
Abstract
Purpose
The knowledge regarding how different soil microbial groups change in subalpine forest areas after clear-cutting is limited. To bridge this gap, we studied the change patterns of the different soil microbial community assemblies and their determining factors in subalpine areas.
Materials and methods
Field measurements and sampling were conducted in three stands at different stages of natural restoration (BF, broadleaf forest; MF, mixed coniferous and broadleaf forest; and PCF, primeval coniferous forest) after clear-cutting in western Sichuan Province, China. Three soil microbial group (bacteria, fungi, and archaea) community assemblies, edaphic properties, soil nematodes, tree diameter at breast height, plant richness, and understory vegetation biomass were examined and statistically analyzed.
Results and discussion
The α-diversity of the three microbial groups in the BF and MF stands was significantly higher than that in the PCF stand. The difference between the stands was greater than the difference within the stands for all three microbial groups. The changes in the three microbial groups were all significantly associated with shifts in soil carbon, soil total nitrogen, soil available nitrogen, and tree species composition. The bacterial and archaeal communities were also closely related to the bacterivore number. Acidobacteria, Actinobacteria, Bacteroidetes, Planctomycetes, Basidiomycota, Ascomycota, and Thaumarchaeota were the main phyla that responded to environmental variation.
Conclusions
The three soil microbial groups all showed regular trends across the three different stands. The changes in the soil microbial community assembly were mainly attributed to the differences in soil nutrients, tree species composition, and feeding traits of soil nematodes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Soil microbes are widely distributed in terrestrial ecosystem and play a critical role in ecological functions (Jiang et al., 2018). For example, bacteria are closely related to soil nutrient turnover (Knelman et al., 2015) and participate in the decomposition of cellulose, hemicellulose, and dead fungal mycelia (Lladó et al., 2017; López-mondéjar et al., 2016). Studies have shown that many archaea are capable of ammonia oxidation and thus are important in the nitrogen cycle (Tripathi et al., 2015). As the primary decomposers, soil fungi produce extracellular enzymes that break down specific forms of carbon (Looby & Treseder, 2018). Therefore, changes in the assembly and activity of soil microbial communities can affect the functionality and thus the biogeochemical processes and productivity of the forest ecosystem. Understanding how the microbial communities change is essential to understanding the dynamic variation in the forest ecosystem (Chauvet et al., 2017; Norden et al., 2015). However, the distribution patterns and diversity of soil microbes at regional and global scale are poorly understood compared with those of aboveground macro-organisms (Kazemi et al., 2016). As with plants, soil microbes are subject to environmental stress and biological interactions; however, the recovery mechanisms of the soil microbial community may differ due to the differences in biology (Sun et al., 2017). Changes in soil microbial communities are often strongly correlated with variations in edaphic properties, especially in substrate availability and pH (Bapiri et al., 2010; Rousk et al., 2010). Plant community composition may also be a main determining factor, particularly for fungal communities, because as plant symbionts, pathogens, and decomposers, fungi are tightly linked to the activities of living plants and saprotrophs (Dassen et al., 2017; Saitta et al., 2018). Tree species composition could affect fungal community composition through substrate quality, symbiotic compatibility, and specific defense mechanisms, such as allelochemicals (Tedersoo et al., 2016). In addition, previous studies have shown that bacteria and archaea are often more sensitive to soil pH, whereas fungi may be more sensitive to soil carbon (Thomson et al., 2015; Tripathi et al., 2013). Therefore, different groups of forest soil microbes may vary in their response to the same environmental variables and show contrasting dynamics after disturbances (Birkhofer et al., 2012).
Clear-cutting involves the felling and removal of all tree stems and was once a common practice in many forests throughout the world (Bergholm et al., 2015; Dong et al., 2018; Fedorov et al., 2016; Johnson et al., 1991). As one of the most severe disturbances to a forest, clear-cutting causes extreme changes in the biotic and abiotic environment (Ding et al., 2017) and indirectly influences the distribution and abundance of soil microbes (Pei et al., 2016). Previous studies have suggested that vegetation communities and soil fertility in the slash tend to recover to a condition that is similar to that of intact forest through natural regeneration (Achat et al., 2015; Ivanova, 2014; Xu et al., 2015). With regard to soil microbes, the extreme environmental conditions and the low resource availability induced by serious disturbance can restrict some sensitive microbial species and reduce the microbial biomass and diversity (Banning et al., 2011; Xiang et al., 2014). As stress decreases with environmental restoration, new ecological niches may contribute to the recovery of the restricted species and eventually facilitate the return of the microbial community to a state is similar to that observed prior to the disturbance (Hart et al., 2005; Xiang et al., 2014). Nevertheless, fungal and archaeal communities may not follow this predictable successional trend ((Zhang et al., 2016b); Banning et al. 2011). Many studies have demonstrated that soil microbial community assembly is simultaneously influenced by both deterministic and stochastic processes, and the result of succession depends on a complex balance between deterministic and stochastic variability (Caruso et al., 2011).
The space-for-time approach has typically been used to examine the changes in different components of the forest ecosystem over time (Cao et al., 2012; Zhang et al., 2016b). However, studies on the differences in the soil microbial community assembly across different stages of natural restoration after clear-cutting in subalpine areas are scarce. The subalpine forest area of western Sichuan is located on the eastern Tibetan Plateau and has suffered long-term and large-scale exploitation from the mid-twentieth century to the end of the twentieth century (Liu et al., 2010). The cutting area is mainly located at an elevation of 2800–3600 m (Zhang et al., 2005). After clear-cutting, natural regeneration commonly occurs and comprises the shrub stage (0–10a), the mixed shrub and broadleaf forest stage (10–20a), the broadleaf forest (BF) stage (20–30a), and the mixed coniferous and broadleaf forest (MF) stage and eventually achieves the dark coniferous forest stage (Shi et al., 1988; Wang et al., 2009). We hypothesized that (1) diversity of three different microbial groups in BF and MF stands would be higher than in the primeval coniferous forest (PCF) stand and the edaphic properties would be the key determinants of the variation in soil microbial community assembly and (2) the bacterial, fungal, and archaeal groups would follow distinct response trajectories, whereas the deterministic processes would dominate only in bacterial community assembly. The present study has verified the above hypotheses and attempted to comprehensively analyze the relationships among plants, soil, nematodes, and microbes. This study contributes to a better understanding of the succession mechanism of soil microbial communities during forest restoration.
2 Materials and methods
2.1 Study site
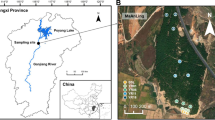
The present study was conducted in Miyaluo (102° 41′–102° 44′ E, 31° 42′–31° 51′ N), which is located in Lixian County, Aba Tibetan and Qiang Autonomous Prefecture, western Sichuan Province, China. This area belongs to the transition zone between the Tibetan Plateau and the Sichuan Basin and has an elevational range of 2200–5500 m (Ma et al., 2007). This area is characterized by a montane monsoon climate with an annual mean temperature of 6–10 °C and an annual precipitation of 600–1100 mm (Xu et al., 2012). The soils at the study sites are classified as a mountain brown soil series in Chinese soil taxonomy.
Because the study area is located in a subalpine valley region, it was very difficult to achieve repetition at the stand level in the present study. Therefore, the field investigation and sampling were repeated on a plot level. Three stands, including broadleaf forest (BF) dominated by Betula albo-sinensis Burk.; mixed coniferous and broadleaf forest (MF) dominated by B. albo-sinensis, dragon spruce (Picea asperata Mast.), and faxon fir (Abies faxoniana Rehd.); and primeval coniferous forest (PCF) dominated by faxon fir, were selected in the present study. The primeval forests in the BF and MF stands were clear-cut in the late 1980s and early 1960s, respectively. The age of the PCF is between 160 and 200 years (Ma et al., 2007). All the selected stands were located on a partially shaded aspect and had similar environmental conditions. The soil depth of the selected stands ranged from 25 to 30 cm. Below the mineral soil horizon, the parent materials are mainly phyllite and slate. The mean slope degrees of the BF, MF, and PCF were 26°, 23°, and 24°, respectively. The densities of the BF, MF, and PCF stands were 767 trees hm−2, 1250 trees hm−2, and 883 trees hm−2, respectively. These stands were rarely disturbed by human activity.
2.2 Sampling design
The field measurements and sampling were conducted in July 2018. Three 20 × 20 m square plots were randomly established in each stand. The diameter at breast height (DBH) was recorded for all trees within each plot, and the relative dominance of conifers (RDC) was calculated for all plots based on basal area at breast height. Three 2 × 2 m subplots were randomly distributed within each plot for shrub sampling. Moreover, three 1 × 1 m microplots were randomly distributed within each plot for herb sampling. Both the shrubs and herbs were destructively excavated and measured for fresh weight. The trees, shrubs, and herbs were identified at the species level. The subsamples of both shrubs and herbs were oven-dried at 65 °C to a constant weight to calculate total biomass. The soil samples were collected at a depth of 0–10 cm at five random points within each plot, and the five soil samples were then combined to create one composite soil sample that was held on ice in an incubator. After removing the rocks, roots, and litter, the soil samples were passed through a 2-mm sieve. Each soil sample was divided into three parts that were used for (1) the analysis of edaphic properties, (2) the analysis of nematode communities, and (3) the extraction of DNA (stored at − 80 °C). The soil samples were collected to determine bulk density and were oven-dried at 105 °C to a constant weight. The mean of three replications represented the bulk density in each plot.
2.3 Edaphic properties and nematode community analyses
Soil organic carbon (SOC) was measured by the potassium dichromate oxidation method, and total nitrogen (TN) was measured by the semimicro-Kjeldahl method (Liu, 1996). The C/N ratio was calculated by dividing the SOC by the TN. The available nitrogen (AN) was measured by alkaline hydrolysis diffusion methods (Bao, 2000). Soil pH was measured using distilled water extracts with a pH meter (Tian et al., 2012). Soil total phosphorus (TP) and available phosphorus (AP) were measured by the Mo-Sb colorimetric method (Liu, 1996).
Nematodes were extracted from a 100 g fresh soil sample using a modified sucrose centrifugal flotation method (Freckman & Virginia, 1993). At least 100 nematodes from each sample were randomly selected and identified at the genus level using an inverted compound microscope according to Yin (Yin, 1998). Nematodes were assigned to one of four groups based on their feeding habit: (1) bacterivores, (2) fungivores, (3) plant parasites, and (4) omnivores/predators.
2.4 Soil DNA extraction and sequencing
Microbial genomic DNA was extracted from approximately 0.5 g of a composite soil sample using the FastDNA Spin Kit (MP Biomedicals, Solon, OH, USA). Extracted DNA was assessed using 1% agarose gel electrophoresis, and the DNA concentration and quality were determined using an ND-2000 UV-Vis spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
The hypervariable regions V4–V5 of the bacterial 16S rRNA genes, the fungal ITS1 region, and the hypervariable regions V4–V5 of the archaeal 16S rRNA genes were amplified using the primers 515F/907R, ITS1/ITS2R, and 524F10extF/Arch958RmodR, respectively, via PCR on an ABI GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA, USA). The PCRs were performed in triplicate 20 μL mixtures containing 0.4 μL of FastPfu polymerase, 4 μL of 5× FastPfu buffer, 0.8 μL (5 μM) of each primer, 2 μL of 2.5 mM dNTPs, and 10 ng of template DNA under the following condition: 95 °C for 3 min, 25 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s and a final extension at 72 °C for 10 min. After amplification, the PCR products were visualized on 2% agarose gels, purified by the AxyPrep DNA gel extraction kit (Axygen Biosciences, Union City, CA, USA), and then quantified by QuantiFluor™-ST (Promega Biotech, Beijing, China). The purified amplicons were pooled in equimolar amounts and then were sequenced on the MiSeq PE300 platform (Illumina, San Diego, CA, USA) by Shanghai Majorbio Biopharm Technology Co., Ltd. in China.
2.5 Bioinformatic and statistical analysis
Raw sequences longer than 200 bp with an average quality score more than 20 and without ambiguous base calls were quality processed using QIIME pipeline (version 1.17). The operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using UPARSE (version 7.1 http://drive5.com/uparse/). The bacterial, fungal, and archaeal sequences were randomly subsampled down to the lowest number of corresponding sequences for all samples prior to further analysis. The bacterial and archaeal sequences were classified using the Silva database (version 128, http://www.arb-silva.de). The fungal sequences were classified using the Unite database (version 6.0, http://unite.ut.ee/index.php). As recommended by the Ribosomal Database Project (RDP), a confidence threshold of 70% was applied to assign the sequences to different taxonomic levels. Alpha diversity was used to analyze the complexity of the species diversity of a sample based on 4 variables (the Chao estimator, coverage estimator, the Simpson and Shannon indices) in Mothur (http://www.mothur.org/wiki). In the present study, a phylum with a relative abundance of greater than 3% in all samples was defined as a dominant phylum, and an order with a relative abundance of greater than 1% in all samples was defined as a dominant order. Principal coordinate analysis (PCoA) and analysis of similarities (ANOSIM) based on a weighted UniFrac distance matrix were applied to determine differences in the microbial community among the three stands. Redundancy analysis (RDA) was performed to identify the effects of the edaphic and biotic factors on the microbial community composition using the R Vegan package (R Core Team, 2013). To assess the relative importance of selection and chance effects, the deterministic and stochastic changes were calculated as variations in community structure using a modified method based on Euclidean distances (Zhang et al., 2011). Compared with the mixed coniferous and broadleaf forest and the dark coniferous forest, the broadleaf forest is an earlier successional stage. Therefore, the variation between the plots of the broadleaf stands was used as a reference point, and the Euclidean distances were calculated based on the relative abundance of microbial communities at the phylum level and nematodes at the genus level, on biomass for herbs and shrubs, and on relative dominance for trees. The deterministic change can be calculated as D = [(mean variation between the reference point and the other stand) − (reference point)], and the stochastic change can be calculated by S = [(mean variation within the other stand) − (reference point)]. For the MF and PCF stands, we calculated the importance of \( \mathrm{chance}=\frac{\left|S\right|}{\left|D\right|+\left|S\right|} \). For the reference point, the importance of chance was 1.
In addition, one-way ANOVA with Tukey’s test (p < 0.05) and Pearson correlation analysis were conducted using the SPSS 18.0 (SPSS, Chicago, IL, USA).
3 Results
3.1 Variation in the environmental variables
The concentrations of SOC, TN, and AN continuously decreased from the MF stand to the PCF stand with decreases of 41%, 43%, and 45%, respectively (Table 1). The concentrations of TP and AP in the MF stand were significantly higher than in the other two stands (Table 1). In contrast, the C/N in the BF and PCF stands was significantly higher than that in the MF stand (Table 1). The soil bulk density (SBD) and pH were not significantly different across the three stands (Table 1). For the biotic variables, the proportion of bacterivores (PB), the proportion of fungivores (PF), the fungivore number (FN), and the plant richness (PR) also appeared to be independent of the change in the stands (Table 1). However, the bacterivore number (BN) in the MF stand was significantly higher than that in the other two stands, whereas the relative dominance of conifers (RDC) showed a significantly increasing trend from the BF stand to the PCF stand (Table 1).
3.2 Microbial community assembly and diversity
All the coverage values were greater than 0.98, which suggests that all three microbial communities were well sampled (Table S1). In total, 2152 bacterial, 2100 fungal, and 747 archaeal nonsingleton OTUs were identified after filtering and removing the chimeras. Based on the Shannon and Simpson indices, the α-diversity of the three microbial groups in the PCF stand was significantly lower than that in the BF and MF stands (Table S1). The bacterial reads were assigned to 5 dominant phyla, and the mean relative abundance of the individual dominant phyla across all the stands was as follows: Proteobacteria (39.82%), Acidobacteria (21.27%), Actinobacteria (18.21%), Bacteroidetes (7.57%), and Planctomycetes (4.00%) (Fig. 1a). For the fungal community, the order is Basidiomycota (47.28%), Ascomycota (29.76%), and Zygomycota (19.77%) (Fig. 1b). Within the archaeal community, Thaumarchaeota was the only dominant phylum and was present in more than 75% in all the stands (Fig. 1c). The relative abundance of Acidobacteria, Actinobacteria, Bacteroidetes, Planctomycetes, Basidiomycota, Ascomycota, and Thaumarchaeota significantly changed among the three stands (p < 0.05) (Fig. 2b, c, d, e, f, g, i). Moreover, the continuous variation in the relative abundance of Planctomycetes, Basidiomycota, and Ascomycota was exhibited from the BF stand to the PCF stand, including decreases in Planctomycetes and Ascomycota and an increase in Basidiomycota (Fig. 2e–g). In contrast, the relative abundance of Proteobacteria and Zygomycota remained relatively stable (Fig. 2a, h). Although the mean relative abundance of Euryarchaeota in the PCF stand was much higher than that in the other two stands, there was no significant difference among the stands (Fig. 1). At the order level, the bacterial communities across the three stands were dominated by Rhizobiales, Norank_c_Acidobacteria, Burkholderiales, Acidobacteriales, Gaiellales, Sphingobacteriales, Xanthomonadales, Rhodospirillales, Planctomycetales, Acidimicrobiales, Solirubrobacterales, and SC-I-84 (Table S2). For the fungal communities, the dominant orders were Mortierellales, Helotiales, Hypocreales, and Tremellales (Table S3). Furthermore, the relative abundance of Russulales in the fungal community sharply increased from 0.20% in the BF stand to 55.42% in the PCF stand (p < 0.05) (Table S3). For the archaeal communities, Norank_c_Soil_Crenarchaeotic_Group_SCG_ was the only dominant order in all stands (Table S4). The pattern of bacterial, fungal, and archaeal β-diversity at the OTUs level are shown in the PCoA plots (Fig. 3). Both the bacterial and fungal communities were differentiated into three clusters corresponding to the three stands (Fig. 3a, b). In contrast, it was difficult to differentiate the archaeal community into three clusters (Fig. 3c). In addition, the r value of the bacterial, fungal, and archaeal communities across the three stands based on ANOSIM were 1, 1, and 0.60, respectively (p < 0.05) (Table 2).

Taxonomic proportions in the a bacterial, b fungal, and c archaeal communities in the three stands

Relative abundance of the major phyla of bacterial, fungal, and archaeal communities in the three stands. a Proteobacteria. b Acidobacteria. c Actinobacteria. d Bacteroidetes. e Planctomycetes. f Basidiomycota. g Ascomycota. h Zygomycota. i Thaumarchaeota

Principal coordinate analysis (PCoA) of the a bacterial, b fungal, and c archaeal communities across the three stands
3.3 The determining factors of the microbial community
The environmental variables that significantly changed within the three stands were selected for further analysis. RDA was performed to examine the relationships between the environmental variables and microbial communities based on the OTUs (Fig. 4, Table 3). Among the selected environmental variables, SOC, TN, and AN were the three dominant factors affecting the communities regardless of the microbial group (Fig. 4). All the selected variables significantly contributed to the variation in the bacterial and archaeal communities across the three stands ((p < 0.05) (Table 3). In contrast, the C/N and AP were not significantly associated with the assembly of the fungal community (Table 3). Using the BF stand as a reference point, the changes in the community assembly of the three microbial groups and in trees were dominated by the deterministic processes in the MF and PCF stands. However, the nematodes and shrubs were strongly driven by stochasticity (Fig. 5).

Redundancy analysis (RDA) showing the effects of environmental variables on the a bacterial, b fungal, and c archaeal communities across the three stands

The relative importance of chance effect in different groups of organisms in the three stands
4 Discussion
Our results showed that the relative abundance of all the dominant phyla in the microbial communities significantly changed among the different stands except for Proteobacteria and Zygomycota. The variation in the relative abundance of different taxa (species sorting) might be a dominant response mechanism of microbial communities to environmental change (Rui et al., 2015). In the present study, the relative abundance of Russulales sharply increased from less than 5% in the BF and MF stands to 55.42% in the PCF stand. This significant increase in the relative abundance of Russulales in the fungal community may indicate that the forests have naturally progressed to the final successional stage after clear-cutting in this subalpine area. A previous study reported that the microbial community can recover much more rapidly than the plant community after suffering serious damage (Xiang et al., 2014). Therefore, the variation in soil microbial community assembly could be used as a bioindicator of a particular stage in the process of soil development or vegetation succession (Jiang et al., 2018).
Although the bacterial community assembly changed within the different stands, Proteobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, and Planctomycetes were always the five dominant phyla, which is similar to a previous finding that Proteobacteria, Acidobacteria, Actinobacteria, Verrucomicrobia, and Bacteroidetes were the five ubiquitous bacterial groups in most soils (Janssen, 2006). In the present study, the α-diversity of the three microbial groups in the BF and MF stands was much higher than that in the PCF stand. Given that the BF and MF stands represent the middle stage of natural succession after clear-cutting, we speculate that variation in the diversity of the soil bacterial, fungal, and archaeal communities in the subalpine area may also be consistent with the intermediate disturbance hypothesis, which suggests that the species diversity is expected to be maximized at an intermediate intensity of disturbance or environmental change (Connell, 1978). This hypothesis has frequently been used to explain community variation across environmental and temporal gradients, particularly for plants (Doležal et al., 2013; Zhang et al., 2016b).
Previous studies have demonstrated that clear-cutting causes significant variation in edaphic properties, particularly in carbon and nitrogen concentrations in subalpine coniferous forests (Liu, 1979; Zhou & Yang, 1984). In the present study, there were always differences in edaphic properties between the different stands, which contribute to changes in the microbial communities. The SOC and TN concentrations decreased from the BF stand to the PCF stand, probably because the organic matter returned to the soil in the middle and late successional stages to a lesser degree than that in the previous stages. In general, the soil directly supports the substrate of the microbial communities, and thus has a greater impact on shaping soil microbial communities (Jiang et al., 2018). The variables related to soil fertility and pH have been suggested to be the critical factors determining the assembly of soil microbial communities (Thomson et al., 2015; Tripathi et al., 2013; Wan et al., 2015; You et al., 2014). However, pH was not identified as a dominant explanatory variable in the variation of different microbial groups due to its narrow range in the present study. Our results indicate that carbon and nitrogen concentrations were the major edaphic factors influencing all three microbial communities in the subalpine forest. We found that bacterivore number was significantly correlated with the assembly of bacterial and archaeal communities, which is consistent with the suggestion that nematodes are an important factor influencing soil microbial assembly (Jiang et al., 2018; Nieminen, 2009). These results suggest that the differences in the feeding characteristics of the nematode community could affect soil microbial community composition.
Previous results on the effects of the plant or tree species composition on the fungal microbial communities are not consistent. Some studies have found that tree species or plant community diversity was the main driver in shaping the fungal community and have suggested that the fungal community is more closely related to plant composition than other microbial groups in some specific ecosystems (Saitta et al., 2018; Tedersoo et al., 2016; Yang et al., 2017a). In contrast, other studies have demonstrated that the fungal community assemblage was determined by highly stochastic processes (Jiang et al., 2018; Yang et al., 2017b). The differences among ecosystems or the complex interactions between biotic and abiotic factors may lead to inconsistent results concerning the critical drivers of microbial communities (Ni et al., 2018; You et al., 2014). In our study, the fungal community changed from Ascomycota-dominant in the BF stand to Basidiomycota-dominant in the PCF stand, suggesting that the assembly of the fungal community may follow a regular pattern along natural vegetation restoration in subalpine areas. Although the Ascomycota are highly adaptable, the soil environment in the coniferous forest may be more conducive to the growth of the Basidiomycota microbes (Sheng, 2018). Previous studies have suggested that the increase in the abundance of Basidiomycota microbes in coniferous forest soil may be related to their ability to degrade complex lignocellulose components (Liu et al., 2018; Lundell & Mäkelä, 2010). In addition to the fungal communities, the bacterial and archaeal communities were also significantly correlated with tree species composition in the present study. The impacts of plant community composition on the soil bacterial and archaeal communities are most likely mediated by changing soil nutrients, particularly by soil carbon and nitrogen concentrations (Dassen et al. 2017; (Fierer et al., 2009; Zhang et al., 2016a)). Our results demonstrated that all three microbial groups were dominated by deterministic process. The deterministic processes include selection imposed by the abiotic and biotic environments; in contrast, the stochastic processes include probabilistic dispersal and random changes in the relative abundance of species (Dini-Andreote et al., 2015; Stegen et al., 2012). Therefore, these results suggest that the assembly of bacterial, fungal, and archaeal communities in the subalpine area was all strongly determined by environmental filtering.
5 Conclusions
The bacterial, fungal, and archaeal community assembly clearly changed across the three subalpine stands at different stages of natural restoration. The variation in the bacterial, fungal, and archaeal community assembly was driven largely by deterministic processes. The relative abundance of sensitive microbial taxa, such as Russulales, could be used to predict the direction and magnitude of natural succession in the study area. In addition to soil nutrients, the tree species composition and the feeding traits of soil nematodes are also key factors influencing microbial community assembly. The information provided by our study will improve the understanding of ecosystem recovery processes in the subalpine area. Due to limitations in the selection of stands, these results do not represent the complete pattern of variation in the microbial community after clear-cutting. Hence, studies including additional restoration stages will be required to further confirm these results.
References
Achat DL, Deleuze C, Landmann G, Pousse N, Ranger J, Augusto L (2015) Quantifying consequences of removing harvesting residues on forest soils and tree growth - a meta-analysis. For Ecol Manag 348:124–141
Banning NC, Gleeson DB, Grigg AH, Grant CD, Andersen GL, Brodie EL, Murphy DV (2011) Soil microbial community successional patterns during forest ecosystem restoration. Appl Environ Microbiol 77:6158–6164
Bao SD (2000) Agricultural soil analysis. China Agriculture Press, Beijing (In Chinese)
Bapiri A, Bååth E, Rousk J (2010) Drying-rewetting cycles affect fungal and bacterial growth differently in an arable soil. Microb Ecol 60:419–428
Bergholm J, Olsson BA, Vegerfors B, Persson T (2015) Nitrogen fluxes after clear-cutting. Ground vegetation uptake and stump/root immobilisation reduce N leaching after experimental liming, acidification and N fertilisation. For Ecol Manag 342:64–75
Birkhofer K, Schöening I, Alt F, Herold N, Klarner B, Maraun M, Marhan S, Oelmann Y, Wubet T, Yurkov A, Begerow D, Berner D, Buscot F, Daniel R, Diekötter T, Ehnes RB, Erdmann G, Fischer C, Foesel B, Groh J, Gutknecht J, Kandeler E, Lang C, Lohaus G, Meyer A, Nacke H, Näther A, Overmann J, Polle A, Pollierer MM, Scheu S, Schloter M, Schulze E-D, Schulze W, Weinert J, Weisser WW, Wolters V, Schrumpf M (2012) General relationships between abiotic soil properties and soil biota across spatial scales and different land-use types. PLoS One 7:e43292
Cao J, Wang X, Tian Y, Wen Z, Zha T (2012) Pattern of carbon allocation across three different stages of stand development of a Chinese pine (Pinus tabulaeformis) forest. Ecol Res 27:883–892
Caruso T, Chan Y, Lacap DC, Lau MCY, McKay CP, Pointing SB (2011) Stochastic and deterministic processes interact in the assembly of desert microbial communities on a global scale. ISME J 5:1406–1413
Chauvet M, Kunstler G, Roy J, Morin X (2017) Using a forest dynamics model to link community assembly processes and traits structure. Funct Ecol 31:1452–1461
Connell JH (1978) Diversity in tropical rain forests and coral reefs - high diversity of trees and corals is maintained only in a non-equilibrium state. Science 199:1302–1310
Dassen S, Cortois R, Martens H, De Hollander M, Kowalchauk GA, Van Der Putten WH, De Deyn GB (2017) Differential responses of soil bacteria, fungi, archaea and protists to plant species richness and plant functional group identity. Mol Ecol 26:4085–4098
Ding Y, Zang R, Lu X, Huang J (2017) The impacts of selective logging and clear-cutting on woody plant diversity after 40 years of natural recovery in a tropical montane rain forest, south China. Sci Total Environ 579:1683–1691
Dini-Andreote F, Stegen JC, Van Elsas JD, Salles FJ (2015) Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci U S A 112:E1326–E1332
Doležal J, Yakubov V, Hara T (2013) Plant diversity changes and succession along resource availability and disturbance gradients in Kamchatka. Plant Ecol 214:477–488
Dong B, Zhao K, Wang Z, Jia Z, Ma L, Xia X (2018) Forest recovery after clear-cutting in Chinese pine (Pinus tabuliformis) plantations of North China. J Arid Land 10:233–248
Fedorov AN, Iwahana G, Konstantinov PY, Machimura T, Argunov RN, Efremov PV, Lopez LMC, Takakai F (2016) Variability of permafrost and landscape conditions following clear cutting of larch forest in central Yakutia. Permafr Periglac Process 28:331–338
Fierer N, Strickland DL, Bradford MA, Cleveland CC (2009) Global patterns in belowground communities. Ecol Lett 12:1238–1249
Freckman DW, Virginia RA (1993) Extraction of nematodes from dry valley Antarctic soils. Polar Biol 13:483–487
Hart SC, DeLuca TH, Newman GS, MacKenzie MD, Boyle SI (2005) Post-fire vegetative dynamics as drivers of microbial community structure and function in forest soils. For Ecol Manag 220:166–184
Ivanova NS (2014) Recovery of tree stand after clear-cutting in the Ural Mountains. Int J Bioresour Stress Manag 5:090–092
Janssen PH (2006) Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72:1719–1728
Jiang Y, Lei Y, Yang Y, Korpelainen H, Niinemets Ü, Li C (2018) Divergent assemblage patterns and driving forces for bacterial and fungal communities along a glacier forefield chronosequence. Soil Biol Biochem 118:207–216
Johnson CE, Johnson AH, Huntington TG, Siccama TG (1991) Whole-tree clear-cutting effects on soil horizons and organic-matter pools. Soil Sci Soc Am J 55:497–502
Kazemi S, Hatam I, Lanoil B (2016) Bacterial community succession in a high-altitude subarctic glacier foreland is a three-stage process. Molecul Ecol 25:5557–5567
Knelman JE, Graham EB, Trahan NA, Schmidt SK, Nemergut DR (2015) Fire severity shapes plant colonization effects on bacterial community structure, microbial biomass, and soil enzyme activity in secondary succession of a burned forest. Soil Biol Biochem 90:161–168
Liu X (1979) A preliminary study on soil fertility of the cut-over area in western Sichuan. Acta Pedol Sin 16:234–243 (In Chinese with English abstract)
Liu GS (1996) Soil physical and chemical analysis and description of soil profiles. Standards Press of China, Beijing (In Chinese)
Liu Y, Zhang Y, Liu S (2010) Aboveground biomass, ANPP and stem volume of birch stands in natural restoration process of subalpine secondary forest in Western Sichuan. Acta Ecol Sin 30:594–601 (In Chinese with English abstract)
Liu D, Liu G, Chen L, Wang J, Zhang L (2018) Soil pH determines fungal diversity along an elevation gradient in Southwestern China. Sci China Life Sci 61:718–726
Lladó S, López-mondéjar R, Baldrian P (2017) Forest soil bacteria: diversity, involvement in ecosystem processes, and response to global change. Microbiol Mol Biol Rev 81:e00063–e00016
Looby CI, Treseder KK (2018) Shifts in soil fungi and extracellular enzyme activity with simulated climate change in a tropical montane cloud forest. Soil Biol Biochem 117:87–96
López-mondéjar R, Zühlke D, Becher D, Riedel K, Baldrian P (2016) Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci Rep 6:25279
Lundell TK, Mäkelä HK (2010) Lignin-modifying enzymes in filamentous basidiomycetes - ecological, functional and phylogenetic review. J Basic Microbiol 50:5–20
Ma J, Shi Z, Zhang Y, Chen B (2007) Quantitative analysis of different restoration stages during natural succession processes of subalpine dark brown coniferous forests in western Sichuan, China. Chinese J Appl Ecol 18:1695–1701 (In Chinese with English abstract)
Ni Y, Yang T, Zhang K, Shen C, Chu H (2018) Fungal communities along a small-scale elevational gradient in an alpine tundra are determined by soil carbon nitrogen ratios. Front Microboil 9:1815
Nieminen JK (2009) Modelling the interactions of soil microbes and nematodes. Nematology 11:619–629
Norden N, Angarita HA, Bongers F, Martínez-Ramos M, Cerda IG, Van Breugel M, Lebrija-Trejos E, Meave JA, Vandrmeer J, Williamson GB, Finegan B, Mesquita R, Chazdon RL (2015) Successional dynamics in Neotropical forests are as uncertain as they are predictable. Proc Natl Acad Sci U S A 112:8013–8018
Pei Z, Eichenberg D, Bruelheide H, Kröber W, Kühn P, Li Y, Von Oheimb G, Purschke O, Scholten T, Buscot F, Gutknecht JLM (2016) Soil and tree species traits both shape soil microbial communities during early growth of Chinese subtropical forests. Soil Biol Biochem 96:180–190
R Core Team (2013) R: a language and environment for statistical computing. http://www.r-project.org. Accessed 1 Sept 2018
Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, Knight R, Fierer N (2010) Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4:1340–1351
Rui J, Li J, Wang S, An J, Liu W, Lin Q, Yang Y, He Z, Li X (2015) Response of bacterial communities to simulated climate changes in alpine meadow soil of the Qing-Tibet plateau. Appl Environ Microbiol 81:6070–6077
Saitta A, Anslan S, Bahram M, Brocca L, Tedersoo L (2018) Tree species identity and diversity drive fungal richness and community composition along an elevational gradient in a Mediterranean ecosystem. Mycorrhiza 28:39–47
Sheng Y (2018) The elevational distribution pattern of forest soil fungal diversity and its formation mechanism in Shennongjia National Park, China. Chinese Academy of Forestry, Beijing, p 26
Shi L, Wang J, Su Y, Hou G (1988) Earlier-stage succession of vegetation on the clear-cuts in Miyaluo forest district in Western Sichuan. Chinese J Plant Ecol 12:306–313
Stegen JC, Lin X, Konopka AE, Fredrickson JK (2012) Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J 6:1653–1664
Sun S, Li S, Avera BN, Strahm BD, Badgley BD (2017) Soil bacterial and fungal communities show distinct recovery patterns during forest ecosystem restoration. Appl Environ Microbiol 83:e00966–e00917
Tedersoo L, Bahram M, Cajthaml T, Põlme S, Hiiesalu I, Anslan S, Harend H, Bugger F, Pritsch K, Koricheva J, Abarenkov K (2016) Tree diversity and species identity effects on soil fungi, protists and animals are context dependent. ISME J 10:346–362
Thomson BC, Tisserant ET, Plassart P, Uroz S, Griffiths RI, Hannula SE, Buée M, Mougel C, Ranjard L, Van Veen JA, Martin F, Bailey MJ, Lemanceau P (2015) Soil conditions and land use intensification effects on soil microbial communities across a range of European field sites. Soil Biol Biochem 88:403–413
Tian Y, Sun X, Li S, Wang H, Wang L, Cao J, Zhang L (2012) Biochar made from green waste as peat substitute in growth media for Calathea rotundifola cv. Fasciata. Sci Hortic 143:15–18
Tripathi BM, Kim M, Lai-Hoe A, Shukor NAA, Rahim RA, Go R, Adams JM (2013) pH dominates variation in tropical soil archaeal diversity and community structure. FEMS Microbiol Ecol 86:303–311
Tripathi BM, Kim M, Tateno R, Kim W, Wang J, Lai-Hoe A, Shukor NAA, Rahim RA, Go R, Adams JM (2015) Soil pH and biome are both key determinants of soil archaeal community structure. Soil Biol Biochem 88:1–8
Wan X, Huang Z, He Z, Yu Z, Wang M, Davis MR, Yang Y (2015) Soil C:N ratio is the major determinant of soil microbial community structure in subtropical coniferous and broadleaf forest plantations. Plant Soil 387:103–116
Wang Q, Wu N, Luo P (2009) Simulation of light regimes in typical subalpine forest succession series of eastern Tibetan Plateau. Pol J Ecol 57:251–260
Xiang X, Shi Y, Yang J, Kong J, Lin X, Zhang H, Zeng J, Chu H (2014) Rapid recovery of soil bacterial communities after wildfire in a Chinese boreal forest. Sci Rep 4:3829
Xu Z, Yin H, Xiong P, Wan C, Liu Q (2012) Short-term responses of Picea asperata seedlings of different ages grown in two contrasting forest ecosystems to experimental warming. Environ Exp Bot 77:1–11
Xu H, Li Y, Liu S, Zang R, He F, Spence JR (2015) Partial recovery of a tropical rain forest a half-century after clear-cut and selective logging. J Appl Ecol 52:1044–1052
Yang T, Adams JM, Shi Y, He J, Jing X, Chen L, Tedersoo L, Chu H (2017a) Soil fungal diversity in natural grasslands of the Tibetan plateau: associations with plant diversity and productivity. New Phytol 215:756–765
Yang T, Adams JM, Shi Y, Sun H, Cheng L, Zhang Y, Chu H (2017b) Fungal community assemblages in a high elevation desert environment: absence of dispersal limitation and edaphic effects in surface soil. Soil Biol Biochem 115:393–402
Yin WY (1998) Pictorial keys to soil animals of China. Science Press, Beijing (In Chinese)
You Y, Wang J, Huang X, Tang Z, Liu S, Sun OJ (2014) Relating microbial community structure to functioning in forest soil organic carbon transformation and turnover. Ecol Evol 4:633–647
Zhang Y, Liu S, Ma J, Shi Z, Liu X (2005) Woodland hydrological effects of birch forests in sub-alpine region of western Sichuan, China. Acta Ecol Sin 25:2939–2946 (In Chinese with English abstract)
Zhang X, Liu W, Bai Y, Zhang G, Han X (2011) Nitrogen deposition mediates the effects and importance of chance in changing biodiversity. Mol Ecol 20:429–438
Zhang C, Liu G, Xue S, Wang G (2016a) Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau. Soil Biol Biochem 97:40–49
Zhang X, Liu S, Li X, Wang J, Ding Q, Wang H, Tian C, Yao M, An J, Huang Y (2016b) Changes of soil prokaryotic communities after clear-cutting in a karst forest: evidences for cutting-based disturbance promoting deterministic processes. FEMS Microbiol Ecol 92:fiw026
Zhou D, Yang Y (1984) The change of ecological conditions on the cutting areas in the west Sichuan province. Sci Silvae Sin 20:132–138 (In Chinese with English abstract)
Acknowledgments
We are very grateful to Dr. Shi Songlin from Chengdu University of Technology for assistance in identifying the plant species.
Funding
This research was financially supported by the National Key R&D Program of China (No. 2017YFC0505001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experiments comply with the current laws of China. This article does not contain any studies with human participants or animals performed by any of the authors. Informed consent was obtained from all individual participants included in the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible editor: Huaiying Yao
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 30 kb)
Rights and permissions
About this article

Cite this article
Cao, J., Pan, H., Chen, Z. et al. Bacterial, fungal, and archaeal community assembly patterns and their determining factors across three subalpine stands at different stages of natural restoration after clear-cutting. J Soils Sediments 20, 2794–2803 (2020). https://doi.org/10.1007/s11368-020-02608-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-020-02608-0