
Abstract
Deafness is the prevailing sensory impairment among humans, impacting every aspect of one's existence. Half of congenital deafness cases are attributed to genetic factors. Studies have shown that Luzp2 is expressed in hair cells (HCs) and supporting cells of the inner ear, but its specific role in hearing remains unclear. To determine the importance of Luzp2 in auditory function, we generated mice deficient in Luzp2. Our results revealed that Luzp2 has predominant expression within the HCs and pillar cells. However, the loss of Luzp2 did not result in any changes in auditory threshold. HCs or synapse number and HC stereocilia morphology in Luzp2 knockout mice did not show any notable distinctions. This was the first study of the role of Luzp2 in hearing in mice, and our results provide important guidance for the screening of deafness genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to the World Health Organization, there is a prevalent sensory disorder known as hearing loss. It is estimated that ~466 million individuals worldwide experience disabling hearing loss [1]. Various factors contribute to the prevalence of hearing loss, including genetic factors, and harmful environments like noise exposure, aging, and the use of ototoxic drugs [2]. At present, for severe and extremely severe sensorineural hearing loss, the main clinical treatment is cochlear implant surgery, but this method depends on the quantity and quality of the patient's remaining spiral ganglion neurons, and the high cost also limits accessibility, particularly in lower-income regions. In recent years, targeted gene therapy for deafness genes has become a trend in the treatment of deafness. Therefore, the investigation of potential genes associated with deafness holds immense importance in managing sensorineural hearing loss [3].
The organ of Corti, which comprises the hair cells (HCs) and supporting cells (SCs), serves as the fundamental structural unit of the cochlea. There are typically >16,000 HCs in the human cochlea, including three rows of outer hair cells (OHCs) and one row of inner hair cells (IHCs) [4]. Their primary role is to convert sound vibrations into electrical signals, facilitating transmission to the brain. At the apex of HCs are stereocilia responsible for transforming mechanical stimulation into essential electrical signals required for optimal auditory perception [5]. The electrical activity of the synapse and the release of neurotransmitters excite the nerve endings of spiral ganglion neurons and generate axon action potentials that then follow the brainstem auditory conduction path to the brain's auditory cortex center to produce hearing. Therefore, the number of HCs, the integrity, and the polarity of the stereocilia structure and ribbon synapses are essential for the normal transmission of sound [6]. SCs maintain the dynamic balance between the cochlear ions and the chemical environment [7]. Previous research has demonstrated that specific categories of SCs located within the inner ear possess the capacity to regenerate HCs in mice [8,9,10,11,12].
Luzp2 has been mapped between the region of Gas2 and HERC2 on mouse chromosome 7, and it encodes the novel leucine zipper protein 2. Studies have shown that Luzp2 is predominantly expressed in the brain, particularly in regions such as the cerebral cortex and hippocampus [13]. Its specific biological function is not yet comprehensively known, but it is speculated to have ties with neurodevelopmental disorders, autism, Alzheimer's disease, schizophrenia, and intellectual impairment, all of which have been documented to be linked with hearing impairment [14,15,16,17]. However, the specific relationship between Luzp2 gene variants and these disorders is still being investigated. Luzp2-deficient mouse embryos exhibit defects in cranial neural tube closure during brain development [18], and overexpression of Luzp2 has been detected in kinds of tumor tissues, and its expression is associated with the prognosis of these patients [19,20,21,22]. Luzp2 could potentially have a crucial impact on the formation of the extracellular matrix within the nervous system, serving as a significant clinical indicator for patients diagnosed with low-grade glioma [23, 24]. Transcript analysis has shown that Luzp2 is strongly expressed in pillar cells, OHCs, and IHCs, which suggests that the auditory system could potentially rely on Luzp2 [25]. Our investigation aimed to understand the involvement of Luzp2 in the function of hearing by creating Luzp2 knockout (KO) mice. Our findings showed that Luzp2 primarily manifested itself within the pillar cells and HCs situated in the cochlea of mice. Nevertheless, the auditory thresholds of the Luzp2 KO mice remained comparable to those of their wild-type (WT) littermates, and minimal variances were found in the quantities of HCs or synapses, as well as the structure of the hair bundles.
Materials and Methods
Animals
We obtained heterozygous Luzp2 mice from Cyagen through a customized purchasing process. To generate homozygous target mice, we proceeded by cross-breeding the heterozygous mice. All experiments involving mice followed the guidelines set by the Animal Care and Use Committee of Nanjing University, ensuring compliance with approved protocols. The experiments adhered strictly to the relevant regulations, including adherence to the ARRIVE guidelines. Our utmost effort was made to minimize the usage of animals throughout the experiments. The genotyping primers for Luzp2 KO mice were as follows: WT reverse, 5’-GGG AAC ACT AAT CAC TGA CCA CTC-3’; Mutant reverse, 5’-GGG AAC ACT AAT CAC TGA CCA CTC-3’; Common forward, 5’-GGC ATC ATT ACT AAC CTC CAT ATC-3’. PCR was conducted in 25 μl volumes using standard conditions for 35 cycles. Each reaction included the primers listed above. For genotyping, the controls comprised a water control (without a DNA template) and a WT control (using 400 ng of WT mouse genomic DNA as the template).
Immunofluorescence Staining
The fixation process for the basilar membranes (BMs) of mice involved immersing them in a 4% solution of paraformaldehyde for 1 h. Following this, the BMs were rinsed using PBS and were then exposed to a blocking medium. Subsequently, the BMs were rinsed with a PBS solution and then exposed to a blocking medium. The next step consisted of overnight incubation of the BMs with primary antibodies at 4°C and three rounds of washing with PBS. Finally, the BMs were fixed on a glass slide with DAKO (Protein Biotechnologies S3023), and images were captured on a Zeiss LSM900 confocal microscope. The antibodies applied in this study were anti-Luzp2 (Novus, NBP2-58798), and other commonly used antibodies have been described in detail in our previous articles.
Ribbon Synapse Quantification
To quantify the synapses, we divided the BM into the apex, middle, and basal turns. To capture an image, the BMs were magnified 63× under identical z-stack conditions. Next, we counted the CtBP2 and PSD95 puncta in the inner hair cells. By discerning the simultaneous occurrence signals of CtBP2 and PSD95, we were able to determine the number of synapses throughout the entire length of the sensory epithelium.
Auditory Brainstem Response (ABR)
To assess ABR, the TDT System III workstation (Tucker Davis Technology, USA) was used. Mice were given 10 mg/kg pentobarbital sodium as an anesthetic, and their body temperature was regulated at 38°C using a heating pad. The positioning of electrodes involved the subcutaneous insertion of the reference electrode behind the ear and the ground electrode under the leg skin. In addition, we put the recording electrode along the scalp towards the tip of the nose. To generate the stimuli and record the responses, we applied TDT hardware and software. The auditory stimulation was as follows: a pitch burst lasting 5 ms, rise and fall times of 0.5 ms, and cos2 gating. A broadband speaker (MF1; TDT) 7 cm in front of the mouse’s head was used to play the stimulus. The ABR threshold was determined at frequencies from 4 kHz to 32 kHz. Every frequency began at 90 dB followed by a decrement of 5 dB until the ABR waves were absent. This minimum wave is commonly accepted as the point where a consistent III-wave response becomes detectable and replicable during an ABR examination.
Scanning Electron Microscopy (SEM)
A solution containing 2.5% glutaraldehyde was used to fix mouse cochleae. To allow adequate fixation, we created a minimal aperture within the apical circle of the cochlea. The cochlea was maintained at 4°C overnight and decalcified using 0.5 mol/L EDTA for 3 h. After fixation in 1% tetroxide at 4°C for 2 h, the tissue was dehydrated in ethanol and then dried using liquid CO2 (CPD300, Leica). Following the application of a conductive coating, the cochlear tissue was imaged using an SEM setup.
RT-qPCR
RNA was extracted using TRIzol reagent (PR910) from Protein Biotechnology. Subsequently, cDNA was synthesized from mRNA using a kit for cDNA synthesis (K1622) following the instructions provided by Thermo Fisher Scientific. The qPCR protocol was set with the following parameters: initial denaturation at 95°C for 15 s and then 40 cycles of denaturing at 95°C for 60 s, annealing at 60°C for 30 s, and extension at 72°C for 15 s. To ensure accurate analysis, Gapdh was selected as the reference gene for normalizing the mRNA expression level. The following primers were utilized for qPCR amplification: Luzp2 forward: 5’- CAG CTC TTG ACA GGG AAT CAC-3’; Luzp2 reverse: 5’- CCA AAC AGG AAC TAC CCT CAT C-3’; Gapdh forward: 5’- GCA AGA GAG AGG CCC TCA G-3’; Gapdh reverse: 5’-TGT GAG GGA GAT GCT CAG TG-3’.
Western Blot
We dissected mouse cochleae in PBS on ice and proceeded to lyse the cells using RIPA lysis buffer in the presence of a cocktail. The protein was quantified using a BCA assay. β-actin (Abcam, ab119716) was served as the internal reference protein, and anti-Luzp2 (1:400, Novus, NBP2-58798) was the primary antibody. The secondary antibody was peroxidase-conjugated with anti-mouse IgG (Abcam, ab7260). The protein samples were centrifuged at 12,000 r/min at 4°C for 10 min. Then, the samples were mixed thoroughly with 5×SDS-PAGE loading buffer, boiled for 15 min, and subjected to SDS-polyacrylamide gel electrophoresis. The protein bands were transferred to a polyvinylidene fluoride membrane, and the target proteins were identified by a Super Signal West Dura Chemiluminescence Substrate Kit. ImageJ software was used to semi-quantify the band densities. The background was diminished, and the signals were adjusted to match the intensity of β-actin. Then the ratio of relative optical density was computed. Every trial was performed thrice.
Statistical Analysis
The mean ± standard deviation is used to present all the data, and every experiment was repeated a minimum of three times. The experimental data were statistically analyzed with GraphPad Prism 7 software. To determine the statistical difference between the two groups, a two-tailed, unpaired Student's t-test was applied for comparison. Statistically significant differences were considered to exist when the P-value was <0.05.
Results
Luzp2 Expression in the Mouse Cochlea
To assess the Luzp2 expression level in the cochlea, we used WT mouse brain tissue as the positive control as previously reported [13, 14, 18]. We immunolabeled Luzp2 in the cochlear epithelia of postnatal day 3 (P3) and P21 WT mice. Using confocal imaging, an organ of Corti whole-mount examination revealed the prominent expression of Luzp2 within the pillar cells and HCs of WT mice at both P3 and P21 (Fig. 1A), consistent with the results of previous studies [25]. To investigate the expression patterns of Luzp2 at various stages of development, q-PCR analysis was conducted on the cochlea of WT specimens. Our findings revealed a significant upregulation of Luzp2 expression at P14 in comparison to P3 and a significant upregulation of Luzp2 in the brain in comparison to the cochlea (Fig. 1B). This expression trend suggested that Luzp2 plays an important role in the development of HCs and the maintenance of hearing function. According to the western blot analysis, Luzp2 exhibited strong expression in both the brain and cochlea (Fig. 1C), demonstrating its stable presence within the mouse’s inner ear.

The expression of Luzp2 in WT mouse cochlea. A In P3 and P21 WT mice, the cochlear epithelium displays Luzp2 expression as demonstrated by immunofluorescence staining. Luzp2 is detectable in both the pillar cells and HCs. Scale bar, 10 μm. We acquired images from the middle turn of the sensory epithelium. B The mouse cochlea's expression of Luzp2 was revealed using RT-qPCR. The data are presented as the mean ± SD. C Western blot showing that expression of Luzp2 (39 kDa) is highly prominent in both the brain and cochlea, β-actin was used as the internal control.
Generation of Luzp2 –/– Mice
To investigate the physiological role of Luzp2 in hearing, we obtained Luzp2+/– mice from Cyagen. These mice were then cross-bred to generate homozygous target mice. The mouse Luzp2 gene contains 12 exons, and exon 2 was selected as the target site. Cas9 and gRNA were co-injected into fertilized eggs for KO mouse production, and the pups were genotyped by PCR followed by sequencing analysis (Fig. 2A). To validate the knockout of the Luzp2 gene in the cochlea, cochleae from P3 WT mice and Luzp2 knockout KO mice were dissected. The immunolabeling of Luzp2 in the cochleae of the KO mice was significantly reduced (Fig. 2B). In the cochlea and brain tissue of WT mice, western blot revealed the presence of a particular band weighing ~39 kDa, but it was not found in the cochlea or brain tissue of KO mice (Fig. 2C). Our results also verified the successful knockdown of the Luzp2 gene in KO mice based on qPCR outcomes (Fig. 2D). Consequently, we confirmed that the cochleae of Luzp2 KO mice effectively showed knockout of the Luzp2 gene.

The Luzp2 gene is knocked out in the transgenic mouse cochlea. A Construction of the heterozygous Luzp2 mice. B The cochleae of mice with P3 KO immunolabel for Luzp2 antibodies. No variation was observed in the immunolabeling of Luzp2 along the BM of the sensory epithelium's middle turn. Scale bar, 10 µm. C Western blots to assess Luzp2 KO mice and protein extractions were obtained from P3 WT and KO mice. Antibodies targeting Luzp2 (39 kDa) were used. The internal control for this analysis was β-actin (42 kDa). D qPCR analysis of Luzp2 levels in KO mice. The expression of the gapdh gene served as an internal control.
Knockout of Luzp2 Does not Change HC Number
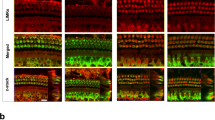
Transcriptome analysis has shown that Luzp2 is strongly expressed in part of SCs and HCs [25]. Consequently, we formulated the hypothesis that the Luzp2 KO mice would exhibit developmental abnormalities in the auditory system. We applied immunofluorescence staining to investigate the cochlear structure of WT and Luzp2 KO mice. P90 and P120 mice basilar membranes were artificially divided into the apex, middle, and basal turn, and myosin7a was used as the HC marker (Fig. 3A–D). We counted the number of HCs by ImageJ, and unexpectedly, no notable distinctions in morphology or HC count were detected when comparing WT and Luzp2 KO mice at P90 and P120 across the basal to apical cochlear turns (Fig. 3E, F).

The number normalcy of the auditory HCs in the Luzp2 KO mice. A–D Under a confocal microscope, the auditory HCs of P90 (A and B) and P120 (C and D) mice are stained with an antibody specific to myosin7a. Scale bar, 10 µm. E–F The numbers of OHCs, IHCs, and total HCs at P90 (E) and P120 (F). Statistical analysis indicates no significant differences. The data obtained are presented as the mean ± SD.
Knockout of Luzp2 Does not Change Ribbon Synapses
We evaluated whether there were potential changes in cochlear ribbon synapse quantity in Luzp2 KO mice using CtBP2 and PSD95 immunolabeled presynapses and postsynapses. PSD95 signals were concentrated in the plasma membrane at the end of IHCs, and CtBP2 interacts with PSD95 and contributes to auditory signal transmission. In comparison to WT mice, Luzp2 KO mice exhibited no significant disparity of CtBP2+ and PSD95+ point number, and almost all CtBP2/PSD95 overlapped spots at P90 (Fig. 4A, B) and P120 (Fig. 4C, D). Furthermore, in both the Luzp2 KO and WT mice, almost all the CtBP2 spots overlapped with PSD95. Our results suggest that the knock-out of Luzp2 does not alter the synapse production in the cochlea.

The ribbon synapse normalcy of the auditory IHCs in the Luzp2 KO mice. A Under a confocal microscope, CtBP2 and PSD95 are used to stain and image ribbon synapses in P90 Luzp2 KO mice (A1 and A2) and WT (A3 and A4) mice, and no observable distinctions between the two groups. (A2) and (A4) are found in a magnified view of the dotted parts of (A1) and (A3). B Ribbon synapse counts in P90 Luzp2 knockout mice show no significant difference compared with WT mice. C Under a confocal microscope, CtBP2 and PSD95 are used to stain and image ribbon synapses in P120 Luzp2 KO (C1 and C2) and WT (C3 and C4) mice and reveal no observable distinctions between the two groups. (C2) and (C4) are magnified views of the dotted parts of (C1) and (C3). (D) Ribbon synapse counts in P120 Luzp2 knockout mice show no significant difference compared with WT mice. Scale bars (A, C), 5 µm. The data are presented as the mean ± SD.
Knockout of Luzp2 Does not Change the Stereocilia Morphology of HCs
Stereocilia, which are protrusions made of actin on the BM of HCs, and movements of the stereocilia in response to sound stimuli transform the mechanical energy into electrical signals that can be interpreted by the brain, and the proper three-dimensional structure of the stereocilia within the cochlea contribute to the ear’s ability to detect and discriminate different frequencies of sound. Our immunofluorescence phalloidin staining showed that the Luzp2 KO mice have normal stereocilia polarity of cochlear HCs at P90 and P120 (Fig. 5A–D). Then, we used SEM to examine the finer structure of stereocilia; the results shown in Fig. 5E–F demonstrate that there was no substantial distinction between WT mice and KO mice at P120. The results indicate that the expression of Luzp2 in mice does not affect stereocilia structure.

No discernible differences in HC stereocilia morphology in KO mice. A–D The auditory middle turn HC stereocilia in P90 and P120 Luzp2 KO mice (A and C), as well as WT mice (B and D), are stained and imaged with FITC-conjugated phalloidin and show normal morphology in both genotypes. E and F SEM images of the finer structure of Luzp2 KO and WT mouse stereocilia. The enlarged images in panels E' and F' correspond to the white dotted boxes in panels E and F, respectively. Scale bars in A -F = 10 µm.
Auditory Functions are Normal in Luzp2 KO Mice
Even though the general health of Luzp2 KO mice has been reported, there remains a lack of knowledge regarding their auditory abilities. As a result, we measured the ABR, which indicates the overall neural activity originating from the cochlea. At P60, P90, and P120, there were no discernible distinctions in hearing ability between the KO mice and WT controls (Fig. 6A–C). In addition to the threshold, we chose the I, III, and V waves of the ABR waveform at a frequency of 16 kHz under a 90 dB intensity stimulus, we analyzed the amplitude and latency of the ABR waveforms in P120 KO and WT mice, and no significant difference was found between mice (Fig. 6D, E). Our results suggest that the maintenance of hearing is not reliant on the indispensability of Luzp2.

No discernible differences in ABR in KO mice. A ABR thresholds for P60 KO and WT mice at frequencies from 4 to 32 kHz. B Similarly, ABR thresholds for P90 KO and WT mice using the identical frequency range. C ABR thresholds for P120 KO and WT mice using the identical frequency range. D–E Representative graphs of ABR waveform, and analysis of I, III, and V wave amplitude and latency. Data are presented as the mean ± SD.
Discussion
Leucine zipper proteins contain leucine zipper motifs, and these motifs are super-secondary structures that function as a dimerization domain [26, 27]. By radiation hybridization, Luzp2 has been mapped to chromosome 11p13–11p14, and this gene deletion has been reported in a subset of individuals diagnosed with Wilms tumor [13]. At present, there are few studies on Luzp2, and its biological function remains unclear, although there is a study showing that targeted disruption of the Luzp2 gene in mice results in no evident phenotypic abnormalities [13]. A few studies have focused on the brain, and some studies have shown that the deletion of this gene can cause abnormal development of the cranial neural tube in mouse embryos [14, 18]. Abnormal expression of this gene is also found in tumors such as breast cancer and glioma [19, 23]. The specific biological role of Luzp2 is still unknown, and research on Luzp2 in the field of hearing is non-existent.
Liu et al. discovered the Luzp2 gene when studying the differences in the transcriptional genomes of HCs and SCs, and they used immunofluorescence to verify that the gene is expressed in the IHCs, OHCs, and pillar cells of the mouse cochlea [25]. We constructed Luzp2 KO mice to study the hearing phenotype of this gene, and we also verified the effects of Luzp2 in terms of the function and morphology of HCs. The hearing of P90 KO and P120 KO mice showed no statistical differences at any frequency, thus it can be concluded that a lack of Luzp2 does not affect cochlear function in mice. In addition, we observed no difference in the morphology of the HCs, synapses, or stereocilia between KO mice and WT controls. Loss of Luzp2 can negatively affect the development of the nervous system, but it does not significantly affect hearing, thus indicating that the roles of Luzp2 in the hearing system and the nervous system are not synchronized. In conclusion, we double-confirmed Luzp2 expression in mouse cochlea, but specific deletion of Luzp2 did not lead to any evident hearing disability phenotypes. The HCs, synapses, and stereocilia of KO mice and control mice did not show any significant differences, and thus Luzp2 does not play a crucial role in the development or function of the auditory system. It appears that Luzp2 might not be a key part of the auditory signal transduction pathway. Such an effect could be compensated for by a similar protein resulting in the loss of Luzp2 not manifesting as an auditory phenotype. Our experiments confirm that Luzp2 KO mice do not show the auditory deficit phenotype and more research is needed on the specific biological function of Luzp2.
Data Availability Statement
The data sets used and analyzed during the present study are available from the corresponding author upon reasonable request.
Change history
02 August 2024
A Correction to this paper has been published: https://doi.org/10.1007/s12264-024-01250-x
References
WHO. Deafness and hearing loss. Homepage Available from: https://www.who.int/health-topics/hearing-loss#tab=tab_2.
Géléoc GS, Holt JR. Sound strategies for hearing restoration. Science 2014, 344: 1241062.
Ahmed H, Shubina-Oleinik O, Holt JR. Emerging gene therapies for genetic hearing loss. J Assoc Res Otolaryngol 2017, 18: 649–670.
Defourny J, Lallemend F, Malgrange B. Structure and development of cochlear afferent innervation in mammals. Am J Physiol Cell Physiol 2011, 301: C750–C761.
Fettiplace R. Hair cell transduction, tuning, and synaptic transmission in the mammalian cochlea. Compr Physiol 2017, 7: 1197–1227.
Schwander M, Kachar B, Müller U. Review series: The cell biology of hearing. J Cell Biol 2010, 190: 9–20.
Raphael Y, Altschuler RA. Structure and innervation of the cochlea. Brain Res Bull 2003, 60: 397–422.
Chen Y, Lu X, Guo L, Ni W, Zhang Y, Zhao L. Hedgehog signaling promotes the proliferation and subsequent hair cell formation of progenitor cells in the neonatal mouse cochlea. Front Mol Neurosci 2017, 10: 426.
Cheng C, Guo L, Lu L, Xu X, Zhang S, Gao J, et al. Characterization of the transcriptomes of Lgr5+ hair cell progenitors and Lgr5- supporting cells in the mouse cochlea. Front Mol Neurosci 2017, 10: 122.
Wu J, Li W, Lin C, Chen Y, Cheng C, Sun S, et al. Co-regulation of the Notch and Wnt signaling pathways promotes supporting cell proliferation and hair cell regeneration in mouse utricles. Sci Rep 2016, 6: 29418.
Cox BC, Chai R, Lenoir A, Liu Z, Zhang L, Nguyen DH, et al. Spontaneous hair cell regeneration in the neonatal mouse cochlea in vivo. Development 2014, 141: 816–829.
Li H, Roblin G, Liu H, Heller S. Generation of hair cells by stepwise differentiation of embryonic stem cells. Proc Natl Acad Sci U S A 2003, 100: 13495–13500.
Wu M, Michaud EJ, Johnson DK. Cloning, functional study and comparative mapping of Luzp2 to mouse Chromosome 7 and human Chromosome 11p13–11p14. Mamm Genome 2003, 14: 323–334.
Stepanov V, Vagaitseva K, Bocharova A, Marusin A, Markova V, Minaycheva L, et al. Analysis of association of genetic markers in the LUZP2 and FBXO40 genes with the normal variability in cognitive performance in the elderly. Int J Alzheimers Dis 2018, 2018: 2686045.
Ralli M, Gilardi A, Stadio AD, Severini C, Salzano FA, Greco A, et al. Hearing loss and Alzheimer’s disease: A Review. Int Tinnitus J 2019, 23: 79–85.
Viertiö S, Perälä J, Saarni S, Koskinen S, Suvisaari J. Hearing loss in persons with psychotic disorder—Findings from a population-based survey. Schizophr Res 2014, 159: 309–311.
Cheng C, Hou Y, Zhang Z, Wang Y, Lu L, Zhang L, et al. Disruption of the autism-related gene Pak1 causes stereocilia disorganization, hair cell loss, and deafness in mice. J Genet Genomics 2021, 48: 324–332.
Hsu CY, Chang NC, Lee MWY, Lee KH, Sun DS, Lai C, et al. LUZP deficiency affects neural tube closure during brain development. Biochem Biophys Res Commun 2008, 376: 466–471.
Jang SY, Jang SW, Ko J. Regulation of ADP-ribosylation factor 4 expression by small leucine zipper protein and involvement in breast cancer cell migration. Cancer Lett 2012, 314: 185–197.
Peng Y, Clark C, Luong R, Tu WH, Lee J, Johnson DT, et al. The leucine zipper putative tumor suppressor 2 protein LZTS2 regulates kidney development. J Biol Chem 2011, 286: 40331–40342.
Kang H, Jang SW, Ko J. Human leucine zipper protein sLZIP induces migration and invasion of cervical cancer cells via expression of matrix metalloproteinase-9. J Biol Chem 2011, 286: 42072–42081.
Feng D, Zhu W, Shi X, Wei W, Han P, Wei Q, et al. Leucine zipper protein 2 serves as a prognostic biomarker for prostate cancer correlating with immune infiltration and epigenetic regulation. Heliyon 2022, 8: e10750.
Li Y, Deng G, Qi Y, Zhang H, Jiang H, Geng R, et al. Downregulation of LUZP2 is correlated with poor prognosis of low-grade glioma. Biomed Res Int 2020, 2020: 9716720.
Feng D, Shi X, Zhu W, Zhang F, Li D, Han P, et al. A pan-cancer analysis of the oncogenic role of leucine zipper protein 2 in human cancer. Exp Hematol Oncol 2022, 11: 55.
Liu H, Chen L, Giffen KP, Stringham ST, Li Y, Judge PD, et al. Cell-specific transcriptome analysis shows that adult pillar and deiters’ cells express genes encoding machinery for specializations of cochlear hair cells. Front Mol Neurosci 2018, 11: 356.
Meissner CS, Suffner S, Schauflinger M, von Einem J, Bogner E. A leucine zipper motif of a tegument protein triggers final envelopment of human cytomegalovirus. J Virol 2012, 86: 3370–3382.
Wemhöner K, Silbernagel N, Marzian S, Netter MF, Rinné S, Stansfeld PJ, et al. A leucine zipper motif essential for gating of hyperpolarization-activated channels. J Biol Chem 2012, 287: 40150–40160.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81970884, 81900941, 81970885, 82371157, 82171145, 82271173, and 81771019), the Natural Science Foundation of Jiangsu Province (BK20190121 and BK20200133), the China Postdoctoral Science Foundation (2020M681555), and a Distinguished Young Scholarship supported by the Medical Science and Technology Development Foundation, Nanjing Department of Health (JQX20003).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cheng, C., Zhu, G., Wang, K. et al. Deletion of Luzp2 Does Not Cause Hearing Loss in Mice. Neurosci. Bull. 40, 1519–1528 (2024). https://doi.org/10.1007/s12264-024-01202-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-024-01202-5