
Abstract
Excessive production and migration of vascular smooth muscle cells (VSMCs) are associated with vascular remodeling that causes vascular diseases, such as restenosis and hypertension. Angiotensin II (Ang II) stimulation is a key factor in inducing abnormal VSMC function. This study aimed to investigate the effects of 6′-sialyllactose (6′SL), a human milk oligosaccharide, on Ang II-stimulated cell proliferation, migration and osteogenic switching in rat aortic smooth muscle cells (RASMCs) and human aortic smooth muscle cells (HASMCs). Compared with the control group, Ang II increased cell proliferation by activating MAPKs, including ERK1/2/p90RSK/Akt/mTOR and JNK pathways. However, 6′SL reversed Ang II-stimulated cell proliferation and the ERK1/2/p90RSK/Akt/mTOR pathways in RASMCs and HASMCs. Moreover, 6′SL suppressed Ang II-stimulated cell cycle progression from G0/G1 to S and G2/M phases in RASMCs. Furthermore, 6′SL effectively inhibited cell migration by downregulating NF-κB-mediated MMP2/9 and VCAM-1 expression levels. Interestingly, in RASMCs, 6′SL attenuated Ang II-induced osteogenic switching by reducing the production of p90RSK-mediated c-fos and JNK-mediated c-jun, leading to the downregulation of AP-1-mediated osteopontin production. Taken together, our data suggest that 6′SL inhibits Ang II-induced VSMC proliferation and migration by abolishing the ERK1/2/p90RSK-mediated Akt and NF-κB signaling pathways, respectively, and osteogenic switching by suppressing p90RSK- and JNK-mediated AP-1 activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vascular smooth muscle cells (VSMCs), the predominant constituents of the vascular wall that provide structural support, play an important role in the biomechanical and biochemical functions of blood vessels in the regulation of blood pressure. Excessive proliferation and migration of VSMCs lead to a reduction in blood vessel lumen diameter, consequently causing vascular remodeling in restenosis and hypertension (Mulvany et al. 1996; Owens et al. 2004). Angiotensin II (Ang II), a crucial mediator of the renin-angiotensin system, can bind to the Ang II type 1 receptor to induce pathological effects, such as vasoconstriction, cell proliferation, migration, and hypertrophy, leading to cardiovascular diseases (Lin et al. 2020).
VSMC proliferation is regulated by many downstream signaling pathways, such as the Ras/mitogen-activated protein kinase (MAPK), and Janus kinase/signal transducer and activator of transcription pathways (Wang et al. 2018). In VSMCs, our previous study showed that the activation of 90 kDa ribosomal S6 kinase (p90RSK) downstream of MAPK extracellular signal-regulated kinase 1/2 (ERK1/2) stimulates the protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling pathway, leading to proliferation both in vitro and in vivo (Huynh et al. 2020). In addition, ERK1/2/p90RSK activation also upregulates nuclear factor kappa-light-chain-enhancer of activated B (NF-κB), which in turn increases VSMC migration by inducing matrix metalloproteinases (MMPs), such as MMP2 and MMP9 (Zhang et al. 2005; Huynh et al. 2020). NF-κB is associated with Ang II-induced aortic thickening by binding to the connexin 43 gene promoter, enhancing the communication between endothelial cells and smooth muscle cells under pathogen stimulation (Alonso et al. 2010). This can be explained by the NF-κB transcription factor, which induces chemokines, pro-inflammatory cytokines, and adhesion molecules, consequently recruiting inflammatory cells as well as VSMCs to the intima (Shimizu et al. 2006). Therefore, modulating p90RSK could contribute to the management of VSMC proliferation and migration during vascular remodeling.
VSMCs exert phenotypic plasticity, characterized by switching from a contractile phenotype to a synthetic phenotype and then an osteogenic stage to adapt to abnormal environmental cues induced by vasoconstrictors, such as Ang II and platelet-derived growth factor (Abedin et al. 2004; Jaminon et al. 2019). The osteogenic phenotype is characterized by increased production of Runt-related transcription factor 2, RY-related HMG-box 9, and osteopontin (OPN) (Jaminon et al. 2019; Grootaert and Bennett 2021). OPN production is regulated by the activator protein 1 (AP-1) transcription complex, formed by the dimerization of c-fos and c-jun (Halazonetis et al. 1988). c-jun N-terminal kinase (JNK) activation leads to upregulation of AP-1-induced OPN production (Taurin et al. 2002; Tang et al. 2018). ERK1/2/p90RSK activation could increase c-fos production (Tagaya and Tamaoki 2007). In addition, JNK activation is also implicated in driving VSMC proliferation (Chen et al. 2016; Hu et al. 2020). Thus, targeting the JNK and p90RSK signaling pathways is one strategy to counteract osteogenic switching and further proliferation in vascular remodeling.
6′-sialyllactose (6′SL), one of the major sialylated oligosaccharides among human milk oligosaccharides, consists of sialic acid which binds to the galactosyl unit of lactose at the 6 position (Martín-Sosa et al. 2003). Recently, several studies have revealed that 6′SL possesses beneficial effects on lipopolysaccharide-induced necrotizing enterocolitis and benign prostatic hyperplasia (Jin et al. 2019; Kim et al. 2019; Sodhi et al. 2021). In addition, it has been studied the safety of 6′SL in human and found that oral administration of 3 g of 6′SL twice a day for 12 weeks showed no significant difference in liver and kidney function-related biochemical values, and no clinical serious adverse reactions between the placebo and 6′SL groups in 60 healthy adults (27–79 years old of man and women) (Kim et al. 2022). However, the effects of 6′SL on vascular diseases remain unclear. In the present study, we investigated the effects of 6′SL on vascular remodeling caused by VSMC proliferation, migration, and osteogenic phenotypic switching.
Materials and methods
Antibodies and reagents
Primary antibodies for p90RSK, phospho-p90RSK (Ser380), ERK1/2, phospho-ERK1/2, Akt, phospho-Akt (Ser473), mTOR, phospho-mTOR, NF-κB p65, phospho-NF-κB p65, JNK, phospho-JNK (Thr183/Tyr185), cyclin D1, cyclin-dependent kinase 6 (CDK6), CDK inhibitor (p27), and phospho-retinoblastoma protein (p-Rb) were purchased from Cell Signaling Technology Inc. (Danvers, MA, USA). OPN and proliferating cell nuclear antigen (PCNA) antibodies were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Anti-MMP2 and anti-MMP9 were purchased from EMD Millipore (Daejeon, South Korea). Anti-α-tubulin antibody, anti-IgG antibody, sulforhodamine B sodium salt (SRB), trichloroacetic acid (TCA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and Ang II (#A9525) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Bovine serum albumin (BSA) was purchased from Bioworld (Dublin, OH, USA). FMK (Axon 1848), a specific p90RSK inhibitor, was purchased from Axon Medchem (Reston, VA, USA). SP600125 (#1496), a JNK inhibitor, was purchased from Tocris Bioscience (Bristol, UK). 6′SL sodium salt powder was purchased from GeneChem Inc. (Daejeon, Korea).
Cell culture
Rat aortic smooth muscle cells (RASMCs) and human aortic smooth muscle cells (HASMCs) were purchased from the American Type Culture Collection (Manassas, VA, USA) and Cell Applications (San Diego, CA, USA), respectively. RASMCs were grown in Dulbecco’s modified Eagle’s medium containing 10% (v/v) fetal bovine serum (Gibco, Carlsbad, CA, USA), and 1% (v/v) 100 IU/mL penicillin and 100 µg/mL streptomycin. HASMCs were grown in Medium 231 containing 5% smooth muscle growth supplement (SMGS) (Gibco, Carlsbad, CA, USA) with 1% (v/v) 100 IU/mL penicillin and 100 µg/mL streptomycin. Cells were cultured at 37 °C and 5% CO2 in a humidified atmosphere (HERAcell 150i, Thermo Electron Corp., Waltham, MA, USA). RASMCs and HASMCs at passages 7–15 and passages 5–10, respectively, were used for experiments.
Cell viability assay
Cell viability was performed using 96-well plates at a density of 1 × 104 cells per well with MTT as previously described (Cai et al. 2021).
Cell proliferation assay
SRB binds to basic amino-acid under a mild acidic condition to form a precipitate. SRB cell proliferation assay was performed as previously described (Jeon et al. 2021). Under a basic condition, this precipitate is solubilized, indicating cell mass. After treatment, cells were fixed with 3.3% TCA for 1 h at 4 °C and rinsed with running tap water. After the cells were dried, the cells were incubated with 0.057% SRB for 30 min at room temperature (RT). The unbound dye then was eliminated by rinsing 3 times with 1% acetic acid. The dried dye subsequently was dissolved by 10 mM Tris base, and then was analyzed by a microplate reader at 510 nm (TECAN, Mannedorf, Switzerland).
Cell cycle assay
Cell cycle analysis was performed using MUSE cell cycle kit (#MCH100106, Merck Millipore, Billerica, MA, USA) as previously described (Jeon et al. 2021). After treatment was finished, the cell suspension was centrifuged at 2000 rpm for 1 min at 4 °C. The cell pellets were collected and then washed with PBS prior to being fixed with 70% ethanol at − 20 °C for at least 3 h. After washing with PBS, the fixed cells subsequently were incubated with Muse cell cycle reagent and analyzed by Muse cell analyzer.
Western blot analysis
Western blotting analysis was performed as described previously (Zheng et al. 2021).
Real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
After treatment, total RNA was isolated using Tri-RNA reagent (Favorgen, Pingtung, China) and 1 µg of total RNA was used to analysis qRT-PCR as previously described (Lee et al., 2021). Briefly, the 1 µg of total RNA was used to synthesize single-stranded DNA by the reverse transcription 5X master mix (Elpisbio, Daejeon, Korea). qRT-PCR was performed with specific primers and iQ™ SYBR green supermix (Bio-rad Inc., Hércules, CA, USA). The mRNA expression levels were analyzed using the 2−∆∆Ct method. β-actin was used as the internal control. All rat primer sequences used in qRT-PCR are listed in Table 1.
In vitro wound-healing assay
Cells were seeded in 12-well plates. Once the cells reached 70% confluence, wounds were created characterized by scratches of 200 µl pipette tips. After treatment, these cells were fixed with 4% formaldehyde in PBS for 10 min. The fixed cells then were stained with 0.5% crystal violet solution for 10 min at RT. Images were captured using an Olympus BX51 microscope (Olympus cooperation, Tokyo, Japan).
Immunofluorescence staining
Immunofluorescence staining was performed as described previously (Nguyen et al. 2021). The cells were observed using a laser scanning confocal spectral microscope (K1-Fluo, Nanoscope systems, Daejeon, Korea). Fluorescence intensity was quantified using Image J software.
Statistical analysis
One-way analysis of variance (ANOVA) was performed and followed by a Bonferroni for multiple comparisons (GraphPad 5.02, GraphPad Software Inc., San Diego, CA, USA). A value of p < 0.05 indicates a statistically significant difference. Data were presented as means ± SEM, and experiments were conducted independently at least 2 times.
Results
Effects of Ang II on cell proliferation and related signaling pathways in VSMCs
First, we explored the effect of Ang II on RASMC and HASMC proliferation. Confluent RASMCs or HASMCs were stimulated with various doses of Ang II for 24 h or 48 h, respectively. MTT and SRB assays showed that Ang II significantly enhanced the viability and proliferation of either RASMCs or HASMCs at a dose of 100 nM (Fig. 1A–C). Thus, 100 nM Ang II was chosen for the subsequent experiments. As the ERK1/2/p90RSK axis and JNK signaling pathways have been reported to upregulate Ang II-induced vascular remodeling (Hu et al. 2020; Huynh and Heo 2021), we explored these signaling pathways to confirm the activity of Ang II. As expected, Ang II-induced phosphorylation of ERK1/2, p90RSK, and JNK peaked at 30 min and remained higher than the control group up to 60 min, but decreased from 2 h after Ang II treatment in RASMCs (Fig. 1D). After 12 h of Ang II challenge, total p90RSK expression increased compared to that in the control group (Fig. 1E). We further confirmed the effects of Ang II by examining the p90RSK downstream targets Akt/mTOR under the same conditions. Consistently, Ang II activated Akt/mTOR at 30 to 60 min after Ang II stimulation (Fig. 1F, H).

Effects of Ang II on cell proliferation and related signaling pathways in VSMCs. A, B RASMCs were exposed to serial concentrations of Ang II for 24 h. Cell viability was determined by MTT assay (A) and SRB assay (B), respectively. C HASMCs were stimulated with indicated concentrations of Ang II for 48 h. Cell proliferation was examined by SRB assay. Data represent means ± SEM (n ≥ 3). D–G RASMCs were treated with 100 nM Ang II for indicated times in each figure. Total cell lysates were subjected to western blot analysis to examine indicated protein expressions. α-tubulin was used as a loading control. H, I The bar graphs indicate densitometric quantifications. Data represent means ± SEM (n ≥ 2). *p < 0.05, **p < 0.01, and ***p < 0.001 versus control group. RASMCs rat aortic smooth muscle cells, HASMCs human aortic smooth muscle cells, Ang II angiotensin II, MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, SRB sulforhodamine B sodium salt, ERK1/2 extracellular signal-regulated protein kinase 1/2, JNK c-jun N-terminal kinase, p90RSK p90 ribosomal S6 kinase, Akt protein kinase B, mTOR mammalian target of rapamycin
6′SL inhibits Ang II-induced activation of the ERK1/2/p90RSK/Akt/mTOR signaling pathway
Next, we explored the effects of 6′SL on VSMC proliferation. As shown in Fig. 2A and B, there was a significant reduction in cell viability in the 6′SL-treated groups at a dose of 100 µM 6′SL compared to the Ang II-stimulated group in RASMCs. Similar effects of 6′SL on HASMC proliferation were observed at the dose of 100 µM 6′SL (Fig. 2C). To determine the underlying molecular mechanisms, we investigated the ERK1/2/p90RSK signaling pathway. In RASMCs, treatment with U0216, an ERK1/2 inhibitor, dramatically inhibited Ang II-induced p90RSK activation (Fig. 2D, fF. However, U0126 treatment abolished the phosphorylation of Akt and mTOR to a lesser degree compared to p90RSK (Fig. 2E, F). Likewise, in HASMCs, inhibiting p90RSK phosphorylation by FMK treatment also dramatically decreased the Ang II-induced activation of Akt/mTOR signaling pathway (Fig. 2G–I). Herein, we found that the inhibitory effects of 100 µM 6′SL on Ang II-induced p90RSK/Akt/mTOR activation were significant but lower than that in the U0126-treated group in RASMCs (Fig. 2D–F) and in the FMK-treated group in HASMCs (Fig. 2G–I). These data reveal that inhibition of the ERK1/2/p90RSK signaling pathway is involved in the anti-proliferative effect of 6′SL in RASMCs and HASMCs.

6′sialyllactose (6′SL) inhibits Ang II-induced activation of ERK1/2/p90RSK/Akt/mTOR signaling pathway. A, B RASMCs were pretreated with indicated concentrations of 6′SL for 3 h followed by treatment with 100 nM Ang II for 24 h. Cell viability and cell proliferation were examined by MTT assay (A) and SRB assay (B), respectively. C HASMCs were treated with 6′SL for 3 h prior to stimulation with 100 nM Ang II for 48 h. Cell proliferation was examined by SRB assay. Data represent means ± SEM (n ≥ 3). D, E RASMCs were pretreated with 100 µM 6′SL or 10 µM U0126 (an ERK1/2 inhibitor) for 1 h followed by 100 nM Ang II treatment for 30 min. Phosphorylated and total forms of indicated proteins were evaluated by western blot analysis. G, H HASMCs were treated with 100 µM 6′SL with or without 20 µM FMK (an irreversible inhibitor of p90RSK) treatment followed by 100 nM Ang II treatment for 30 min. Expressions of indicated protein were examined by western blot analysis. α-tubulin was used as a loading control. F, I The bar graphs indicate densitometric quantifications. Data represent means ± SEM (n = 2). *p < 0.05, **p < 0.01 and ***p < 0.001 versus control group; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus Ang II-treated group
6′SL suppresses Ang II-stimulated cell cycle progression
To confirm the anti-proliferative effect of 6′SL, we employed a cell cycle assay to explore the cell cycle distribution. Herein, we found that the cell population of the G0/G1 in the Ang II-treated group was lower than that in the vehicle group. However, this reduction in the cell population of these phages was significantly alleviated by 6′SL treatment (Fig. 4A). These data illustrate that 6′SL treatment inhibited the Ang II-triggered shift of RASMCs from G0/G1 to S and G2/M phases.
Next, we confirmed the effect of 6′SL by examining the cell cycle-related molecules. The cell cycle is regulated by the binding of cyclins to CDKs during specific phases. These complexes stimulate cell cycle progression by phosphorylating transcription regulators, such as Rb (Malumbres 2014). Consistent with the cell cycle assay results, the elevated protein expression and mRNA levels of cell cycle progression molecules including cyclin D1, CDK6 and p-Rb in response to Ang II indicated that Ang II induced the transition from G0 to G1 (Fig. 3B, C). In the later phase, Ang II led to upregulation of cyclin E and CDK2, associated with the transition from G1 phase to S phase (Fig. 3C). Moreover, there was a concomitant downregulation of p27, a cell cycle checkpoint arrestor, in the Ang II-treated group compared to that in the control group. As expected, 6′SL reversed these effects of Ang II on the cell cycle shift from the G0 to G1/S phase (Fig. 3B, C). These data indicated that 6′SL inhibited Ang II-induced cell cycle progression.

6′SL suppresses Ang II-stimulated cell cycle progression. RASMCs were treated with 100 µM 6′SL for 3 h prior to 100 nM Ang II treatment for 24 h (A, B). A Cell cycle profiles were examined by the Muse® Cell Cycle Kit. B Western blot data of cell growth-related proteins including cyclin D1, cyclin-dependent kinase (CDK6) and phospho-retinoblastoma protein (p-Rb), and CDK inhibitor (p27). C Relative mRNA levels of cell cycle molecules were examined by qRT-PCR. Data represent means ± SEM (n = 2). *p < 0.05, **p < 0.01, and ***p < 0.001 versus control group; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus Ang II-treated group
6′SL attenuates Ang II-induced proliferation accompanied by osteogenic switching
Osteogenic switching has been reported to contribute to the progression of vascular remodeling and OPN is one of the markers characterizing osteogenic differentiation (Abedin et al. 2004; Jaminon et al. 2019). Binding of c-fos and c-jun to AP-1 sites leads to osteogenic switching characterized by increased OPN transcription (Renault et al. 2003; Liu et al. 2021). Thus, we further examined whether VSMC proliferation occurs with osteogenic switching and the effect of 6′SL on this process. Both OPN and the proliferation marker PCNA were increased by Ang II treatment in terms of protein expression and mRNA levels. However, 6′SL treatment remarkably reversed the effects of Ang II (Fig. 4A–D). These findings suggest that 6′SL exerts a protective effect on Ang II-induced osteogenic switching in VSMCs.

6′SL attenuates Ang II-induced proliferation accompanied by osteogenic switching. RASMCs were pretreated with 100 µM or 200 µM of 6′SL for 3 h before treatment with 100 nM Ang II for 24 h (A–C) or 12 h (D). A Red fluorescence represents immunofluorescence staining of proliferating cell nuclear antigen (PCNA). B Green fluorescence illustrates immunofluorescence staining of osteopontin (OPN). Blue fluorescence indicates nuclei stained by DAPI. The scale bar indicates 30 μm. Fluorescence intensity was analyzed using Image J software. Data represent means ± SEM (n ≥ 6) C Western blot analysis for PCNA and OPN. D Relative mRNA levels were examined by qRT-PCR. Data represent means ± SEM of two independent experiments. **p < 0.01 and ***p < 0.001 versus control group; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus Ang II-treated group
6′SL diminishes NF-κB pathway in Ang II-stimulated VSMCs
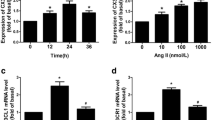
NF-κB signaling pathway is involved in migration by increasing MMPs production (Bond et al. 2001; Lee et al. 2008). Therefore, we determined the effect of 6′SL on NF-κB transcription factor. Immunofluorescence data showed that 6′SL markedly inhibited Ang II-induced translocation of NF-κB from the cytosol to the nucleus in RASMCs (Fig. 5A, B). We also examined NF-κB phosphorylation and found that the Ang II-induced NF-κB activation was significantly decreased by 6′SL treatment not only in RASMCs (Fig. 5C) but also in HASMCs (Fig. 5D). Consistently, 6′SL reduced Ang II-induced NF-κB mRNA (Fig. 5E). In addition, the expression of vascular cell adhesion molecule-1 (VCAM-1), an NF-κB target adhesion molecule, is prevalent in VSMCs in intimal neovessels (Braun et al. 1999). Herein, we found that 6′SL significantly downregulated Ang II-induced VCAM-1 expression (Fig. 5E). These results suggest that 6′SL might inhibit NF-κB-mediated VSMC migration stimulated by Ang II.

6′SL diminishes NF-κB signaling pathway in Ang II-stimulated VSMCs. A RASMCs were pretreated with 100 µM 6′SL for 3 h prior to 100 nM Ang II stimulation for 12 h. Immunofluorescence staining of NF-κB. DAPI was co-indicates for nuclei. The scale bar indicates 30 μm. B Nuclear NF-κB quantification data were analysis using Image J software. Data are represented as means ± SEM (n = 5). RASMCs (C) and HASMCs (D) were treated with 100 µM 6′SL for 1 h followed by 100 nM Ang II treatment for 30 min. Western blot data of phosphorylated and total forms of NF-κB (upper panels). The bar graphs indicate densitometric quantification (lower panels). Data represent means ± SEM (n = 2). E Cellular mRNA levels of NF-κB and vascular cell adhesion molecule-1 (VCAM-1) were measured by qRT-PCR. Data represent means ± SEM (n = 3). **p < 0.01 and ***p < 0.001 versus control group; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus Ang II-treated group
6′SL inhibits Ang II-induced migration of VSMCs
Next, we confirmed the effects of 6′SL on VSMC migration. The wound healing assay showed that Ang II resulted in remarkable migration compared to the control group, whereas 6′SL treatment significantly suppressed this Ang II effect in a dose-dependent manner in RASMCs (Fig. 6A, B). Similarly, 100 µM 6′SL treatment significantly inhibited Ang II-induced HASMC migration but there was no difference between low and high doses of 6′SL treatment (Fig. 6C, D). MMPs, which are target molecules of NF-κB, are considered factors causing the excessive migration of VSMCs (Newby 2006). Thus, we investigated the expression of MMP2 and MMP9 and found that 6′SL treatment significantly reduced Ang II-induced expression of these molecules in RASMCs (Fig. 6E–G). These data suggest that 6′SL exerts anti-migration effects on VSMCs by downregulating NF-κB-mediated MMP2 and MMP9 production.

6′SL inhibits Ang II-induced migration of VSMCs. RASMCs (A, B) and HASMCs (C, D) were treated with 100 or 200 µM 6′SL for 3 h before 100 nM Ang II challenge for 24 h. A, C Wound-healing assay illustrates cell migration. The yellow dotted line indicates scratches at time 0 h. B, D The migration rates were determined by the number of cells that migrated into the wound. The migration area was quantified using Image J software. Data represent means ± SEM (n = 5). E RASMCs were pretreated with 100 µM 6′SL for 3 h prior to 100 nM Ang II challenge for 12 h. Western blot data of matrix metallopeptidase (MMP2) and MMP9. F The bar graph indicates densitometric quantifications. G RASMCs were treated with 100 µM 6′SL for 3 h followed by 100 nM Ang II stimulation for 12 h. Relative mRNA levels of MMP2 and MMP9 were examined by qRT-PCR. Data represent means ± SEM (n = 2).*p < 0.05, **p < 0.01 and ***p < 0.001 versus control group; #p < 0.05, ##p < 0.01 and ###p < 0.001 versus to Ang II group, $p < 0.05 compared to indicated group
6′SL decreases Ang II-induced JNK and p90RSK-mediated AP-1 activity
Regulation of the AP-1 complex, formed by c-fos and c-jun components, is affected by NF-κB and JNK activation (Taurin et al. 2002; Fujioka et al. 2004). Based on these studies, we further investigated the effects of 6′SL on the JNK pathway in RASMCs and found that 6′SL treatment significantly abolished JNK phosphorylation induced by Ang II, and that 6′SL remarkably decreased c-jun mRNA levels compared to Ang II treatment (Fig. 7A–C). In addition, we also checked c-fos production and found that Ang II-induced excessive production of c-fos was considerably downregulated by 6′SL treatment (Fig. 7D). Next, we examined whether there was crosstalk between JNK and p90RSK, and found that there was no change in the phosphorylation of JNK or p90RSK under FMK (an irreversible inhibitor of p90RSK) or SP600125 (a JNK inhibitor), respectively (Fig. 7E). However, FMK pre-treatment significantly inhibited the Ang II-stimulated c-fos and c-jun transcription (Fig. 7F, G). These data suggest that 6′SL has the potential to reduce proliferation and AP-1-mediated phenotypic switching by inhibiting the p90RSK and JNK signaling pathways.

6′SL decreases Ang II-induced JNK and p90RSK-mediated AP-1 activity. A RASMCs were treated with 100 µM 6′SL for 1 h before 30 min exposure to 100 nM Ang II. Protein expressions of phospho-JNK and JNK were examined by western blot analysis. B The bar graph illustrates densitometric quantifications. C, D RASMCs were treated with 100 µM 6′SL for 3 h before stimulation with 100 nM Ang II treatment for 12 h. Relative mRNA levels of activator protein-1 (AP-1) components, including c-jun and c-fos, were examined by qRT-PCR. E RASMCs were pretreated with 100 nM SP600125 (a JNK inhibitor) or 20 µM FMK (a p90RSK inhibitor) for 1 h followed by 100 nM Ang II stimulation for 30 min. Cell lysates were subjected to western blot analysis for detecting indicated molecules. F, G RASMCs were treated with 100 nM SP600125 or 20 µM FMK for 3 h before 100 nM Ang II stimulation for 12 h. Relative mRNA levels of c-fos and c-jun were examined by qRT-PCR. Data represent means ± SEM (n = 3). *p < 0.05 and **p < 0.01 versus control group; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus Ang II-treated group
Discussion
Vascular remodeling is associated with the pathological effects of vasoconstrictors such as Ang II, causing vascular diseases such as restenosis and hypertension (Owens et al. 2004). 6′SL has been reported to exert various protective effects (Jin et al. 2019; Kim et al. 2019; Sodhi et al. 2021). However, there are no studies on the effect of 6′SL on vascular diseases. In this study, we demonstrated that 6′SL had inhibitory effects on Ang II-induced VSMC proliferation and migration in RASMCs by attenuating ERK1/2/p90RSK-mediated Akt and NF-κB signaling pathways, respectively. In addition, 6′SL inhibited osteogenic switching by suppressing AP-1-mediated OPN production by abolishing p90RSK and JNK signaling pathways (Fig. 8).

A schematic diagram illustrates anti-proliferation, anti-migration, and anti-osteogenic switching effects of 6′SL in RASMCs. In general, Ang II binds to AT1R and subsequently activates ERK1/2/p90RSK signaling pathway. The phosphorylation of p90RSK leads to the Akt activation, resulting in the downregulation of a cell cycle checkpoint arrestor p27 and the upregulation of cell cycle progression molecules including cyclins, CDKs, p-Rb, and PCNA. Moreover, p90RSK activation also induces nuclear translocation of NF-κB transcription factor, leading to the increased transcription of MMP2, MMP9 and VCAM-1 to increase VSMC migration. In addition, p90RSK activation further stimulates c-fos production. On the other hand, Ang II triggers JNK and NF- κB-mediated c-jun production, thereby increasing AP-1 transcriptional activity and in turn enhancing OPN. In this study, 6′SL not only decreased Ang II-induced p90RSK-mediated proliferation and migration, but also effectively inhibited AP-1-mediated osteogenic phenotypic switching. 6′SL 6′-sialyllactose, RASMCs rat aortic smooth muscle cells, Ang II angiotensin II, AT1R Ang II type 1 receptor, MAP3K mitogen-activated protein kinase kinase kinase, ERK1/2 extracellular signal regulated protein kinase 1/2, p90RSK p90 ribosomal S6 kinase, JNK c-jun N-terminal kinase, Akt protein kinase B, mTOR mammalian target of rapamycin, NF-κB nuclear factor kappa-light-chain-enhancer of activated B, AP-1 activator protein-1, VCAM-1 vascular cell adhesion molecule-1, MMP matrix metallopeptidase, CDK cyclin-dependent kinase, OPN osteopontin, PCNA proliferating cell nuclear antigen, Rb retinoblastoma protein, p27 cyclin-dependent kinase inhibitor, P phosphorylation
VSMC hyperplasia is closely related to vascular remodeling. The Akt/mTOR signaling pathway has been reported to be involved in promoting VSMC proliferation both in vitro and in vivo (Stabile et al. 2003; Yin et al. 2017). In our previous study, we found that blocking p90RSK effectively reduced Ang II-induced Akt/mTOR-mediated VSMC proliferation (Huynh et al. 2020). In the current study, we observed that 100 nM Ang II induced the phosphorylation of ERK1/2/p90RSK and its downstream target Akt/mTOR, resulting in an increase in VSMC proliferation (Fig. 1). The ERK1/2/p90RSK/Akt/mTOR axis was confirmed by inhibiting ERK1/2 or p90RSK with U0126 or FMK, individually, in RASMCs or HASMCs (Fig. 2). Interestingly, inhibition of ERK1/2 totally blocked Ang II-accelerated p90RSK activation (Fig. 2). In HASMCs, inhibiting p90RSK activation totally eliminated the effect of Ang II on Akt/mTOR phosphorylation (Fig. 2). Herein, we found that 100 µM 6′SL significantly blunted Ang II-induced activation of ERK1/2/p90RSK/Akt/mTOR, but 6′SL was less effective than in U0126 treatment in RASMCs and in FMK-treated HASMCs (Fig. 2). These data reveal that inhibition of the ERK1/2/p90RSK pathway is involved in the anti-proliferative effect of 6′SL in VSMCs. This effect was further confirmed by examining cell cycle profiles. As expected, 6′SL significantly reduced the elevated levels of cycle progression molecules, which are Akt/mTOR target molecules, such as cyclin D1, CDK6, cyclin E, CDK2, and p-Rb in response to Ang II in RASMCs (Fig. 3). Furthermore, 6′SL enhanced Ang II-induced p27 reduction, inhibiting the cell cycle shift from G0 to G1/S phase (Fig. 3). These data revealed that 6′SL inhibited VSMC proliferation by inhibiting the p90RSK-mediated Akt/mTOR signaling pathway.
The transcription factor NF-κB is a characteristic of vascular remodeling owing to its effect on VSMC migration (Zahradka et al. 2002). In our previous study, we showed that Ang II stimulated NF-κB activation by phosphorylating ERK1/2/p90RSK in VSMCs, contributing to the increase in MMPs (Huynh et al. 2020). In agreement with this report, here, immunofluorescence data showed that there was an increase in NF-κB nuclear translocation in Ang II group compared to the control group (Fig. 5A, B). In addition, Ang II increased NF-κB phosphorylation in RASMCs (Fig. 5C) and HASMCs (Fig. 5D). As expected, the effects of Ang II were effectively reversed by treatment with 6′SL in both these cell lines (Fig. 5C, D). In previous report, the genetic deficiency of MMP2 or MMP9 in mice leads to reduction of VSMC invasion and MMP2 deficiency inhibits this event more effectively than MMP9. However, MMP9 but not MMP2 plays a vital role in the attachment of VMSCs to collagen thereby VSMCs degrade collagen and consequently facilitate VSMC migration (Johnson and Galis 2004). Herein, we found that 6′SL inhibited NF-κB-mediated MMP2 and MMP9 production and expression, thereby suppressing Ang II-induced migration (Fig. 6). In addition, the expression of NF-κB target adhesion molecules such as VCAM-1 is prevalent in VSMCs in intimal neovessels (Braun et al. 1999). The presence of adhesion molecules in turn recruits immune cells including macrophages and leukocytes, resulting in excessive production of pro-inflammatory mediators and subsequent inflammation and late neointimal thickening (Welt and Rogers 2002). In the present study, Ang II increased VCAM-1 production, whereas 6′SL considerably diminished this effect (Fig. 5E). Collectively, these data suggest that p90RSK/NF-κB signaling pathway is involved in the anti-migratory effect of 6′SL.
Accumulating evidence has demonstrated that osteogenic switching plays an important role in the progression of vascular remodeling, and OPN is one of the markers of osteogenic differentiation (Abedin et al. 2004; Jaminon et al. 2019). Therefore, in this study, we examined whether VSMC proliferation occurs during osteogenic switching. As expected, we found that Ang II increased both OPN and the proliferation marker PCNA in terms of mRNA level and protein expression, suggesting that VSMC proliferation is accompanied by osteogenic phenotypic switching (Fig. 4). Interestingly, this effect of Ang II was considerably mitigated by treatment with 100 µM 6′SL.
Since the activity of AP-1 transcription factor in VSMCs plays an important role in regulating OPN production, we further examined two members of the AP-1 transcription complex, c-fos and JNK-mediated c-jun (Halazonetis et al. 1988; Renault et al. 2003). Here, we found that Ang II remarkably augmented JNK phosphorylation and c-jun mRNA levels, whereas 6′SL completely inhibited Ang II-induced JNK activation and reversed c-jun transcription (Fig. 7A-C). Concurrently, Ang II also induced an increase in c-fos production, but 6′SL treatment significantly reversed this effect of Ang II (Fig. 7D). These data suggest that 6′SL reduces the activity of AP-1 transcription factor and subsequently abolishes OPN production.
To investigate whether there was a relationship between p90RSK and JNK, we pretreated RASMCs with SP600125, a JNK inhibitor, or FMK, a p90RSK inhibitor, before stimulation with Ang II. We found that no crosstalk between JNK and p90RSK activation (Fig. 7E). However, FMK treatment significantly reduced Ang II-induced c-jun transcription to a lesser extent than SP600125 treatment. This may be explained by the involvement of p90RSK-mediated NF-κB activation in the enhancement of AP-1 activity and the consequent production of c-jun (Fujioka et al. 2004). Interestingly, pre-treatment with FMK, but not SP600125 significantly reduced c-fos production (Fig. 7G). This may result from the effect of ERK1/2/p90RSK activation on c-fos as previously described (Tagaya and Tamaoki 2007; He et al. 2010). In addition, there is accumulating evidence indicating that JNK activation results in VSMC proliferation by downregulating p27 and upregulating cyclins and CDKs (Banes et al. 2001; Zhan et al. 2003; Chen et al. 2016). Thus, our findings suggest that 6′SL exerts an inhibitory effect on anti-osteogenic switching and anti-proliferation of VSMCs by attenuating the activation of JNK and p90RSK-mediated AP-1 activity.
In conclusion, our data suggest that 6′SL exerts a protective effect against Ang II-induced vascular remodeling by inhibiting p90RSK/Akt-mediated proliferation and p90RSK/NF-κB-mediated migration of RASMCs and HASMCs. In addition, 6′SL diminished Ang II-induced osteogenic phenotypic switching via inhibition of JNK and p90RSK signaling pathways, thereby downregulating AP-1-mediated OPN production. Thus, 6′SL is a potential therapeutic candidate for vascular remodeling.
References
Abedin M, Tintut Y, Demer LL (2004) Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol 24:1161–1170. https://doi.org/10.1161/01.Atv.0000133194.94939.42
Alonso F, Krattinger N, Mazzolai L, Simon A, Waeber G, Meda P, Haefliger JA (2010) An angiotensin II- and NF-kappaB-dependent mechanism increases connexin 43 in murine arteries targeted by renin-dependent hypertension. Cardiovasc Res 87:166–176. https://doi.org/10.1093/cvr/cvq031
Banes AK, Loberg RD, Brosius FC 3rd, Watts SW (2001) Inability of serotonin to activate the c-Jun N-terminal kinase and p38 kinase pathways in rat aortic vascular smooth muscle cells. BMC Pharmacol 1:8. https://doi.org/10.1186/1471-2210-1-8
Bond M, Chase AJ, Baker AH, Newby AC (2001) Inhibition of transcription factor NF-kappaB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc Res 50:556–565. https://doi.org/10.1016/s0008-6363(01)00220-6
Braun M, Pietsch P, Schrör K, Baumann G, Felix SB (1999) Cellular adhesion molecules on vascular smooth muscle cells. Cardiovasc Res 41:395–401. https://doi.org/10.1016/s0008-6363(98)00302-2
Cai H, Zheng Y, Wen Z, Yang Y, Yang S, Zhang Q (2021) LncRNA AIRN influences the proliferation and apoptosis of hepatocellular carcinoma cells by regulating STAT1 ubiquitination. Arch Pharm Res 44:414–426. https://doi.org/10.1007/s12272-021-01317-7
Chen CC, Liang CJ, Leu YL, Chen YL, Wang SH (2016) Viscolin inhibits in vitro smooth muscle cell proliferation and migration and neointimal hyperplasia in vivo. PLoS ONE 11:e0168092. https://doi.org/10.1371/journal.pone.0168092
Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL, Chiao PJ (2004) NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol 24:7806–7819. https://doi.org/10.1128/mcb.24.17.7806-7819.2004
Grootaert MOJ, Bennett MR (2021) Vascular smooth muscle cells in atherosclerosis: time for a re-assessment. Cardiovasc Res 117:2326–2339. https://doi.org/10.1093/cvr/cvab046
Halazonetis TD, Georgopoulos K, Greenberg ME, Leder P (1988) c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 55:917–924. https://doi.org/10.1016/0092-8674(88)90147-x
He Q, Harding P, LaPointe MC (2010) PKA, Rap1, ERK1/2, and p90RSK mediate PGE2 and EP4 signaling in neonatal ventricular myocytes. Am J Physiol Heart Circ Physiol 298:H136-143. https://doi.org/10.1152/ajpheart.00251.2009
Hu C, Zuo K, Li K, Gao Y, Chen M, Hu R, Liu Y, Chi H, Wang H, Qin Y, Liu X, Zhong J, Cai J, Yang X, Li J (2020) p38/JNK is required for the proliferation and phenotype changes of vascular smooth muscle cells induced by L3MBTL4 in essential hypertension. Int J Hypertens 2020:3123968. https://doi.org/10.1155/2020/3123968
Huynh DTN, Heo KS (2021) Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch Pharm Res 44:1051–1061. https://doi.org/10.1007/s12272-021-01360-4
Huynh DTN, Jin Y, Myung CS, Heo KS (2020) Inhibition of p90RSK is critical to abolish Angiotensin II-induced rat aortic smooth muscle cell proliferation and migration. Biochem Biophys Res Commun 523:267–273. https://doi.org/10.1016/j.bbrc.2019.12.053
Jaminon A, Reesink K, Kroon A, Schurgers L (2019) The role of vascular smooth muscle cells in arterial remodeling: focus on calcification-related processes. Int J Mol Sci. https://doi.org/10.3390/ijms20225694
Jeon H, Jin Y, Myung CS, Heo KS (2021) Ginsenoside-Rg2 exerts anti-cancer effects through ROS-mediated AMPK activation associated mitochondrial damage and oxidation in MCF-7 cells. Arch Pharm Res 44:702–712. https://doi.org/10.1007/s12272-021-01345-3
Jin BR, Kim HJ, Kim EY, Chung TW, Ha KT, An HJ (2019) 6’-Sialyllactose ameliorates in vivo and in vitro benign prostatic hyperplasia by regulating the E2F1/pRb-AR pathway. Nutrients. https://doi.org/10.3390/nu11092203
Johnson C, Galis ZS (2004) Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol 24:54–60. https://doi.org/10.1161/01.Atv.0000100402.69997.C3
Kim EY, Jin BR, Chung TW, Bae SJ, Park H, Ryu D, Jin L, An HJ, Ha KT (2019) 6-sialyllactose ameliorates dihydrotestosterone-induced benign prostatic hyperplasia through suppressing VEGF-mediated angiogenesis. BMB Rep 52:560–565. https://doi.org/10.5483/BMBRep.2019.52.9.113
Kim JH, Yong SY, Kim SH, Baek A, Go TH, Kang DR (2022) Randomized, triple-blind, placebo-controlled study to evaluate the safety of 6’-Sialyllactose in healthy adults. Regul Toxicol Pharmacol 129:105110. https://doi.org/10.1016/j.yrtph.2021.105110
Lee SJ, Seo KW, Yun MR, Bae SS, Lee WS, Hong KW, Kim CD (2008) 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-kappaB signaling pathways. Free Radic Biol Med 45:1487–1492. https://doi.org/10.1016/j.freeradbiomed.2008.08.022
Lee S, Pham DV, Park PH (2021) Sestrin2 induction contributes to anti-inflammatory responses and cell survival by globular adiponectin in macrophages. Arch Pharm Res 45:38–50. https://doi.org/10.1007/s12272-021-01364-0
Lin QY, Bai J, Liu JQ, Li HH (2020) Angiotensin II stimulates the proliferation and migration of lymphatic endothelial cells through angiotensin Type 1 receptors. Front Physiol 11:560170. https://doi.org/10.3389/fphys.2020.560170
Liu Q, Xiang P, Chen M, Luo Y, Zhao Y, Zhu J, Jing W, Yu H (2021) Nano-sized hydroxyapatite induces apoptosis and osteogenic differentiation of vascular smooth muscle cells via JNK/c-JUN pathway. Int J Nanomed 16:3633–3648. https://doi.org/10.2147/ijn.S303714
Malumbres M (2014) Cyclin-dependent kinases. Genome Biol 15:122. https://doi.org/10.1186/gb4184
Martín-Sosa S, Martín MJ, García-Pardo LA, Hueso P (2003) Sialyloligosaccharides in human and bovine milk and in infant formulas: variations with the progression of lactation. J Dairy Sci 86:52–59. https://doi.org/10.3168/jds.S0022-0302(03)73583-8
Mulvany MJ, Baumbach GL, Aalkjaer C, Heagerty AM, Korsgaard N, Schiffrin EL, Heistad DD (1996) Vascular remodeling. Hypertension 28:505–506
Newby AC (2006) Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res 69:614–624. https://doi.org/10.1016/j.cardiores.2005.08.002
Nguyen TLL, Huynh DTN, Jin Y, Jeon H, Heo KS (2021) Protective effects of ginsenoside-Rg2 and -Rh1 on liver function through inhibiting TAK1 and STAT3-mediated inflammatory activity and Nrf2/ARE-mediated antioxidant signaling pathway. Arch Pharm Res 44:241–252. https://doi.org/10.1007/s12272-020-01304-4
Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84:767–801. https://doi.org/10.1152/physrev.00041.2003
Renault MA, Jalvy S, Belloc I, Pasquet S, Sena S, Olive M, Desgranges C, Gadeau AP (2003) AP-1 is involved in UTP-induced osteopontin expression in arterial smooth muscle cells. Circ Res 93:674–681. https://doi.org/10.1161/01.Res.0000094747.05021.62
Shimizu K, Mitchell RN, Libby P (2006) Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 26:987–994. https://doi.org/10.1161/01.ATV.0000214999.12921.4f
Sodhi CP, Wipf P, Yamaguchi Y, Fulton WB, Kovler M, Niño DF, Zhou Q, Banfield E, Werts AD, Ladd MR, Buck RH, Goehring KC, Prindle T Jr, Wang S, Jia H, Lu P, Hackam DJ (2021) The human milk oligosaccharides 2’-fucosyllactose and 6’-sialyllactose protect against the development of necrotizing enterocolitis by inhibiting toll-like receptor 4 signaling. Pediatr Res 89:91–101. https://doi.org/10.1038/s41390-020-0852-3
Stabile E, Zhou YF, Saji M, Castagna M, Shou M, Kinnaird TD, Baffour R, Ringel MD, Epstein SE, Fuchs S (2003) Akt controls vascular smooth muscle cell proliferation in vitro and in vivo by delaying G1/S exit. Circ Res 93:1059–1065. https://doi.org/10.1161/01.Res.0000105086.31909.1b
Tagaya E, Tamaoki J (2007) Mechanisms of airway remodeling in asthma. Allergol Int 56:331–340. https://doi.org/10.2332/allergolint.R-07-152
Tang L, Dai F, Liu Y, Yu X, Huang C, Wang Y, Yao W (2018) RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol Res 133:201–212. https://doi.org/10.1016/j.phrs.2018.05.011
Taurin S, Dulin NO, Pchejetski D, Grygorczyk R, Tremblay J, Hamet P, Orlov SN (2002) c-Fos expression in ouabain-treated vascular smooth muscle cells from rat aorta: evidence for an intracellular-sodium-mediated, calcium-independent mechanism. J Physiol 543:835–847. https://doi.org/10.1113/jphysiol.2002.023259
Wang D, Uhrin P, Mocan A, Waltenberger B, Breuss JM, Tewari D, Mihaly-Bison J, Huminiecki Ł, Starzyński RR, Tzvetkov NT, Horbańczuk J, Atanasov AG (2018) Vascular smooth muscle cell proliferation as a therapeutic target. Part 1: molecular targets and pathways. Biotechnol Adv 36:1586–1607. https://doi.org/10.1016/j.biotechadv.2018.04.006
Welt FG, Rogers C (2002) Inflammation and restenosis in the stent era. Arterioscler Thromb Vasc Biol 22:1769–1776. https://doi.org/10.1161/01.atv.0000037100.44766.5b
Yin Q, Jiang D, Li L, Yang Y, Wu P, Luo Y, Yang R, Li D (2017) LPS promotes vascular smooth muscle cells proliferation through the TLR4/Rac1/Akt signalling pathway. Cell Physiol Biochem 44:2189–2200. https://doi.org/10.1159/000486024
Yoon YC, Fang Z, Lee JE, Park JH, Ryu JK, Jung KH, Hong SS (2020) Selonsertib inhibits liver fibrosis via downregulation of ASK1/ MAPK pathway of hepatic stellate cells. Biomol Ther (Seoul) 28:527–536. https://doi.org/10.4062/biomolther.2020.016
Zahradka P, Werner JP, Buhay S, Litchie B, Helwer G, Thomas S (2002) NF-kappaB activation is essential for angiotensin II-dependent proliferation and migration of vascular smooth muscle cells. J Mol Cell Cardiol 34:1609–1621. https://doi.org/10.1006/jmcc.2002.2111
Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao T, Miyazaki H, Iwao H (2003) Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol 23:795–801. https://doi.org/10.1161/01.Atv.0000066132.32063.F2
Zhang L, Ma Y, Zhang J, Cheng J, Du J (2005) A new cellular signaling mechanism for angiotensin II activation of NF-kappaB: An IkappaB-independent, RSK-mediated phosphorylation of p65. Arterioscler Thromb Vasc Biol 25:1148–1153. https://doi.org/10.1161/01.ATV.0000164624.00099.e7
Zheng X, Liu H, Ma M, Ji J, Zhu F, Sun L (2021) Anti-thrombotic activity of phenolic acids obtained from Salvia miltiorrhiza f. alba in TNF-α-stimulated endothelial cells via the NF-κB/JNK/p38 MAPK signaling pathway. Arch Pharm Res 44:427–438. https://doi.org/10.1007/s12272-021-01325-7
Acknowledgements
This research was funded by National Research Foundation of Korea (KNRF-2019R1C1C1007331 and 2022R1A2C4001776).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nguyen, T.L.L., Jin, Y., Kim, L. et al. Inhibitory effects of 6′-sialyllactose on angiotensin II-induced proliferation, migration, and osteogenic switching in vascular smooth muscle cells. Arch. Pharm. Res. 45, 658–670 (2022). https://doi.org/10.1007/s12272-022-01404-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-022-01404-3