Ding, Q., et al. (2015). Community assembly of ectomycorrhizal fungi along a subtropical secondary forest succession. The New Phytologist, 205, 771–785.
Gao, Y., Song, H., Zhou, F., Chen, S., He, G., Yan, J., Sun, Q., Long, H., Zhai, Z., Hu, D., et al. (2022). Community of soil-inhabiting myxomycetes shares similar assembly mechanisms with fungi, and is affected by bacterial community in subtropical forests of China. Soil Biology and Biochemistry, 175, 108854.
Hermans, S. M., Buckley, H. L., Case, B. S., Curran-Cournane, F., Taylor, M., & Lear, G. (2017). Bacteria as emerging indicators of soil condition. Applied and Environmental Microbiology, 83, e02826-16.
Hobbie, E. A., & Agerer, R. (2010). Nitrogen isotopes in ectomycorrhizal sporocarps correspond to belowground exploration types. Plant and Soil, 327, 71–83.
Hu, Y., Wang, Z., Zhang, Z., Song, N., Zhou, H., Li, Y., Wang, Y., Li, C., & Hale, L. (2021). Alteration of desert soil microbial community structure in response to agricultural reclamation and abandonment. CATENA, 207, 105678.
Hua, X., Jiang, C., & Liu, G. (1995). Floristic survey of ectomycorrhizal fungi for the southern pine in China. Journal of Nanjing Forestry University, 19, 29–36.
Huo, X., Ren, C., Wang, D., Wu, R., Wang, Y., Li, Z., Huang, D., & Qi, H. (2023). Microbial community assembly and its influencing factors of secondary forests in Qinling Mountains. Soil Biology and Biochemistry, 184, 109075.
Izumi, H., Cairney, J. W., Killham, K., Moore, E., Alexander, I. J., & Anderson, I. C. (2008). Bacteria associated with ectomycorrhizas of slash pine (Pinus elliottii) in south-eastern Queensland, Australia. FEMS Microbiology Letters, 282, 196–204.
Jiang, S., Xing, Y., Liu, G., Hu, C., Wang, X., Yan, G., & Wang, Q. (2021). Changes in soil bacterial and fungal community composition and functional groups during the succession of boreal forests. Soil Biology and Biochemistry, 161, 108393.
Jörgensen, K., Clemmensen, K. E., Wallander, H., & Lindahl, B. D. (2023). Do ectomycorrhizal exploration types reflect mycelial foraging strategies? The New Phytologist, 237, 576–584.
Kang, P., Pan, Y., Yang, P., Hu, J., Zhao, T., Zhang, Y., Ding, X., & Yan, X. (2022). A comparison of microbial composition under three tree ecosystems using the stochastic process and network complexity approaches. Frontiers in Microbiology, 13, 1018077.
Koizumi, T., Hattori, M., & Nara, K. (2018). Ectomycorrhizal fungal communities in alpine relict forests of Pinus pumila on Mt. Norikura. Japan. Mycorrhiza, 28, 129–145.
Kranabetter, J. M., Durall, D. M., & MacKenzie, W. H. (2009). Diversity and species distribution of ectomycorrhizal fungi along productivity gradients of a southern boreal forest. Mycorrhiza, 19, 99–111.
Kyaschenko, J., Clemmensen, K. E., Hagenbo, A., Karltun, E., & Lindahl, B. D. (2017). Shift in fungal communities and associated enzyme activities along an age gradient of managed Pinus sylvestris stands. The ISME Journal, 11, 863–874.
Liu, C., Cui, Y., Li, X., & Yao, M. (2021a). microeco: An R package for data mining in microbial community ecology. FEMS Microbiology Ecology, 97, fiaa255.
Liu, D., Wang, H., An, S., Bhople, P., & Davlatbekov, F. (2019a). Geographic distance and soil microbial biomass carbon drive biogeographical distribution of fungal communities in Chinese Loess Plateau soils. Science of the Total Environment, 660, 1058–1069.
Liu, G., Chen, L., Shi, X., Yuan, Z., Yuan, L. Y., Lock, T. R., & Kallenbach, R. L. (2019b). Changes in rhizosphere bacterial and fungal community composition with vegetation restoration in planted forests. Land Degradation & Development, 30, 1147–1157.
Liu, L., Zhu, K., Krause, S. M., Li, S., Wang, X., Zhang, Z., Shen, M., Yang, Q., Lian, J., Wang, X., et al. (2021b). Changes in assembly processes of soil microbial communities during secondary succession in two subtropical forests. Soil Biology and Biochemistry, 154, 108144.
Liu, Y., Chen, L., Ma, T., Li, X., Zheng, M., Zhou, X., Chen, L., Qian, X., Xi, J., Lu, H., et al. (2023). EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research. iMeta, 2, e83.
Long, H., Wu, X., Wang, Y., Yan, J., Guo, X., An, X., Zhang, Q., Li, Z., & Huo, G. (2021). Effects of revegetation on the composition and diversity of bacterial and fungal communities of sandification land soil, in Southern China. Environmental Monitoring and Assessment, 193, 706.
Louca, S., Parfrey, L. W., & Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science, 353, 1272–1277.
Maechler, M. (2019). Finding groups in data: Cluster analysis extended Rousseeuw et al. R package version, 2.
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., Schilling, J. S., & Kennedy, P. G. (2016). FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecology, 20, 241–248.
Nilsson, R. H., Larsson, K. H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., Kennedy, P., Picard, K., Glöckner, F. O., Tedersoo, L., et al. (2019). The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Research, 47, D259–D264.
Ning, C., Egerton-Warburton, L. M., Mueller, G. M., Xiang, W., Yan, W., & Liu, S. (2021). Shifts in ectomycorrhizal fungal community composition during the early establishment of native and exotic pine seedlings. Applied Soil Ecology, 157, 103722.
Ning, C., Mueller, G. M., Egerton-Warburton, L. M., Xiang, W., & Yan, W. (2019a). Host phylogenetic relatedness and soil nutrients shape ectomycorrhizal community composition in native and exotic pine plantations. Forests, 10, 263.
Ning, C., Xiang, W., Mueller, G. M., Egerton-Warburton, L. M., Yan, W., & Liu, S. (2019b). Differences in ectomycorrhizal community assembly between native and exotic pines are reflected in their enzymatic functional capacities. Plant and Soil, 446, 179–193.
Oksanen, J., Kindt, R., Legendre, P., O’Hara, B., Stevens, M., Oksanen, M., & Suggests, M. (2007). The Vegan Package. Community Ecology Package, 10, 631–637.
Peay, K. G., Kennedy, P. G., & Bruns, T. D. (2011). Rethinking ectomycorrhizal succession: Are root density and hyphal exploration types drivers of spatial and temporal zonation? Fungal Ecology, 4, 233–240.
Pennisi, E., & Cornwall, W. (2020). Hidden web of fungi could shape the future of forests. Science, 369, 1042–1043.
Policelli, N., Bruns, T. D., Vilgalys, R., & Nunez, M. A. (2019). Suilloid fungi as global drivers of pine invasions. The New Phytologist, 222, 714–725.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., Peplies, J., & Glöckner, F. O. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research, 41, D590–D596.
Sebastiana, M., Corrêa, A., Castro, P., & Ramos, M. (2020). Pisolithus. In N. Amaresan, M. Senthil Kumar, K. Annapurna, K. Kumar, & A. Sankaranarayanan (Eds.), Beneficial microbes in agro-ecology (pp. 707–726). Academic Press.
Sloan, W. T., Lunn, M., Woodcock, S., Head, I. M., Nee, S., & Curtis, T. P. (2006). Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environmental Microbiology, 8, 732–740.
Stegen, J. C., Lin, X., Fredrickson, J. K., Chen, X., Kennedy, D. W., Murray, C. J., Rockhold, M. L., & Konopka, A. (2013). Quantifying community assembly processes and identifying features that impose them. The ISME Journal, 7, 2069–2079.
Strickland, M. S., Callaham, M. A., Gardiner, E. S., Stanturf, J. A., Leff, J. W., Fierer, N., & Bradford, M. A. (2017). Response of soil microbial community composition and function to a bottomland forest restoration intensity gradient. Applied Soil Ecology, 119, 317–326.
Stuart, E. K., Castañeda-Gómez, L., Macdonald, C. A., Wong-Bajracharya, J., Anderson, I. C., Carrillo, Y., Plett, J. M., & Plett, K. L. (2022). Species-level identity of Pisolithus influences soil phosphorus availability for host plants and is moderated by nitrogen status, but not CO2. Soil Biology and Biochemistry, 165, 108520.
Tai, X. S., Mao, W. L., Liu, G. X., Chen, T., Zhang, W., Wu, X. K., Long, H. Z., Zhang, B. G., & Zhang, Y. (2013). High diversity of nitrogen-fixing bacteria in the upper reaches of the Heihe River, northwestern China. Biogeosciences, 10, 5589–5600.
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T., & Singh, B. K. (2020). Plant-microbiome interactions: From community assembly to plant health. Nature Reviews Microbiology, 18, 607–621.
Wang, B., Huang, S., Li, Z., Zhou, Z., Huang, J., Yu, H., Peng, T., Song, Y., & Na, X. (2022a). Factors driving the assembly of prokaryotic communities in bulk soil and rhizosphere of Torreya grandis along a 900-year age gradient. Science of the Total Environment, 837, 155573.
Wang, C., Zheng, Y., Sakai, Y., Toyoda, A., Minakuchi, Y., Abe, K., Yokota, A., & Yabe, S. (2019a). Tengunoibacter tsumagoiensis gen. nov., sp. nov., Dictyobacter kobayashii sp. nov., Dictyobacter alpinus sp. nov., and description of Dictyobacteraceae fam. nov. within the order Ktedonobacterales isolated from Tengu-no-mugimeshi, a soil-like granular mass of micro-organisms, and emended descriptions of the genera Ktedonobacter and Dictyobacter. International Journal of Systematic and Evolutionary Microbiology, 69, 1910–1918.
Wang, D. D., Zhao, W., Reyila, M., Huang, K. C., Liu, S., & Cui, B. K. (2022b). Diversity of microbial communities of Pinus sylvestris var. mongolica at Spatial Scale. Microorganisms, 10, 371.
Wang, K., Zhang, Y., Tang, Z., Shangguan, Z., Chang, F., Jia, F., Chen, Y., He, X., Shi, W., & Deng, L. (2019b). Effects of grassland afforestation on structure and function of soil bacterial and fungal communities. Science of the Total Environment, 676, 396–406.
Wang, X., Kou, Y., Liu, J., Zhao, W., & Liu, Q. (2023a). Soil microbial legacy determines mycorrhizal colonization and root traits of conifer seedlings during subalpine forest succession. Plant and Soil, 485, 361–375.
Wang, Y., Dong, L., Zhang, M., Cui, Y., Bai, X., Song, B., Zhang, J., & Yu, X. (2023b). Dynamic microbial community composition, co-occurrence pattern and assembly in rhizosphere and bulk soils along a coniferous plantation chronosequence. CATENA, 223, 106914.
Wang, Y. L., Zhang, X., Xu, Y., Babalola, B. J., Xiang, S. M., Zhao, Y. L., & Fan, Y. J. (2021). Fungal diversity and community assembly of ectomycorrhizal fungi associated with five pine species in Inner Mongolia. China. Frontiers in Microbiology, 12, 646821.
Wu, N., Li, Z., Meng, S., & Wu, F. (2021). Soil properties and microbial community in the rhizosphere of Populus alba var. pyramidalis along a chronosequence. Microbiological Research, 250, 126812.
Wu, Z., Haack, S. E., Lin, W., Li, B., Wu, L., Fang, C., & Zhang, Z. (2015). Soil microbial community structure and metabolic activity of Pinus elliottii plantations across different stand ages in a subtropical area. PLoS ONE, 10, e0135354.
Xu, L., & Coleman-Derr, D. (2019). Causes and consequences of a conserved bacterial root microbiome response to drought stress. Current Opinion in Microbiology, 49, 1–6.
Zhang, X., Liu, S., Li, X., Wang, J., Ding, Q., Wang, H., Tian, C., Yao, M., An, J., & Huang, Y. (2016). Changes of soil prokaryotic communities after clear-cutting in a karst forest: Evidences for cutting-based disturbance promoting deterministic processes. FEMS Microbiology Ecology, 92, fiw026.
Zhao, P. S., Guo, M. S., Gao, G. L., Zhang, Y., Ding, G. D., Ren, Y., & Akhtar, M. (2020). Community structure and functional group of root-associated Fungi of Pinus sylvestris var. mongolica across stand ages in the Mu Us Desert. Ecology and Evolution, 10, 3032–3042.
Zhou, J., & Ning, D. (2017). Stochastic community assembly: Does it matter in microbial ecology? Microbiology and Molecular Biology Reviews, 81, e00002-17.
Zhou, X., Guo, Z., Chen, C., & Jia, Z. (2017). Soil microbial community structure and diversity are largely influenced by soil pH and nutrient quality in 78-year-old tree plantations. Biogeosciences, 14, 2101–2111.
Acknowledgements
The authors would like to thank Dr. Jennifer Smith (University of Otago) for language editing, and the two anonymous reviewers for their valuable insights and suggestions. This study was supported by grants from the National Natural Science Foundation of China (32160029), and the Foundation of Key Laboratory of Extreme Environmental Microbial Resources and Engineering, Gansu Province (EEMRE201803).
Author information
Authors and Affiliations
Contributions
XZ and SYX conceived and designed the study with the help of HL and YG; XZ, SYX, QW, BBZ, YMM, and ZCD performed the experiments; XZ, SYX, XW, and HL analyzed the data, and XZ drafted the manuscript; LC, JL, and HL helped to revise the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this study.
Ethical statements and consent to participate
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, X., Xiong, SY., Wu, X. et al. Dynamics of Microbial Community Structure, Function and Assembly Mechanism with Increasing Stand Age of Slash Pine (Pinus elliottii) Plantations in Houtian Sandy Area, South China. J Microbiol. 61, 953–966 (2023). https://doi.org/10.1007/s12275-023-00089-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12275-023-00089-7