
Abstract
The aim of this study was to investigate the effects of paroxetine, a potent inhibitor of CYP2D6, on the pharmacokinetics of atomoxetine and its two metabolites, 4-hydroxyatomoxetine and N-desmethylatomoxetine, in different CYP2D6 genotypes. Twenty-six healthy subjects were recruited and divided into CYP2D6*wt/*wt (*wt=*1 or *2, n = 10), CYP2D6*wt/*10 (n = 9), and CYP2D6*10/*10 groups (n = 7). In atomoxetine phase, all subjects received a single oral dose of atomoxetine (20 mg). In paroxetine phase, after administration of a single oral dose of paroxetine (20 mg) for six consecutive days, all subjects received a single oral dose of atomoxetine with paroxetine. Plasma concentrations of atomoxetine and its metabolites were determined up to 24 h after dosing. During atomoxetine phase, there were significant differences in Cmax and AUC0−24 of atomoxetine and N-desmethylatomoxetine among three genotype groups, whereas significant differences were not found in relation to CYP2D6*10 allele after administration of paroxetine. AUC ratios of 4-hydroxyatomoxetine and N-desmethylatomoxetine to atomoxetine were significantly different among three genotype groups during atomoxetine phase (all, P < 0.001), but after paroxetine treatment significant differences were not found. After paroxetine treatment, AUC0−24 of atomoxetine was increased by 2.3-, 1.7-, and 1.3-fold, in CYP2D6*wt/*wt, CYP2D6*wt/*10, and CYP2D6*10/*10 groups in comparison to atomoxetine phase, respectively. AUC ratio of 4-hydroxyatomoxetine to atomoxetine in each group was significantly decreased, whereas AUC ratio of N-desmethylatomoxetine to atomoxetine significantly increased after administration of paroxetine. In conclusion, paroxetine coadministration significantly affected pharmacokinetic parameters of atomoxetine and its two metabolites, 4-hydroxyatomoxetine and N-desmethylatomoxetine. When atomoxetine was administered alone, Cmax, AUC0-24 and CL/F of atomoxetine were significantly different among the three CYP2D6 genotype groups. However, after paroxetine coadministration, no significant differences in these pharmacokinetic parameters were observed among the CYP2D6 genotype groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atomoxetine (ATX) is a highly selective and potent norepinephrine reuptake inhibitor with low affinity for other noradrenergic receptors or for other neurotransmitter transporters or receptors (Wong et al. 1982; Gehlert et al. 1993). ATX is a nonstimulant and is indicated to treat attention-deficit/hyperactivity disorder (ADHD) in children, adolescents, and adults.
After oral administration of ATX, it is rapidly and completely absorbed and predominantly metabolized by cytochrome P450 2D6 (CYP2D6) through oxidative metabolism (Ring et al. 2002; Sauer et al. 2003). The primary oxidative metabolite of atomoxetine is 4-hydroxyatomoxetine (4-HAT) and 4-HAT is subsequently conjugated forming 4-HAT-O-glucuronide, which is excreted into urine and feces (Sauer et al. 2003). ATX is also metabolized by CYP2C19 to minor metabolite of ATX, N-desmethylatomoxetine (NAT) (Ring et al. 2002). It was found that 4-HAT is a selective inhibitor of the presynaptic norepinephrine transporter, similar to ATX but NAT appeared to be pharmacologically inactive relative to ATX (Sauer et al. 2003). CYP2D6 is one of the highly polymorphic enzymes and previous studies demonstrated the polymorphic expression of CYP2D6 had significantly affected the pharmacokinetics of ATX (Cui et al. 2007; Matsui et al. 2012; Byeon et al. 2015).
Paroxetine is a selective serotonin reuptake inhibitor (SSRI) and is clinically used to treat depression and other mental illnesses. Paroxetine is almost completely absorbed and undergoes extensive first pass metabolism (Heydorn 1999). CYP2D6 is predominantly involved in the metabolism of paroxetine and it has been shown that paroxetine is a potent CYP2D6 inhibitor and inhibits the activity of CYP2D6 in a concentration-dependent manner (Crewe et al. 1992; Sindrup et al. 1992; Jeppesen et al. 1996).
Thus, we intended to investigate the effects of paroxetine on the pharmacokinetic parameters of ATX and its two metabolites, 4-HAT and NAT, in relation to CYP2D6 genotype status.
Materials and methods
Subjects
Twenty-six healthy subjects who genotyped as CYP2C19*1/*1 (24 males and 2 females) participated in this study and they were divided into three different groups: CYP2D6*wt/*wt (*wt=*1 or *2, n = 10), CYP2D6*wt/*10 (n = 9), and CYP2D6*10/*10 (n = 7). The variant alleles for CYP2C19 (CYP2C19*2, CYP2C19*3, and CYP2C19*17) and the variant alleles for CYP2D6, CYP2D6*2 and CYP2D6*10 were identified using polymerase chain reaction-restriction fragment length polymorphisms (PCR-RFLP) and CYP2D6*5 and gene duplication (*XN) were identified using long-PCR method, as previously described (Desta et al. 2002; Sim et al. 2006; Byeon et al. 2018c). All subjects were asked to abstain from taking other medications, caffeine, grapefruit products, alcoholic beverages, and any products that can affect the results of the study and smoking for at least 1 week before and throughout the study period. Each subject was confirmed to be healthy before participating in the study by checking their medical histories, physical examinations, and routine laboratory tests (blood chemistry, hematology, and urine analysis). All participants provided verbal and written informed consent before enrollment to the study. The present study was conducted according to the guidelines of the Declaration of Helsinki and the protocol and informed consent document was approved by the institutional ethics committee of the School of Pharmacy, Sungkyunkwan University, Suwon, Republic of Korea.
Study design
This was an open-label, two-period study. During the atomoxetine phase with atomoxetine alone, two capsules of 10 mg atomoxetine (Strattera®, Eli Lilly and Company, Seoul, Korea) were administered to all subjects with 240 mL of water after an overnight fast. During the paroxetine phase with atomoxetine and paroxetine coadministration, each subject received a single oral dose of 20 mg paroxetine (Seroxat®, Handok Pharm., Seoul, Korea) once a day for six consecutive days. On day 7, an oral dose of atomoxetine (20 mg) was coadministered with paroxetine (20 mg) to all participants with 240 mL of water. During each study, all participants maintained fasting and standard meals were provided at 4 h and 10 h after administration of atomoxetine. Venous blood samples (7 mL) were taken in heparinized tubes before and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, and 24 h after administration of atomoxetine during each study. Blood samples were centrifuged at 3,000 rpm for 10 min and the plasma fractions were kept at -70ºC until needed.
Analysis of ATX, 4-HAT and NAT
Plasma concentrations of ATX, 4-HAT, and NAT were determined by employing high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) analytical method developed in our laboratory (Choi et al. 2012b, 2013).
The lower limit of quantifications of ATX, 4-HAT, and NAT were 1 ng/mL, 0.05 ng/mL, and 0.1 ng/mL, respectively. The calibration curves of ATX, 4-HAT, and NAT were linear over the range of 1–750, 0.05–20, and 0.1–20 ng/mL, respectively. The intraday and interday precisions were less than 6.8 and 9.6% for ATX, 5.3 and 7.4% for 4-HAT, and 7.5 and 7.8% for NAT.
Pharmacokinetic analysis
Noncompartmental methods with the BA Calc 2007 analysis program (KFDA, Seoul, Korea) was used to estimate the pharmacokinetic parameters of ATX, 4-HAT, and NAT. The peak plasma concentration (Cmax) and time to reach Cmax (tmax) were obtained from the observed values. The area under the plasma concentration-time curve (AUC) was calculated by the trapezoidal rule. AUC ratio was calculated as the AUC0−24 (nM h) of 4-HAT and NAT divided by the AUC0−24 (nM h) of ATX. The elimination rate constant (ke) was estimated from the least squares regression slope of the terminal plasma concentration. The AUC from 0 to infinity (AUC0−∞) was calculated as AUC0−∞ = AUC + Ct/ke, where Ct is the last plasma concentration measured. The half-life (t1/2) was calculated as ln 2/ke and the apparent oral clearance (CL/F) of atomoxetine was calculated as CL/F = dose/AUC0−∞.
Statistical analysis
All pharmacokinetic data are expressed as the mean ± SD, except for tmax and tmax is expressed as median value (range). One-way analysis of variance with Bonferroni’s t-test or Kruskal–Wallis one-way analysis of variance with Mann–Whitney rank-sum test were used to compare differences in pharmacokinetic parameters between the genotype groups after normality and equal variance tests. Two-sided paired t-tests or Wilcoxon signed-rank sum tests were used for comparisons of pharmacokinetic parameters of ATX with and without paroxetine treatment. Data were analyzed using the statistical program SigmaPlot® (version 12.0, Systat Software Inc., Chicago, IL, USA). A P-value < 0.05 was considered statistically significant.
Results
None of the subjects experienced undesirable symptoms and/or signs during the study and demographic characteristics of the three CYP2D6 genotype groups did not differ significantly (Table 1).
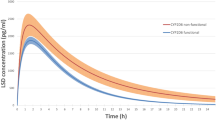
Plasma concentration-time curves of ATX, 4-HAT, and NAT in relation to CYP2D6*10 allele are shown in Fig. 1, and pharmacokinetic parameters of each analyte are summarized in Table 2. In atomoxetine phase, Cmax and AUC0−24 of ATX and NAT were significantly different among different CYP2D6 genotype groups, whereas those of 4-HAT were not significantly different among different CYP2D6 genotype groups.

Plasma concentration-time profiles for atomoxetine (ATX), 4-hydroxyatomoxetine (4-HAT), and N-desmethylatomoxetine (NAT) during atomoxetine phase and paroxetine phase in CYP2D6*wt/*wt (*wt=*1 or *2, n = 10), CYP2D6*wt/*10 (n = 9), and CYP2D6*10/*10 (n = 7) groups. Each value represents the mean ± SD. The data during atomoxetine phase and paroxetine phase are indicated as open blue circles and closed red circles, respectively
After administration of paroxetine, in CYP2D6*wt/*wt group, Cmax and AUC0−24 of ATX were 2.3- and 9.8-fold higher than those in atomoxetine phase (all, P < 0.001). Significant differences were also found in Cmax and AUC0−24 of 4-HAT and NAT and paroxetine treatment prolonged tmax of NAT by 9.7-fold (P < 0.01). In CYP2D6*wt/*10 group, Cmax of ATX, 4-HAT and NAT were significantly higher and tmax of NAT was prolonged by 3.6-fold after paroxetine treatment (P < 0.001). In CYP2D6*10/*10 group, paroxetine treatment increased Cmax and AUC0−24 of ATX by 1.3- (P < 0.05) and 2.9-fold (P < 0.01), respectively, and Cmax and AUC0−24 of 4-HAT and NAT were also significantly changed.
AUC ratio of 4-HAT and NAT to ATX was significantly different in relation to CYP2D6*10 allele (all, P < 0.001) after administration of atomoxetine alone, whereas there were no significant differences among three different groups after paroxetine treatment. Paroxetine treatment decreased the AUC ratio of 4-HAT to ATX by 93% in CYP2D6*wt/*wt group, 77% in CYP2D6*wt/*10 group, and 71% in CYP2D6*10/*10 group. Conversely, AUC ratio of NAT to ATX was increased by 3.1-fold in CYP2D6*wt/*wt group, 2.2-fold in CYP2D6*wt/*10 group, and 1.5-fold in CYP2D6*10/*10 group.
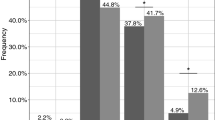
When atomoxetine was administered alone, the AUC, Cmax, and CL/F of plasma atomoxetine showed significant differences depending on the CYP2D6 genotype, but when atomoxetine was administered with paroxetine, plasma atomoxetine AUC, Cmax, and CL/F were not different in the three CYP2D6 genotypes (Fig. 2).

Individual values for the area under plasma concentration-time curves from time 0 to 24 h and the maximum plasma concentration of atomoxetine during atomoxetine phase and paroxetine phase in CYP2D6*wt/*wt (*wt=*1 or *2, n = 10), CYP2D6*wt/*10 (n = 9), and CYP2D6*10/*10 (n = 7) groups. The three genotype groups were compared by one-way ANOVA. The horizontal line indicates the mean of individual values. *P < 0.05, **P < 0.01, and ***P < 0.001, compared between two groups. N.S, not significant
Discussion
ADHD is the most common neurobehavioural disorder of childhood and it can persist into adulthood in 10–60% of cases (Spencer et al. 1996; Swanson et al. 1998; Pary et al. 2002). ATX is the first nonstimulant agent approved by the US Food and Drug Administration to treat ADHD and CYP2D6 is primarily involved in the oxidative metabolism of ATX (Ring et al. 2002; Sauer et al. 2003).
The disposition of the drug in the body is greatly affected by the activity of the drug metabolizing enzymes. Also, since drug metabolizing enzymes are genetically very polymorphic, many studies have been conducted to investigate the effects of genetic variants of drug metabolizing enzymes on the pharmacokinetics of clinically used drugs (Choi et al. 2014; Lee et al. 2014, 2016, 2018; Kim et al. 2017, 2018a; Byeon et al. 2018a, b, d, 2019).
CYP2D6 is a highly polymorphic drug metabolizing enzyme and genetic polymorphisms of CYP2D6 show interethnic differences; CYP2D6*3 and CYP2D6*4 alleles are common in Caucasians, whereas CYP2D6*10 allele is more prevalent in Asians (Teh and Bertilsson 2012; Byeon et al. 2018c). CYP2D6*10 (rs1065852) allele, which leads to a p.Pro34Ser substitution, results in an unstable enzyme with reduced substrate affinity and comprises at least 50% of all CYP2D6 alleles in Asians, which is approximately 10-fold higher than that in Caucasians (Choi et al. 2012a; Teh and Bertilsson 2012; Byeon et al. 2018c). It implies that CYP2D6 genetic polymorphism can affect the plasma exposure of ATX and previous studies have been shown that CYP2D6*10/*10 genotype had significant effects on the pharmacokinetics of ATX (Cui et al. 2007; Matsui et al. 2012; Byeon et al. 2015). ATX is also metabolized by CYP2C19 to NAT, which is a minor metabolite of ATX. CYP2C19 is one of the polymorphic enzymes and it has been found that CYP2C19 genetic polymorphisms can affect the plasma exposure of ATX (Choi et al. 2014). Thus, in this study, subjects genotyped as CYP2C19*1/*1 were recruited to exclude the effects of CYP2C19 genetic polymorphism, and they were classified into three groups in relation to CYP2D6*10 allele.
Patients with ADHD are at increased risk for various comorbidities, including depression (Munir et al. 1987, Atomoxetine ADHD and Comorbid MDD Study Group et al. 2007). SSRIs are widely used to treat anxiety disorders and major depression and in vitro paroxetine was the most potent SSRI at inhibiting the CYP2D6-catalysed oxidation of sparteine (Crewe et al. 1992; Sindrup et al. 1992). Belle et al. (2002) reported that steady-state ATX plasma concentrations were higher after coadministration with paroxetine in CYP2D6 extensive metabolizers. In this study, paroxetine treatment increased the steady-state Cmax, AUC0 − 12 and t1/2 of ATX by approximately 3.5-, 6.5-, and 2.5-fold, respectively. Although the effect of paroxetine treatment on the pharmacokinetics of 4-HAT could not be quantified in this study, steady-state Cmax and AUC0 − 12 of NAT were 15.5- and 21.0-fold higher, respectively, and tmax of NAT was significantly increased after administration of paroxetine. Based on such drug interaction, we used 20 mg of ATX rather than the usual adult starting dose of 40 mg/d in order to avoid any potential adverse effects of ATX from the potent CYP2D6 inhibition of paroxetine. Our study showed drug interaction of ATX with paroxetine in relation to CYP2D6 genotype status. During administration of ATX alone, the pharmacokinetics of ATX was significantly different among three different genotype groups like previous studies (Cui et al. 2007; Matsui et al. 2012; Byeon et al. 2015); Cmax and AUC0−24 of ATX in CYP2D6*10/*10 group were 1.6- and 3.0-fold higher than those in CYP2D6*wt/*wt group, respectively. Cmax and AUC0−24 of NAT in CYP2D6*10/*10 were significantly higher than those in CYP2D6*wt/*wt and CYP2D6*wt/*10 group, although no significant change was found in the pharmacokinetic parameters of 4-HAT in relation to CYP2D6*10 allele. It is speculated that the pharmacokinetics of 4-HAT might be largely affected by uridine diphosphate glucuronosyltransferases (UGTs) as 4-HAT is subsequently conjugated by UGTs to form 4-HAT-O-glucuronide (Sauer et al. 2003), resulting in very low plasma concentrations of 4-HAT in human plasma after ATX treatment. In addition, 4-HAT is equipotent to ATX (Sauer et al. 2003) but it is speculated that 4-HAT may have little impact on clinical response due to very low plasma concentrations of 4-HAT.
During paroxetine phase, when all subjects received oral dose of atomoxetine with paroxetine, there were no significant differences in the pharmacokinetic parameters of ATX, 4-HAT, and NAT in relation to CYP2D6 genotype status. This suggested that administration of paroxetine, a potent CYPD6 inhibitor, makes the plasma exposure of ATX in CYP2D6*wt/*wt subjects similar to that in CYP2D6*10/*10 subjects who have functional but has decreased CYP2D6 activity. Due to relatively shorter plasma sampling time, AUC from 0 to infinity could not be calculated during paroxetine phase. Cmax and AUC0−24 of ATX in paroxetine phase was increased compared with atomoxetine phase in three different groups: 2.3-fold and 9.8-fold higher in CYP2D6*wt/*wt group, 1.7-fold and 4.7-fold higher in CYP2D6*wt/*10 group, and 1.3-fold and 2.9-fold higher in CYP2D6*10/*10 group, respectively. Cmax of 4-HAT was significantly decreased by paroxetine treatment, otherwise Cmax, AUC0−24, and tmax of NAT was increased, suggesting CYP2D6-mediated biotransformation of ATX was potently inhibited by paroxetine.
As we did not collet urine sample of each subject, we calculated the AUC ratio of 4-HAT and NAT to ATX using plasma concentrations. During atomoxetine phase, AUC ratio of 4-HAT to ATX in CYP2D6*wt/*wt group was 2.4-fold higher than that of CYP2D6*10/*10 group, and conversely AUC ratio of NAT to ATX in CYP2D6*wt/*wt group was 44.8% lower compared with CYP2D6*10/*10 group. After treatment with paroxetine, no significant change was found among different three genotype groups and compared to atomoxetine phase.
There are a few limitations in our study. ATX is predominantly metabolized by CYP2D6, but when CYP2D6 is not present, other CYP450 isozymes such as CYP2C19, CYP3A, CYP1A2, CYP2A6, and CYP2E1 are capable of forming 4-HAT at a substantially slower rate (Sauer et al. 2003). Paroxetine is a potent CYP2D6 inhibitor, however, it does not seem to affect CYP1A2, CYP2C19, and CYP3A4 (Belle et al. 2002). Thus, genetic polymorphisms of other CYP450 isozymes could have a little effect on the study results. Also, Belle et al. (2002) demonstrated coadministration of ATX with paroxetine to CYP2D6 extensive metabolizers resulted in higher standing heart rate and orthostatic heart rate changes than predicted based on circulating ATX concentrations alone. Although we did not evaluate safety parameters such as heart rate in all subjects in our study, but based on these results, it is speculated the pharmacodynamic interaction between ATX and paroxetine could occur in all three different groups, therefore, further study will be needed.
In conclusion, the present study evaluated the effect of paroxetine, a potent CYP2D6 inhibitor, on the pharmacokinetic parameters of ATX in relation to CYP2D6*10 allele. After paroxetine treatment, pharmacokinetic parameters of ATX and its two metabolites, 4-HAT and NAT, have been affected significantly. When ATX was administered alone, Cmax, AUC0-24 and CL/F were significantly different among the three CYP2D6 genotype groups. However, after paroxetine coadministration, no significant differences in these pharmacokinetic parameters were observed among the CYP2D6 genotype groups.
References
Atomoxetine ADHD, Comorbid MDDS, Group, Bangs ME, Emslie GJ, Spencer TJ, Ramsey JL, Carlson C, Bartky EJ, Busner J, Duesenberg DA, Harshawat P, Kaplan SL, Quintana H, Allen AJ, Sumner CR (2007) Efficacy and safety of atomoxetine in adolescents with attention-deficit/hyperactivity disorder and major depression. J Child Adolesc Psychopharmacol 17:407–420. doi:https://doi.org/10.1089/cap.2007.0066
Belle DJ, Ernest CS, Sauer JM, Smith BP, Thomasson HR, Witcher JW (2002) Effect of potent CYP2D6 inhibition by paroxetine on atomoxetine pharmacokinetics. J Clin Pharmacol 42:1219–1227. doi:https://doi.org/10.1177/009127002762491307
Byeon JY, Kim YH, Na HS, Jang JH, Kim SH, Lee YJ, Bae JW, Kim IS, Jang CG, Chung MW, Lee SY (2015) Effects of the CYP2D6*10 allele on the pharmacokinetics of atomoxetine and its metabolites. Arch Pharm Res 38:2083–2091. doi:https://doi.org/10.1007/s12272-015-0646-z
Byeon JY, Kim YH, Kim SH, Lee CM, Jung EH, Chae WK, Jang CG, Lee SY, Lee YJ (2018a) The influences of CYP2C9*1/*3 genotype on the pharmacokinetics of zolpidem. Arch Pharm Res 41(9):931–936. doi:https://doi.org/10.1007/s12272-018-1070-y
Byeon JY, Kim YH, Kim SH, Lee CM, Jung EH, Chae WK, Jang CG, Lee SY, Lee YJ (2018b) Effects of genetic polymorphisms of CYP2C19 on the pharmacokinetics of zolpidem. Arch Pharm Res 41(8):861–866. doi:https://doi.org/10.1007/s12272-018-1065-8
Byeon JY, Kim YH, Lee CM, Kim SH, Chae WK, Jung EH, Choi CI, Jang CG, Lee SY, Bae JW, Lee YJ (2018c) CYP2D6 allele frequencies in Korean population, comparison with East Asian, Caucasian and African populations, and the comparison of metabolic activity of CYP2D6 genotypes. Arch Pharm Res 41(9):921–930. doi:https://doi.org/10.1007/s12272-018-1075-6
Byeon JY, Lee YJ, Kim YH, Kim SH, Lee CM, Bae JW, Jang CG, Lee SY, Choi CI (2018d) Effects of diltiazem, a moderate inhibitor of CYP3A4, on the pharmacokinetics of tamsulosin in different CYP2D6 genotypes. Arch Pharm Res 41(5):564–570. doi:https://doi.org/10.1007/s12272-018-1030-6
Byeon JY, Lee CM, Lee YJ, Kim YH, Kim SH, Jung EH, Chae WK, Lee YJ, Jang CG, Lee SY (2019) Influence of CYP2D6 genetic polymorphism on pharmacokinetics of active moiety of tolterodine. Arch Pharm Res 42(2):182–190. https://doi.org/10.1007/s12272-018-1099-y
Choi CI, Bae JW, Lee HI, Jang CG, Sohn UD, Lee SY (2012b) Determination of atomoxetine metabolites in human plasma by liquid chromatography/tandem mass spectrometry and its application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci 885–886:103–108. doi:https://doi.org/10.1016/j.jchromb.2011.12.023
Choi CI, Bae JW, Jang CG, Lee SY (2012a) Tamsulosin exposure is significantly increased by the CYP2D6* 10/* 10 genotype. J Clin Pharmacol 52:1934–1938. doi:https://doi.org/10.1177/0091270011432168
Choi CI, Jang CG, Bae JW, Lee SY (2013) Validation of an analytical LC–MS/MS method in human plasma for the pharmacokinetic study of atomoxetine. J Anal Chem 68:986–991. https://doi.org/10.1134/S1061934813110051
Choi CI, Bae JW, Lee YJ, Lee HI, Jang CG, Lee SY (2014) Effects of CYP2C19 genetic polymorphisms on atomoxetine pharmacokinetics. J Clin Psychopharmacol 34:139–142. doi:https://doi.org/10.1097/JCP.0b013e3182a608a2
Crewe HK, Lennard MS, Tucker GT, Woods FR, Haddock RE (1992) The effect of selective serotonin re-uptake inhibitors on cytochrome P4502D6 (CYP2D6) activity in human liver microsomes. Br J Clin Pharmacol 34:262–265. doi:https://doi.org/10.1111/j.1365-2125.1992.tb04134.x
Cui YM, Teng CH, Pan AX, Yuen E, Yeo KP, Zhou Y, Zhao X, Long AJ, Bangs ME, Wise SD (2007) Atomoxetine pharmacokinetics in healthy Chinese subjects and effect of the CYP2D6*10 allele. Br J Clin Pharmacol 64:445–449. doi:https://doi.org/10.1111/j.1365-2125.2007.02912.x
Desta Z, Zhao X, Shin J-G, Flockhart D (2002) Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 41:913–958. https://doi.org/10.2165/00003088-200241120-00002
Gehlert DR, Gackenheimer SL, Robertson DW (1993) Localization of rat brain binding sites for [3H] tomoxetine, an enantiomerically pure ligand for norepinephrine reuptake sites. Neurosci Lett 157:203–206. doi:https://doi.org/10.1016/0304-3940(93)90737-6
Heydorn WE (1999) Paroxetine: a review of its pharmacology, pharmacokinetics and utility in the treatment of a variety of psychiatric disorders. Expert Opin Investig Drugs 8:417–441. doi:https://doi.org/10.1517/13543784.8.4.417
Jeppesen U, Gram L, Vistisen K, Loft S, Poulsen H, Brøsen K (1996) Dose-dependent inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. Eur J Clin Pharmacol 51:73–78. doi:https://doi.org/10.1007/s002280050163
Kim SH, Kim DH, Byeon JY, Kim YH, Kim DH, Lim HJ, Lee CM, Whang SS, Choi CI, Bae JW, Lee YJ, Jang CG, Lee SY (2017) Effects of CYP2C9 genetic polymorphisms on the pharmacokinetics of celecoxib and its carboxylic acid metabolite. Arch Pharm Res 40:382–390. doi:https://doi.org/10.1007/s12272-016-0861-2
Kim MJ, Byeon JY, Kim YH, Kim SH, Lee CM, Jung EH, Chae WK, Lee YJ, Jang CG, Lee SY, Choi CI (2018a) Effect of the CYP2D6*10 allele on the pharmacokinetics of clomiphene and its active metabolites. Arch Pharm Res 41:347–353. doi:https://doi.org/10.1007/s12272-018-1005-7
Kim SH, Byeon JY, Kim YH, Lee CM, Lee YJ, Jang CG, Lee SY (2018b) Physiologically based pharmacokinetic modelling of atomoxetine with regard to CYP2D6 genotypes. Sci Rep 8:12405. doi:https://doi.org/10.1038/s41598-018-30841-8
Lee HI, Bae JW, Choi CI, Lee YJ, Byeon JY, Jang CG (2014) Lee SY (2014) Strongly increased exposure of meloxicam in CYP2C9*3/*3 individuals. Pharmacogenet Genomics. 24(2):113–7. https://doi.org/10.1097/FPC.0000000000000025
Lee HJ, Kim YH, Kim SH, Lee CM, Yang AY, Jang CG, Lee SY, Bae JW, Choi CI (2016) Effects of CYP2C9 genetic polymorphisms on the pharmacokinetics of zafirlukast. Arch Pharm Res 39:1013–1019. doi:https://doi.org/10.1007/s12272-016-0785-x
Lee HI, Byeon JY, Kim YH, Lee CM, Choi CI, Jang CG, Bae JW, Lee YJ, Lee SY (2018) Effects of CYP2C19 and CYP3A5 genetic polymorphisms on the pharmacokinetics of cilostazol and its active metabolites. Eur J Clin Pharmacol 74(11):1417–1426. doi:https://doi.org/10.1007/s00228-018-2522-5
Matsui A, Azuma J, Witcher JW, Long AJ, Sauer JM, Smith BP, DeSante KA, Read HA, Takahashi M, Nakano M (2012) Pharmacokinetics, safety, and tolerability of atomoxetine and effect of CYP2D6*10/*10 genotype in healthy Japanese men. J Clin Pharmacol 52:388–403. doi:https://doi.org/10.1177/0091270011398657
Munir K, Biederman J, Knee D (1987) Psychiatric comorbidity in patients with attention deficit disorder: a controlled study. J Am Acad Child Adolesc Psychiatry 26:844–848. doi:https://doi.org/10.1097/00004583-198726060-00008
Pary R, Lewis S, Matuschka PR, Rudzinskiy P, Safi M, Lippmann S (2002) Attention deficit disorder in adults. Ann Clin Psychiatry 14:105–111. doi:https://doi.org/10.1023/a:1016807021779
Ring BJ, Gillespie JS, Eckstein JA, Wrighton SA (2002) Identification of the human cytochromes P450 responsible for atomoxetine metabolism. Drug Metab Dispos 30:319–323. doi:https://doi.org/10.1124/dmd.30.3.319
Sauer JM, Ponsler GD, Mattiuz EL, Long AJ, Witcher JW, Thomasson HR, Desante KA (2003) Disposition and metabolic fate of atomoxetine hydrochloride: the role of CYP2D6 in human disposition and metabolism. Drug Metab Dispos 31:98–107. doi:https://doi.org/10.1124/dmd.31.1.98
Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M (2006) A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 79:103–113. doi:https://doi.org/10.1016/j.clpt.2005.10.002
Sindrup SH, Brosen K, Gram LF, Hallas J, Skjelbo E, Allen A, Allen GD, Cooper SM, Mellows G, Tasker TCG, Zussman BD (1992) The relationship between paroxetine and the sparteine oxidation polymorphism. Clin Pharmacol Ther 51:278–287. doi:https://doi.org/10.1038/clpt.1992.23
Spencer T, Biederman J, Wilens T, Harding M, O’Donnell D, Griffin S (1996) Pharmacotherapy of attention-deficit hyperactivity disorder across the life cycle. J Am Acad Child Adolesc Psychiatry 35:409–432. doi:https://doi.org/10.1080/10826084.2016.1273955
Swanson JM, Sergeant JA, Taylor E, Sonuga-Barke EJ, Jensen PS, Cantwell DP (1998) Attention-deficit hyperactivity disorder and hyperkinetic disorder. Lancet 351:429–433. https://doi.org/10.12968/hmed.2010.71.11.79663
Teh LK, Bertilsson L (2012) Pharmacogenomics of CYP2D6: molecular genetics, interethnic differences and clinical importance. Drug Metab Pharmacokinet 27:55–67. https://doi.org/10.2133/dmpk.dmpk-11-rv-121
Wong DT, Threlkeld PG, Best KL, Bymaster FP (1982) A new inhibitor of norepinephrine uptake devoid of affinity for receptors in rat brain. J Pharmacol Exp Ther 222:61–65
Acknowledgements
This research was supported the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, & Future Planning (NRF-2019R1A2C1004582).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Eui Hyun Jung and Yun Jeong Lee contributed equally to this study.
Rights and permissions
About this article

Cite this article
Jung, E.H., Lee, Y.J., Kim, DH. et al. Effects of paroxetine on the pharmacokinetics of atomoxetine and its metabolites in different CYP2D6 genotypes. Arch. Pharm. Res. 43, 1356–1363 (2020). https://doi.org/10.1007/s12272-020-01300-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-020-01300-8