
Abstract
Celecoxib is a non-steroidal anti-inflammatory drug (NSAID) and a representative selective cyclooxygenase (COX)-2 inhibitor, which is commonly prescribed for osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, acute pain, and primary dysmenorrhea. It is mainly metabolized by CYP2C9 and partly by CYP3A4 after oral administration. Many studies reported that CYP2C9 genetic polymorphism has significant effects on the pharmacokinetics of celecoxib and the occurrence of adverse drug reactions. The aim of this study was to develop a physiologically based pharmacokinetic (PBPK) model of celecoxib according to CYP2C9 genetic polymorphism for personalized pharmacotherapy. Initially, a clinical pharmacokinetic study was conducted where a single dose (200 mg) of celecoxib was administered to 39 healthy Korean subjects with CYP2C9*1/*1 or CYP2C9*1/*3 genotypes to obtain data for PBPK development. Based on the conducted pharmacokinetic study and a previous pharmacokinetic study involving subjects with CYP2C9*1/*13 and CYP2C9*3/*3 genotype, PBPK model for celecoxib was developed. A PBPK model for CYP2C9*1/*1 genotype group was developed and then scaled to other genotype groups (CYP2C9*1/*3, CYP2C9*1/*13 and CYP2C9*3/*3). After model development, model validation was performed with comparison of five pharmacokinetic studies. As a result, the developed PBPK model of celecoxib successfully described the pharmacokinetics of each CYP2C9 genotype group and its predicted values were within the acceptance criterion. Additionally, all the predicted values were within two-fold error range in comparison to the previous pharmacokinetic studies. This study demonstrates the possibility of determining the appropriate dosage of celecoxib for each individual through the PBPK modeling with CYP2C9 genomic information. This approach could contribute to the reduction of adverse drug reactions of celecoxib and enable precision medicine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Celecoxib is a representative cyclooxygenase (COX)-2 selective inhibitor and a non-steroidal anti-inflammatory drug (NSAID) commonly prescribed for osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, acute pain, and primary dysmenorrhea (Goldstein et al. 2001; Pfizer Inc. 2016). Like other NSAIDs, celecoxib is highly protein bound (> 97%), especially to albumin (Davies et al. 2000). Celecoxib undergoes hepatic metabolism mainly by methyl-hydroxylation to hydroxy celecoxib and further by oxidation to carboxy celecoxib, and this hepatic metabolism is the main pathway for elimination of celecoxib. CYP2C9 plays a major role in the methyl-hydroxylation of celecoxib and CYP3A4 is also involved in the hydroxylation process, albeit to a lesser extent (Davies et al. 2000; Paulson et al. 2000; Sandberg et al. 2002). CYP2C9 is one of the most important metabolizing enzymes for many of the prescribed drugs (Rettie et al. 2005). The human CYP2C9 gene is highly polymorphic in its promoter and coding regions, and more than 71 variant alleles of CYP2C9 have been reported (https://www.pharmvar.org/gene/CYP2C9). The allelic variants vary in frequency among different ethnic groups, and CYP2C9 *2 and *3 are major variant alleles in most ethnic groups. The CYP2C9*2 and *3 variants have been reported to be significantly prevalent in Caucasians (8.0–19.1% and 3.3–16.2%, respectively). However, CYP2C9*2 is absent in East Asians (Chinese, Japanese, and Korean), and CYP2C9*3 and *13 occur at frequencies of 1.0–6.0% and 0.2–1.0%, respectively (Kimura et al. 1998; Bae et al. 2005; Dai et al. 2014; Ding et al. 2015; Kim et al. 2017). CYP2C9*2, *3 and *13 alleles are associated with significant reductions in intrinsic clearance of a variety of CYP2C9 substrates compared with CYP2C9* 1; however, the degree of these reductions appear to be highly substrate-dependent (Lee et al. 2002).
Although one clinical study indicated that CYP2C9 genotype does not affect the steady-state systemic exposure (area under the curve, AUC) and elimination rate (Brenner et al. 2003), most of the pharmacogenetic studies for celecoxib have reported that subjects with CYP2C9*3 or CYP2C9*13 allele including CYP2C9*1/*3, CYP2C9*1/*13 and CYP2C9*3/*3 genotype resulted in significantly increased AUC and maximum plasma concentration (Cmax) compared to subjects with CYP2C9*1/*1 genotype (extensive metabolizers, EMs) (Tang et al. 2001; Kirchheiner et al. 2003; Stempak et al. 2005; Lundblad et al. 2006; Prieto-Perez et al. 2013; Liu et al. 2015; Kim et al. 2017; Park et al. 2018). Administration of celecoxib is associated with gastrointestinal (GI), cardiovascular, and renal adverse events (Mohammed et al. 1999; Moore et al. 2005; Caldwell et al. 2006; Pfizer Inc. 2016; Kim et al. 2017). Especially, one study demonstrated that subjects with higher AUC and Cmax resulted in more adverse events than those with lower values (Liu et al. 2015). Furthermore, another study reported that one patient identified as an intermediate metabolizer (IM) of CYP2C9 had gastropathy after taking celecoxib (Gupta et al. 2015).
Physiologically based pharmacokinetic (PBPK) modeling is a tool that enables a priori simulation of drug concentration–time profiles through a mechanistic approach to the pharmacokinetics of drug by integrating biological and physiological information at the organism level (Kuepfer et al. 2016). Based on the pharmacokinetic data from a clinical study, PBPK modeling can be applied to simulate the pharmacokinetic profile of different administration protocols and drug-drug interactions (Zhuang and Lu 2016). Due to these characteristics, utilization of PBPK modeling for drug development and discovery has rapidly developed (Jones et al. 2015). Moreover, several studies indicated that PBPK modeling can also be used to apply genetic polymorphism effects on the pharmacokinetics of drugs (Yeo et al. 2013; Vieira et al. 2014; Emoto et al. 2015; Djebli et al. 2015; Duan et al. 2017; Futatsugi et al. 2018; Gong et al. 2018).
In this study, a PBPK model of celecoxib was developed according to CYP2C9 genetic polymorphism based on reported in vitro metabolic rate data (Tang et al. 2001) with CYP2C9 and CYP3A4 enzymes. The objective of this study was to develop a PBPK model to obtain the optimal dosage of celecoxib related to CYP2C9 genetic polymorphism.
Methods
Subjects
Twenty-four healthy subjects with CYP2C9*1/*1 genotype and fifteen subjects with CYP2C9*1/*3 genotype were recruited for the pharmacokinetic study. Subjects were healthy according to medical history, physical examination, and routine laboratory tests including urine analysis, hematology, and blood chemistry. Their CYP2C9 genotypes were confirmed by polymerase chain reaction restriction fragment length polymorphism (PCR–RFLP) methods as previously described (Bae et al. 2005).
Study design
All subjects provided informed consent for the study. The study complied with the Declaration of Helsinki and was approved by the Institutional Review Board of Metro hospital (Anyang, Republic of Korea). Pharmacokinetic study of celecoxib was conducted as an open-label, single-phase study. After overnight fasting, participants were administered a single oral dose of 200 mg celecoxib (CelebrexⓇ, Pfizer Korea, Seoul, Republic of Korea). Blood samples were collected in EDTA tubes before and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24, 36, and 48 h after administration. Meals were provided at 4, 10, 24, and 30 h after drug administration. Collected blood samples were centrifuged at 3000 rpm for 10 min. Then, the supernatant plasma samples from the EDTA tubes were stored at − 70 °C until needed.
Determination of plasma concentration
HPLC–MS/MS (High Performance Liquid Chromatography-tandem mass spectrometry) analysis of celecoxib and valdecoxib (Internal standard, IS) was determined and validated based on following published method (Kim et al. 2017). HPLC was operated on an Agilent 1200 series HPLC system (Santa Clara, CA, USA) and mass spectrometry was performed by Applied Biosystems SCIEX API 3200 series (Toronto, ON, Canada). Phenomenex Luna phenyl hexyl column (100 × 2.0 mm, 3 μm, Torrance, CA, USA) was selected for analyte separation. The mobile phase, a mixture of 10 mM ammonium formate (pH 3.5, adjusted with formic acid) with HPLC grade water and acetonitrile (10: 90, v/v) was used for analysis and set to 0.2 mL/min in HPLC system. In quantitative analysis, MRM (Multiple Reaction Monitoring) mode with a dwell time of 250 ms was selected for analyzing celecoxib (m/z 380.2 → 316.1) and IS (m/z 313.3 → 118.0). The calibration curve was linear over the range of 2–1000 ng/mL for celecoxib.
Data and statistical analysis
All pharmacokinetic parameter calculations for celecoxib analysis was based on BA calc 2007 software from MFDS (Ministry of Food and Drugs Safety, Republic of Korea). Maximum plasma concentration values of celecoxib (Cmax) and the time to reach maximum plasma concentration Cmax (Tmax) were experimentally observed values. Area under the curve (AUC: AUC0-t, AUCinf) parameters were assessed based on trapezoidal rule, and t was the last time of measured concentration. AUC0-t was area under the plasma concentration time curve from time 0 to t and AUCinf was area under the plasma concentration time curve from time 0 to infinite. The clearance was calculated as CL/F = dose/AUCinf and half-life was calculated as T1/2 = ln 2/ke and ke was the elimination constant which was derived from terminal data from a concentration–time plot. All of the calculated pharmacokinetic data results were expressed as mean ± SD (standard deviation). After consideration of normality and equal variance, differences in pharmacokinetic parameters between CYP2C9 genotype groups were evaluated using the student t-test. All results were analyzed by SigmaPlotⓇ version 12 (Systat Software Inc., Chicago, IL, USA). In this study, P values < 0.05 were considered statistically significant.
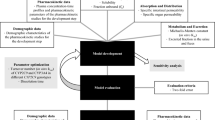
PBPK model construction and workflow
The PBPK modeling of celecoxib was developed and optimized using the latest PK-SimⓇ software (Version 7.2, Bayer AG, Wuppertal, Germany). The pharmacokinetic data used for model development was based on the pharmacokinetic information involving subjects with CYP2C9*1/*1 and CYP2C9*1/*3 genotypes conducted in this study. Basic information for modeling population group including gender, age, weight, height, and BMI was based on subjects who participated in the pharmacokinetic study. In the case of CYP2C9*1/*13 (n = 5) and CYP2C9*3/*3 (n = 2) genotype groups, pharmacokinetic information from a previously published research was applied (Kim et al. 2017).
Basic physico-chemistry data for celecoxib was collected from published literature (Paulson et al. 1999; Baek et al. 2015), PubChem (https://pubchem.ncbi.nlm.nih.gov), and Drug Bank (https://www.drugbank.ca). Adjusted lipophilicity for celecoxib was 3.9 per data provided by Drug Bank. Fraction unbound (fu) value was 3.5% based on literature (Paulson et al. 1999). ADME properties used in PK-SimⓇ were adjusted based on a latest human compartmental GI model including fluid secretion and absorption, which was used to simulate the absorption (Thelen et al. 2011, 2012).
Poulin and Theil method was used to calculate the organ-plasma partition coefficients. In this method, lipophilicity (log P), pKa, and fu were the main input parameters for calculation of partition coefficients (Poulin and Theil 2000; Poulin et al. 2001; Kuepfer et al. 2016).
For metabolism, metabolic enzymes were primarily considered for the description of celecoxib metabolism. When considering the metabolic pathway of celecoxib, both CYP2C9 and CYP3A4 activities were applied to the model development. Input values for in vitro metabolic rate in the presence of recombinant sub-enzymes were determined according to a previously published study (Tang et al. 2001). In addition, in vitro metabolic rate constant for recombinant CYP2C9 used for the initial model development was applied to the CYP2C9*1/*1 genotype group. Modified in vitro metabolic rate values for other genotype groups (CYP2C9*1/*3, CYP2C9*1/*13, and CYP2C9*3/*3) were applied in sequence.
In the case of excretion, celecoxib is mainly eliminated by the hepatic metabolism (Davies et al. 2000). Therefore, kidney excretion was partially applied by optimizing plasma clearance for kidney and fu. Protein gene expression data used in the PBPK modeling was derived from published RT-PCR assay data (Nishimura et al. 2003; Nishimura et al. 2005; Nishimura et al. 2006). Additionally, solubility, kidney plasma clearance and 80% dissolution time were optimized by adjusting parameter identification tool in PK-SimⓇ version 7.2 for better goodness of fit. The optimization was performed using the Levenberg–Marquardt algorithm of the parameter identification tool in PK-SimⓇ software.
After generation of the PBPK model for CYP2C9*1/*1, simulation for the CYP2C9*1/*3, CYP2C9*1/*13, and CYP2C9*3/*3 genotype groups were generated by adjusting the biometric data and in vitro metabolic rate of recombinant CYP2C9 for each genotype.
In this study, a model acceptance criterion based on the variance of observed PK data (Cmax, AUC0-48, and AUCinf) was applied to evaluate the suitability of the developed PBPK model for celecoxib. Acceptance criterion for modeling was calculated by a previously reported method (Abduljalil et al. 2014). Equation 1 and Eq. 2 were used to calculate the model assessment as follows:
where \(\overline{x}\) is the mean value of the pharmacokinetic values from this study; CV% is the coefficient variation of the pharmacokinetic values from this study; N is the number of subjects; Ub is the upper limit of boundary; Lb is the lower limit of boundary.
In this study, the model acceptance range was calculated by converting the Cmax, AUC0-48, and AUCinf values from each CYP2C9 genotype group obtained from the pharmacokinetic study into Ub and Lb values, respectively. Thereby, all the predicted pharmacokinetic values in the model development were used as an index to determine the suitability of the comparison with observed pharmacokinetic values.
Validation of the PBPK model
PBPK model validation was performed by comparing the predicted values with observed values from the pharmacokinetic studies. In this study, comparison between predicted value and observed value was evaluated by two-fold error which is widely used for acceptable prediction in PBPK model evaluation (Jones et al. 2012; Guo et al. 2015; Rasool et al. 2017; Park et al. 2017). Two-fold error was calculated using Eq. 3:
Celecoxib pharmacokinetic studies used for comparison in the validation process consisted of different ethnic groups including Asian (China, Korea) and Caucasian (Spain, Sweden, USA) population. These studies also included available celecoxib single/multiple pharmacokinetic data with genotype groups including CYP2C9*1/*1, CYP2C9*1/*3, and CYP2C9*3/*3 (Tang et al. 2001; Lundblad et al. 2006; Prieto-Perez et al. 2013; Liu et al. 2015; Park et al. 2018). Demographic data of these pharmacokinetic studies used for validation are summarized in Table 3.
Results
Clinical pharmacokinetic study
During the pharmacokinetic study, no unexpected adverse symptoms and/or signs related to celecoxib administration were observed in any of the 39 subjects. There were no significant differences in demographic characteristics between the CYP2C9*1/*1 group and the CYP2C9*1/*3 group. The pharmacokinetic parameters and plasma concentration–time profile of celecoxib in each genotype group are shown in Fig. 1 and Table 1.

Mean plasma concentration–time profile of celecoxib after administration of single 200 mg oral dose of celecoxib in CYP2C9*1/*1 and CYP2C9*1/*3 genotype groups
Among the pharmacokinetic parameters, Cmax, AUC0-48, AUCinf, and CL/F were significantly different between the two genotype groups. Compared with the CYP2C9*1/*1 group, the CYP2C9*1/*3 group had 1.3 fold higher Cmax, 1.8 fold higher AUC0-48, 1.8 fold higher AUCinf, 1.8 lower CL/F, while T1/2 and Tmax were not significantly different between the two genotype groups.
PBPK model construction for celecoxib
Based on the conducted pharmacokinetic study and a previously reported study (Kim et al. 2017), the PBPK model for celecoxib was developed. Predefined physico-chemical parameters and ADME properties used in the PBPK model development are shown in Table 2. Data set for model development included CYP2C9*1/*1, CYP2C9*1/*3, CYP2C9*1/*13, and CYP2C9*3/*3 genotype groups.
At first, the PBPK model for celecoxib with regards to the CYP2C9*1/*1 genotype was developed. Initial simulation reflected celecoxib elimination only by metabolism of CYP enzymes including CYP2C9 and CYP3A4. This study assumed that celecoxib was eliminated mainly by hepatic metabolism with little urine excretion of unchanged celecoxib according to Davies et al. (2000).
Thereby, urine excretion was applied to kidney plasma clearance, and parameter identification tool in PK-SimⓇ software was utilized to perform optimization. Investigated dissolution rate of celecoxib capsule over 1–2 h, which was determined using the paddle method (described in the Korea Pharamacopeia), was 80% or more. Thus, dissolution rate was optimized within 1–2 h and was determined to be 90 min (Baek et al. 2015).
After optimization, predicted values of Cmax, AUC0-48, and AUCinf, in the CYP2C9*1/*1 genotype group met the predefined acceptance criterion range (Table 3) and demonstrated more optimal goodness of fit (Fig. 2).

The observed and predicted celecoxib pharmacokinetic profile in CYP2C9*1/*1 genotype group: (A) predicted pharmacokinetic profile by initial input values which represent the physico-chemical and ADME properties of celecoxib, (B) predicted pharmacokinetic profile by applying optimized kidney plasma clearance
Compared with the reported volume of distribution at steady state (Vss) value following oral administration (approximately 400 L) for celecoxib (Pfizer Inc. 2016), predicted Vss of oral administration from this study was 467.5 L. Absolute bioavailability information for celecoxib is not known because of low solubility in aqueous media (Pfizer Inc. 2016). In our prediction, the oral bioavailability was 76% and the predicted Vss of intravenous drug administration was 354.0 L.
Then, the developed model was scaled to CYP2C9*1/*3, CYP2C9*1/*13, and CYP2C9*3/*3 genotype groups by modifying biometric data and value of in vitro metabolic rate of recombinant CYP2C9 (Table 2).
As a result, the predicted pharmacokinetic data of CYP2C9*1/*3, CYP2C9*1/*13, and CYP2C9*3/*3 genotype groups also met the acceptance criterion range (Table 3, Fig. 3).

The observed and predicted celecoxib pharmacokinetic profile in (A) CYP2C9*1/*3, (B) CYP2C9*1/*13, and (C) CYP2C9*3/*3 genotype groups
Sensitivity analysis
A sensitivity analysis was conducted to confirm which input parameters had significant impact on the pharmacokinetic simulation of celecoxib. Sensitivity analysis was performed in PK-SimⓇ and target pharmacokinetic parameters were AUC and Cmax. The sensitivity ratio of input parameters are represented in Fig. 4.

Sensitivity ratio of input parameters for celecoxib on the AUC (A) and Cmax (B)
Among the input parameters for model development, lipophilicity (log P) showed the highest sensitivity ratio in all target pharmacokinetic parameters. The fu, celecoxib dose, and solubility were significantly sensitive to AUC and Cmax. In vitro Vmax and Km, which represent the enzyme activity of CYP2C9, also showed to be significantly sensitive to AUC and Cmax.
Model validation
After the model development, model validation was conducted by comparison of data from previous celecoxib pharmacokinetic studies, which assessed the role of CYP2C9 polymorphism. Model validation was performed by comparing the observed and predicted values of the PK parameters (AUC and Cmax) of each study.
Each simulation for validation was performed considering the respective ethnic group, biometric data and the CYP2C9 genotype. As a result, all the predicted pharmacokinetic values of each study laid within the two-fold error range and the results are shown in Table 4.
Discussion
Most drug metabolizing enzymes and transporters are genetically polymorphic, and these genetic polymorphisms influence pharmacokinetics and pharmacodynamics of drugs to varying degrees (Byeon et al. 2019; Bae et al. 2020; Jung et al. 2020; Shin et al. 2020). Drug interactions also have a significant effect on drug action (Lee et al. 2019). PBPK modeling may enable an optimized drug administration strategy for each individual patient by reflecting all of the characteristics such as the patient's physical characteristics, genetic polymorphisms of drug metabolizing enzymes and transporters, drug interactions, diseases, etc. (Duan et al. 2017; Kim et al. 2018).
Through this study, a celecoxib PBPK model was developed based on the genetic polymorphism of CYP2C9. Celecoxib is primarily metabolized by CYP2C9 and partly by CYP3A4 after oral administration. CYP2C9 is most abundantly expressed in the CYP2C subfamily, accounting for approximately 20% of total hepatic cytochrome P450 protein (Daly et al. 2017).
Most of NSAIDs including diclofenac, ibuprofen, lornoxicam, tenoxicam, and meloxicam are metabolized by CYP2C9, and the genetic polymorphism of CYP2C9 has been reported to affect the metabolism of these drugs (Vianna et al. 2004; Choi et al. 2011; Lee et al. 2014; Zhang et al. 2014; Krasniqi et al. 2016; Daly et al. 2017).
Likewise, most clinical studies, including the present one, have reported that CYP2C9 genetic polymorphism significantly affects the metabolism of celecoxib (Tang et al. 2001; Kirchheiner et al. 2003; Stempak et al. 2005; Prieto-Perez et al. 2013; Liu et al. 2015; Kim et al. 2017; Park et al. 2018). Besides, in our clinical study, AUC0-48, AUCinf, Cmax and CL/F parameters were significantly different between CYP2C9*1/*1 and CYP2C9*1/*3 genotype groups.
Thus, PBPK model for celecoxib in this study was developed with regards to the genetic polymorphism of the CYP2C9 enzyme. The workflow of this study was to utilize our clinical pharmacokinetic data for model development, and to validate the model with comparison studies. In the development of the PBPK model, input values for physico-chemical parameter and ADME properties of celecoxib were adapted from previously reported studies, and these were calculated and optimized by PK-SimⓇ software.
Input values of in vitro metabolic rate of recombinant CYP2C9 (in vitro Vmax and Km) for CYP2C9*1/*1, CYP2C9*1/*3, and CYP2C9*3/*3 genotype were adapted from the literature (Tang et al. 2001). In case of the CYP2C9*1/*13 group, values were optimized by PK-SimⓇ based on the pharmacokinetic data by Kim et al. (2017) as in vitro metabolic rate data for CYP2C9*1/*13 genotype group was not available otherwise. Through the change of these input parameter values, we intended to demonstrate pharmacokinetic differences between four genotype groups. As expected, sensitivity analysis indicated that in vitro Vmax (CYP2C9) and Km (CYP2C9) parameter differences between four genotype groups had a significant impact on the pharmacokinetics of celecoxib.
The evaluation criteria for model development and validation were separated into model acceptance criterion (99.998% confidence interval) and two-fold error. Model acceptance criterion used to evaluate model development was based on a previously presented literature (Abduljalil et al. 2014). According to Abduljalil et al. (2014), the presented model acceptance criterion has a 99.998% confidence interval, which can more accurately reflect all the inter-individual variabilities of observed pharmacokinetic values than the two-fold error criterion. The ranges of the proposed criterion were 1.58-, 1.33-, 1.79-, and 1.81-fold for CYP2C9*1/*1, CYP2C9*1/*3, CYP2C9*1/*13, and CYP2C9*3/*3 genotype groups, respectively. Moreover, all the predicted pharmacokinetic parameters for model development met the two-fold error range, as well as proposed model acceptance criterion.
There are eight studies that have reported CYP2C9 genetic polymorphism affecting the pharmacokinetics of celecoxib (Tang et al. 2001; Kirchheiner et al. 2003; Stempak et al. 2005; Lundblad et al. 2006; Prieto-Perez et al. 2013; Liu et al. 2015; Kim et al. 2017; Park et al. 2018). Among them, five pharmacokinetic studies were selected for comparison in the validation process. Kirchheiner et al. (2003) and Stempak et al. (2005) were excluded because biometric information for the subject group was not specified. Kim et al. (2017) was also excluded due to the use of pharmacokinetic data from the CYP2C9*1/*13 and CYP2C9*3/*3 genotype groups for model development.
In this model validation, predicted target pharmacokinetic values including AUC and Cmax for Korean (Park et al. 2018), Chinese (Liu et al. 2015), and Caucasian population (Tang et al. 2001; Lundblad et al. 2006; Prieto‐Pérez et al. 2013) studies met the two-fold error criterion. These results indicated that the PBPK model for celecoxib according to CYP2C9 genetic polymorphism has successfully described the pharmacokinetics of celecoxib after single or multiple oral administration.
Of note, the observed pharmacokinetic values between East Asian population (Chinese and Korean) and Caucasian population were significantly different. These results had a considerable impact on the validation for model development of celecoxib. The ratio scores (between observed and predicted pharmacokinetic values) in the Korean and Chinese population simulation through this model were within 0.67–0.94, and 0.72–1.18, respectively. In the Caucasian population, however, the ratio scores (between observed and predicted pharmacokinetic values) were within 0.54–0.88, which was significantly lower compared to Asian population (P < 0.01) (Table 4).
Shu et al. (2001) suggested that CYP2C9 abundance was not significantly related to ethnicity. However, this study strongly suggests that there is a limitation in simulating with same CYP2C9 enzyme abundance (3.84 µM in liver tissue) to all races. Despite these limitations, this model can be useful in determining optimal dosage considering the patient’s demographic data (age, height, weight, and BMI) and CYP2C9 genotype. Additionally, this model may also be applied to construct a drug-drug interaction model with celecoxib and CYP2C9 inhibitor or inducer.
In conclusion, a PBPK model for celecoxib with regard to CYP2C9 genetic polymorphism was developed, which predicted the pharmacokinetics of celecoxib, considering demographic data of subjects, physico-chemical parameters, ADME properties, and CYP2C9 genotype. Although further development is required, this PBPK model is the first attempt to demonstrate in silico prediction of celecoxib pharmacokinetics that reflects the pharmacogenetic effects. These results will be beneficial in prescribing the appropriate dosage of celecoxib considering inter-individual differences.
References
Abduljalil K, Cain T, Humphries H, Rostami-Hodjegan A (2014) Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: an analysis based on in vivo observations. Drug Metab Dispos 42(9):1478–1484. https://doi.org/10.1124/dmd.114.058099
Bae JW, Kim HK, Kim JH, Yang SI, Kim MJ, Jang CG, Lee SY (2005) Allele and genotype frequencies of CYP2C9 in a Korean population. Br J Clin Pharmacol 60(4):418–422. https://doi.org/10.1111/j.1365-2125
Bae JW, Oh KY, Yoon SJ, Shin HB, Jung EH, Cho CK, Lim CW, Kang P, Choi CI, Jang CG, Lee SY, Lee YJ (2020) Effects of CYP2D6 genetic polymorphism on the pharmacokinetics of metoclopramide. Arch Pharm Res 43(11):1207–1213. https://doi.org/10.1007/s12272-020-01293-4
Baek NH, Song YG, Choi MS, Seo KS, Woo JY, Sohn JH (2015) Republic of Korea Patent No. 10–2015–0049649. http://kpat.kipris.or.kr/kpat/biblioa.do?method=biblioFrame. Accessed 10 June 2021
Brenner SS, Herrlinger C, Dilger K, Murdter TE, Hofmann U, Marx C, Klotz U (2003) Influence of age and cytochrome P450 2C9 genotype on the steady-state disposition of diclofenac and celecoxib. Clin Pharmacokinet 42(3):283–292. https://doi.org/10.2165/00003088-200342030-00003
Byeon JY, Lee CM, Lee YJ, Kim YH, Kim SH, Jung EH, Chae WK, Lee YJ, Jang CG, Lee SY (2019) Influence of CYP2D6 genetic polymorphism on pharmacokinetics of active moiety of tolterodine. Arch Pharm Res 42(2):182–190. https://doi.org/10.1007/s12272-018-1099-y
Caldwell B, Aldington S, Weatherall M, Shirtcliffe P, Beasley R (2006) Risk of cardiovascular events and celecoxib: a systematic review and meta-analysis. J R Soc Med 99(3):132–140. https://doi.org/10.1258/jrsm.99.3.132
Choi CI, Kim MJ, Jang CG, Park YS, Bae JW, Lee SY (2011) Effects of the CYP2C9*1/*13 genotype on the pharmacokinetics of lornoxicam. Basic Clin Pharmacol Toxicol 109(6):476–480. https://doi.org/10.1111/j.1742-7843.2011.00751.x
Dai DP, Xu RA, Hu LM, Wang SH, Geng PW, Yang JF, Cai JP (2014) CYP2C9 polymorphism analysis in Han Chinese populations: building the largest allele frequency database. Pharmacogenomics J 14(1):85–92. https://doi.org/10.1038/tpj.2013.2
Daly AK, Rettie AE, Fowler DM, Miners JO (2017) Pharmacogenomics of CYP2C9: functional and clinical considerations. J Pers Med 8(1):1. https://doi.org/10.3390/jpm8010001
Davies NM, McLachlan AJ, Day RO, Williams KM (2000) Clinical pharmacokinetics and pharmacodynamics of celecoxib: a selective cyclo-oxygenase-2 inhibitor. Clin Pharmacokinet 38(3):225–242. https://doi.org/10.2165/00003088-200038030-00003
Ding Y, Yang D, Zhou L, He P, Yao J, Xie P, Jin T (2015) Cytochrome P450 2C9 (CYP2C9) polymorphisms in Chinese Li population. Int J Clin Exp Med 8(11):21024–21033
Djebli N, Fabre D, Boulenc X, Fabre G, Sultan E, Hurbin F (2015) Physiologically based pharmacokinetic modeling for sequential metabolism: effect of CYP2C19 genetic polymorphism on clopidogrel and clopidogrel active metabolite pharmacokinetics. Drug Metab Dispos 43(4):510–522. https://doi.org/10.1124/dmd.114.062596
Duan P, Zhao P, Zhang L (2017) Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug-drug interactions (DDIs). Eur J Drug Metab Pharmacokinet 42(4):689–705. https://doi.org/10.1007/s13318-016-0383-9
Emoto C, Fukuda T, Venkatasubramanian R, Vinks AA (2015) The impact of CYP3A5*3 polymorphism on sirolimus pharmacokinetics: insights from predictions with a physiologically-based pharmacokinetic model. Br J Clin Pharmacol 80(6):1438–1446. https://doi.org/10.1111/bcp.12743
Futatsugi A, Toshimoto K, Yoshikado T, Sugiyama Y, Kato Y (2018) Evaluation of alteration in hepatic and intestinal BCRP function in vivo due to ABCG2 c.421C>A polymorphism based on PBPK analysis of rosuvastatin. Drug Metab Dispos 46(5):749–757. https://doi.org/10.1124/dmd.117.078816
Goldstein JL, Correa P, Zhao WW, Burr AM, Hubbard RC, Verburg KM, Geis GS (2001) Reduced incidence of gastroduodenal ulcers with celecoxib, a novel cyclooxygenase-2 inhibitor, compared to naproxen in patients with arthritis. Am J Gastroenterol 96(4):1019–1027. https://doi.org/10.1111/j.1572-0241.2001.03740.x
Gong J, Iacono L, Iyer RA, Humphreys WG, Zheng M (2018) Physiologically based pharmacokinetic modeling of a CYP2C19 substrate, BMS-823778, utilizing pharmacogenetic data. Br J Clin Pharmacol 84(6):1335–1345. https://doi.org/10.1111/bcp.13565
Guo J, Zhou D, Li Y, Khanh BH (2015) Physiologically based pharmacokinetic modeling to predict complex drug-drug interactions: a case study of AZD2327 and its metabolite, competitive and time-dependent CYP3A inhibitors. Biopharm Drug Dispos 36(8):507–519. https://doi.org/10.1002/bdd.1962
Gupta A, Zheng L, Ramanujam V, Gallagher J (2015) Novel use of pharmacogenetic testing in the identification of CYP2C9 polymorphisms related to NSAID-induced gastropathy. Pain Med 16(5):866–869. https://doi.org/10.1111/pme.12654
Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, Hall SD (2015) Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther 97(3):247–262. https://doi.org/10.1002/cpt.37
Jones HM, Dickins M, Youdim K, Gosset JR, Attkins NJ, Hay TL, Gardner IB (2012) Application of PBPK modeling in drug discovery and development at Pfizer. Xenobiotica 42(1):94–106. https://doi.org/10.3109/00498254.2011.627477
Jung EH, Lee YJ, Kim DH, Kang P, Lim CW, Cho CK, Jang CG, Lee SY, Bae JW (2020) Effects of paroxetine on the pharmacokinetics of atomoxetine and its metabolites in different CYP2D6 genotypes. Arch Pharm Res 43(12):1356–1363. https://doi.org/10.1007/s12272-020-01300-8
Jung EH, Lee CM, Byeon JY, Shin HB, Oh KY, Cho CK, Lim CW, Jang CG, Lee SY, Lee YJ (2002) Relationship between plasma exposure of zolpidem and CYP2D6 genotype in healthy Korean subjects. Arch Pharm Res 43(9):976–981. https://doi.org/10.1007/s12272-020-01250-1
Kim SH, Kim DH, Byeon JY, Kim YH, Kim DH, Lim HJ, Lee SY (2017) Effects of CYP2C9 genetic polymorphisms on the pharmacokinetics of celecoxib and its carboxylic acid metabolite. Arch Pharm Res 40(3):382–390. https://doi.org/10.1007/s12272-016-0861-2
Kim S-H, Byeon J-Y, Kim Y-H, Lee C-M, Lee Y-J, Jang C-G, Lee S-Y (2018) Physiologically based pharmacokinetic modelling of atomoxetine with regard to CYP2D6 genotypes. Sci Rep 8(1). https://doi.org/10.1038/s41598-018-30841-8
Kimura M, Ieiri I, Mamiya K, Urae A, Higuchi S (1998) Genetic polymorphism of cytochrome P450s, CYP2C19, and CYP2C9 in a Japanese population. Ther Drug Monit 20(3):243–247. https://doi.org/10.1097/00007691-199806000-00001
Kirchheiner J, Stormer E, Meisel C, Steinbach N, Roots I, Brockmoller J (2003) Influence of CYP2C9 genetic polymorphisms on pharmacokinetics of celecoxib and its metabolites. Pharmacogenetics 13(8):473–480. https://doi.org/10.1097/00008571-200308000-00005
Krasniqi V, Dimovski A, Domjanovic IK, Bilic I, Bozina N (2016) How polymorphisms of the cytochrome P450 genes affect ibuprofen and diclofenac metabolism and toxicity. Arh Hig Rada Toksikol 67(1):1–8. https://doi.org/10.1515/aiht-2016-67-2754
Kuepfer L, Niederalt C, Wendl T, Schlender JF, Willmann S, Lippert J, Teutonico D (2016) Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst Pharmacol 5(10):516–531. https://doi.org/10.1002/psp4.12134
LC Laboratories Inc. (2018) Certificate of analysis, celecoxib [Online] Available from: https://www.lclabs.com/products/lots-certificate-of-analysis/celecoxib-CXB-104.pdf. Accessed 15 June, 2021
Lee CM, Jung EH, Byeon JY, Kim SH, Jang CG, Lee YJ, Lee SY (2019) Effects of steady-state clarithromycin on the pharmacokinetics of zolpidem in healthy subjects. Arch Pharm Res 42(12):1101–1106. https://doi.org/10.1007/s12272-019-01201-5
Lee CR, Goldstein JA, Pieper JA (2002) Cytochrome P4502C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics 12(3):251–263. https://doi.org/10.1097/00008571-200204000-00010
Lee HI, Bae JW, Choi CI, Lee YJ, Byeon JY, Jang CG, Lee SY (2014) Strongly increased exposure of meloxicam in CYP2C9*3/*3 individuals. Pharmacogenet Genomics 24(2):113–117. https://doi.org/10.1097/FPC.0000000000000025
Liu R, Gong C, Tao L, Yang W, Zheng X, Ma P, Ding L (2015) Influence of genetic polymorphisms on the pharmacokinetics of celecoxib and its two main metabolites in healthy Chinese subjects. Eur J Pharm Sci 79:13–19. https://doi.org/10.1016/j.ejps.2015.09.005
Lundblad MS, Ohlsson S, Johansson P, Lafolie P, Eliasson E (2006) Accumulation of celecoxib with a 7-fold higher drug exposure in individuals homozygous for CYP2C9*3. Clin Pharmacol Ther 79:287–288. https://doi.org/10.1016/j.clpt.2005.11.007
Mohammed S, Croom DW (1999) Gastropathy due to celecoxib, a cyclooxygenase-2 inhibitor. N Engl J Med 340(25):2005–2006. https://doi.org/10.1056/NEJM199906243402516
Moore RA, Derry S, Makinson GT, McQuay HJ (2005) Tolerability and adverse events in clinical trials of celecoxib in osteoarthritis and rheumatoid arthritis: systematic review and meta-analysis of information from company clinical trial reports. Arthritis Res Ther 7(3):R644-665. https://doi.org/10.1186/ar1704
Nishimura M, Naito S (2005) Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet 20(6):452–477. https://doi.org/10.2133/dmpk.20.452
Nishimura M, Naito S (2006) Tissue-specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab Pharmacokinet 21(5):357–374. https://doi.org/10.2133/dmpk.21.357
Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T (2003) Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi 123(5):369–375. https://doi.org/10.1248/yakushi.123.369
Park MH, Shin SH, Byeon JJ, Lee GH, Yu BY, Shin YG (2017) Prediction of pharmacokinetics and drug-drug interaction potential using physiologically based pharmacokinetic (PBPK) modeling approach: A case study of caffeine and ciprofloxacin. Korean J Physiol Pharmacol 21(1):107–115. https://doi.org/10.4196/kjpp.2017.21.1.107
Park SI, Park JY, Park MJ, Yim SV, Kim BH (2018) Effects of Ojeok-san on the pharmacokinetics of celecoxib at steady-state in healthy volunteers. Basic Clin Pharmacol Toxicol 123(1):51–57. https://doi.org/10.1111/bcpt.12971
Paulson SK, Hribar JD, Liu NW, Hajdu E, Bible RH Jr, Piergies A, Karim A (2000) Metabolism and excretion of [(14)C]celecoxib in healthy male volunteers. Drug Metab Dispos 28(3):308–314
Paulson SK, Kaprak TA, Gresk CJ, Fast DM, Baratta MT, Burton EG, Karim A (1999) Plasma protein binding of celecoxib in mice, rat, rabbit, dog and human. Biopharm Drug Dispos 20(6):293–299. https://doi.org/10.1002/(sici)1099-081x(199909)20:6%3c293::aid-bdd188%3e3.0.co;2-x
Pfizer Inc. (2016) U.S Prescribing information. [Online] Available from https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/020998s048lbl.pdf. Accessed 30 June 2021
Poulin P, Schoenlein K, Theil FP (2001) Prediction of adipose tissue: plasma partition coefficients for structurally unrelated drugs. J Pharm Sci 90(4):436–447. https://doi.org/10.1002/1520-6017(200104)90:4%3c436::aid-jps1002%3e3.0.co;2-p
Poulin P, Theil FP (2000) A priori prediction of tissue:plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J Pharm Sci 89(1):16–35. https://doi.org/10.1002/(SICI)1520-6017(200001)89:1%3c16::AID-JPS3%3e3.0.CO;2-E
Prieto-Perez R, Ochoa D, Cabaleiro T, Roman M, Sanchez-Rojas SD, Talegon M, Abad-Santos F (2013) Evaluation of the relationship between polymorphisms in CYP2C8 and CYP2C9 and the pharmacokinetics of celecoxib. J Clin Pharmacol 53(12):1261–1267. https://doi.org/10.1002/jcph.169
Rasool MF, Khalil F, Laer S (2017) Optimizing the Clinical Use of Carvedilol in Liver Cirrhosis Using a Physiologically Based Pharmacokinetic Modeling Approach. Eur J Drug Metab Pharmacokinet 42(3):383–396. https://doi.org/10.1007/s13318-016-0353-2
Rettie AE, Jones JP (2005) Clinical and toxicological relevance of CYP2C9: drug-drug interactions and pharmacogenetics. Annu Rev Pharmacol Toxicol 45:477–494. https://doi.org/10.1146/annurev.pharmtox.45.120403.095821
Sandberg M, Yasar U, Stromberg P, Hoog JO, Eliasson E (2002) Oxidation of celecoxib by polymorphic cytochrome P450 2C9 and alcohol dehydrogenase. Br J Clin Pharmacol 54(4):423–429. https://doi.org/10.1046/j.1365-2125.2002.01660.x
Shin HB, Jung EH, Kang P, Lim CW, Oh KY, Cho CK, Lee YJ, Choi CI, Jang CG, Lee SY, Bae JW (2020) ABCB1 c2677G>T/c3435C>T diplotype increases the early-phase oral absorption of losartan. Arch Pharm Res 43(11):1187–1196. https://doi.org/10.1007/s12272-020-01294-3
Shu Y, Cheng ZN, Liu ZQ, Wang LS, Zhu B, Huang SL, Zhou HH (2001) Interindividual variations in levels and activities of cytochrome P-450 in liver microsomes of Chinese subjects. Acta Pharmacol Sin 22(3):283–288
Stempak D, Bukaveckas BL, Linder M, Koren G, Baruchel S (2005) Cytochrome P450 2C9 genotype: impact on celecoxib safety and pharmacokinetics in a pediatric patient. Clin Pharmacol Ther 78(3):309–310. https://doi.org/10.1016/j.clpt.2005.06.005
Tang C, Shou M, Rushmore TH, Mei Q, Sandhu P, Woolf EJ, Rodrigues AD (2001) In-vitro metabolism of celecoxib, a cyclooxygenase-2 inhibitor, by allelic variant forms of human liver microsomal cytochrome P450 2C9: correlation with CYP2C9 genotype and in-vivo pharmacokinetics. Pharmacogenetics 11(3):223–235. https://doi.org/10.1097/00008571-200104000-00006
Thelen K, Coboeken K, Willmann S, Burghaus R, Dressman JB, Lippert J (2011) Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: oral solutions. J Pharm Sci 100(12):5324–5345. https://doi.org/10.1002/jps.22726
Thelen K, Coboeken K, Willmann S, Dressman JB, Lippert J (2012) Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part II: extension to describe performance of solid dosage forms. J Pharm Sci 101(3):1267–1280. https://doi.org/10.1002/jps.22825
Vianna-Jorge R, Perini JA, Rondinelli E, Suarez-Kurtz G (2004) CYP2C9 genotypes and the pharmacokinetics of tenoxicam in Brazilians. Clin Pharmacol Ther 76(1):18–26. https://doi.org/10.1016/j.clpt.2004.03.002
Vieira MD, Kim MJ, Apparaju S, Sinha V, Zineh I, Huang SM, Zhao P (2014) PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin Pharmacol Ther 95(5):550–557. https://doi.org/10.1038/clpt.2014.43
Yeo KR, Kenny JR, Rostami-Hodjegan A (2013) Application of in vitro-in vivo extrapolation (IVIVE) and physiologically based pharmacokinetic (PBPK) modeling to investigate the impact of the CYP2C8 polymorphism on rosiglitazone exposure. Eur J Clin Pharmacol 69(6):1311–1320. https://doi.org/10.1007/s00228-012-1467-3
Zhang M, Yang Y, Zhao G, Di X, Xu L, Jiang N, Xu X (2014) Effect of CYP2C9*3 mutant variants on meloxicam pharmacokinetics in a healthy Chinese population. Genet Mol Res 13(1):831–837. https://doi.org/10.4238/2014
Zhuang X, Lu C (2016) PBPK modeling and simulation in drug research and development. Acta Pharm Sin B 6(5):430–440. https://doi.org/10.1016/j.apsb.2016.04.004
Acknowledgements
This research was supported the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, & Future Planning (NRF-2019R1A2C1004582).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kim, YH., Kang, P., Cho, C. et al. Physiologically based pharmacokinetic (PBPK) modeling for prediction of celecoxib pharmacokinetics according to CYP2C9 genetic polymorphism. Arch. Pharm. Res. 44, 713–724 (2021). https://doi.org/10.1007/s12272-021-01346-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-021-01346-2