
Abstract
Gene therapy, or the treatment of human disease using genetic material, for inner ear dysfunction is coming of age. Recent progress in developing gene therapy treatments for genetic hearing loss has demonstrated tantalizing proof-of-principle in animal models. While successful translation of this progress into treatments for humans awaits, there is growing interest from patients, scientists, clinicians, and industry. Nonetheless, it is clear that a number of hurdles remain, and expectations for total restoration of auditory function should remain tempered until these challenges have been overcome. Here, we review progress, prospects, and challenges for gene therapy in the inner ear. We focus on technical aspects, including routes of gene delivery to the inner ear, choice of vectors, promoters, inner ear targets, therapeutic strategies, preliminary success stories, and points to consider for translating of these successes to the clinic.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Hearing loss is the most common neurological disorder on the planet. In the USA alone, the cost to society runs well into the billions of dollars. About 2 to 3 of every 1000 children in the USA are born with a clinically significant hearing loss in one or both ears. Estimates suggest that 50 % of congenital hearing loss has a genetic etiology with approximately 4000 babies affected each year. Furthermore, with improved genetic testing and expanded focus on moderate to severe forms of hearing loss, additional genetic contributions have been identified (Imtiaz et al. 2016; Naz et al. 2017). Unfortunately, there are currently no biological treatments for hearing loss. Current state-of-art treatments focus on sound amplification and implanted electrodes that stimulate the auditory nerve. These strategies offer partial recovery of function for a limited patient population but do not come close to restoring natural hearing. Clearly, there is strong need for development of biological treatments for restoration of auditory function, and gene therapy has emerged as a possible strategy for treatment of some forms of genetic deafness (Géléoc and Holt, 2014). Ultimately, which forms of genetic deafness and which gene therapy strategies, vectors, promoters, routes of delivery, genetic sequences of the therapeutic agent, time points of administration, patient populations, and costs will need to be determined before clinical trials can commence. Here, we review the current state of the field and the prospects for development of gene therapy for treatment of genetic deafness in humans.
ROUTES FOR DELIVERY
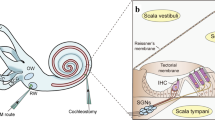
The inner ear is an attractive target for gene therapy. Like the eye, the inner ear is a relatively accessible, enclosed fluid-filled space. This provides several distinct advantages, as well as a few disadvantages. Because there are significant physical and diffusion barriers, including the blood-labyrinthine barrier, systemic distribution of therapeutic agents is limited (Harris and Ryan 1995). While the blood-labyrinthine barrier is not as tight as the blood-brain barrier, it provides an impediment that may slow distribution of large molecules, such as viral vectors and other gene therapy reagents. These features can be advantageous, because the blood-labyrinthine barrier allows therapeutic agents injected into the cochlea to remain within the target organ at high concentration while minimizing distribution, and perhaps toxicity, in off target organs and tissues.
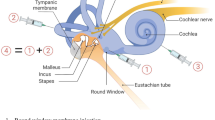
Multiple routes of delivery to the inner ear have been explored. These include injection into the perilymphatic spaces via the round window membrane (RWM) and via the oval window and injection into the scala tympani or scala vestibule via cochleostomy (Rivera et al. 2012; Chien et al. 2015a, b). Distribution throughout the perilymphatic spaces has been demonstrated for all these routes of delivery. Furthermore, it has been demonstrated that advection flow through the cochlea and vestibular organs can facilitate distribution of therapeutic agents from the site of injection to more distant regions of the inner ear (Borkholder et al. 2014).
Delivery into the endolymphatic spaces has also been explored via cochleostomy into the scala media, via canalostomy (Suzuki et al. 2017) and by injection into the endolymphatic sac (Yamasoba et al. 1999; Kitahara et al. 2003; Rivera et al. 2012). These approaches have also yielded broad distribution but face the added challenge of breaching the barrier between high potassium endolymph and perilymph. Disruption of the barrier poses two potential problems. First, leakage of high potassium into the perilymphatic spaces that bathe the basolateral surface of hair cells and neurons can chronically depolarize these cells and lead to cell death. Second, breach of the tight junctions between endolymph and perilymph can lead to rundown of the endocochlear potential which typically ranges between +80 and +120 mV. Rundown of the endocochlear potential reduces the driving force for sensory transduction in hair cells and therefore leads to reduced cochlear sensitivity and elevated auditory thresholds. Avoiding these complications is particularly challenging in the adult cochlea. However, by targeting endolymphatic spaces in the vestibular system, which does not have an endolymphatic potential, but are continuous with cochlear endolymph spaces, these confounding issues may be minimized while still providing sufficient distribution within the cochlea.
The choice of delivery route may largely be dependent on the cell type to be targeted. For example, if the stria vascularis is to be targeted, as might be the case for mutations in KCNQ1 and KCNE1 genes which cause Jervell and Lange-Nielsen syndrome (combination of sensorineural hearing loss and cardiac abnormalities), injection in to the endoymphatic spaces may be required (Chang et al. 2015). On the other hand, if spiral ganglion neurons are the target, a perilymph injection would be needed. For some cell types, particularly those that reside at the junction between endolymph and perilymph, both delivery routes may need to be explored to identify the optimal approach (Fig. 1).

Schematic diagram illustrating several routes of delivery for injecting gene therapy vectors into the inner ear. Fluorescent images showing expression of green fluorescent protein were selected from the literature to illustrate use of several AAV vector serotypes and delivery routes from the top right, moving clockwise, they are as follows: cochleostomy, exo-AAV-GFP (György et al. 2017); cochleostomy, AAV8-GFP (Kilpatrick et al., 2011); round window membrane, AAV2-GFP (Akil et al. 2015); posterior semicircular canal, Anc80-GFP (Suzuki et al. 2017). Each fluorescence image is oriented with a single row of inner hair cells at the bottom and three rows of outer hair cells at the top.
VECTORS
A number of viral and nonviral vectors have been developed for delivery of genetic material in various tissues and organs (Sacheli et al. 2013; Fukui and Raphael 2013; Kohrman and Raphael 2013). In most cases, these vectors are replication incompetent and pose little threat of viral-induced disease. Rather, the viral genome has been partly or fully deleted, expanding the capacity to allow inclusion of therapeutic DNA cargo within the viral capsid. Some vectors include single-stranded DNA, while others include double-stranded DNA. All viral vectors have a limited delivery capacity and some have limited expression duration. Choice of delivery vector can affect the outcome of a gene therapy approach. However, provided that the target cell type is transduced, the nature of the viral capsid may be of little consequence. By analogy, as long as she makes it to the operating room, does it really matter if your neurosurgeon drives a Chevy or a Ford? In other words, the quality of the intervention is likely to have a greater influence on the outcome than the delivery vehicle. Several of the more common delivery vectors have been tested in the inner ear and are reviewed below.
Lentivectors
Lentiviruses form a genus of the Retroviridae family and have a single-stranded DNA genome of approximately 9 kb packaged into enveloped virions. The lentivectors (LVs) used for gene therapy are based on human immune deficiency virus type 1 or feline immunodeficiency virus. LVs are usually pesudotyped, meaning the vector is combined with foreign viral envelope proteins to expand cell tropism, such as glycoproteins from viruses with broader tropism, i.e., vesicular stomatitis virus (Cronin et al. 2005). To date, several generations of LVs have been developed with the ability to deliver up to 8 kb of foreign DNA (Table 1). Apart from large cargo capacity, the major advantage of LVs is their ability to efficiently transduce both dividing and nondividing cells. To enhance biosafety and prevent consequences of LV integration and replication, self-inactivating and integration-deficient LVs were developed (Logan et al. 2004; Wanisch and Yáñez-Muñoz 2009; Yang et al. 2013a). Since the integration-deficient LVs persist in host cells as nonreplicative episomes, they are applicable for stable transgene expression in nondividing cells, like cells of the inner ear.
In recent years, several studies have demonstrated LV’s ability to transduce inner ear cells. In vitro LV transduction in cochlear explants from neonatal rats yielded numerous transduced cell types, including spiral ganglion neurons, glial cells, and supporting cells, although transduction of the sensory cells was not observed. In vivo, via in utero injection into the developing otocyst, LVs showed low-level transgene expression in outer hair cells (OHCs) and supporting cells, but no expression in inner hair cells (IHCs) was detected (Bedrosian et al. 2006). At more mature stages, in vivo intracochlear perfusion of LV into guinea pig inner ear revealed restricted virus dissemination limited to the periphery of the perilymphatic space (Han et al. 1999). Likewise, introduction of LV into perilymph via the round window membrane led to transgene expression in cells lining the scala tympani and scala vestibuli without affecting cells of the organ Corti or the spiral ganglion (Pietola et al. 2008). LV injection into endolymph via scala media lateral wall cochleostomy resulted in expression of transgene in the marginal cells of the stria vascularis, the organ of Corti, and spiral ganglion cells, although was accompanied by hearing loss caused by the surgical approach (Wei et al. 2013). However, LV injection into scala media of neonatal mice did not induce hearing loss and provided transgene expression in Hensen’s cells in the organ of Corti, marginal cells, and intermediate cells in the stria vascularis. It is of note that in all the described cases, IHCs and OHCs were minimally transduced (Wang et al. 2013). Although some concerns about LV ototoxicity have been raised, the possible ototoxicity can be eliminated by decreasing the LV dosage (Bedrosian et al. 2006). Thus, other than their lack of ability to target hair cells, LVs may be broadly useful vectors for inner ear gene delivery.
Adenovirus
Adenoviruses (AdVs) are members of the Adenoviridae family. They represent nonenveloped icosahedral nucleocapsids of 90–100 nm in diameter and contain a 36-kb double-stranded DNA genome (Table 1). Although humans can be infected by more than 50 serotypes of AdVs, those used for gene transfer are based on the most-studied serotypes, 2 and 5. AdVs can broadly infect a large number of different cell types, both dividing and nondividing, with high-level and possibly long-term transgene expression. These properties have made AdVs promising gene transfer vectors for inner ear gene therapy (Shu et al. 2016).
Several generations of replication-deficient AdVs have been developed with increased biosafety and cargo capacity due to removal of regions that include the E1/3 genes (first generation, 7 kb of capacity) or the E1/2–4 genes (second generation, 14 kb of capacity) (Räty et al. 2008). At the same time, due to high transduction efficiency and expression of residual viral genes, these vectors induce considerable innate and adaptive immune responses. The third generation of “gutless” AdVs does not contain any viral DNA except for packaging signals and inverted terminal repeats. Production of advanced generation AdVs is facilitated by providing the deleted AdV gene sequences in packaging cell lines. Deletion of AdV genome allows increased cargo capacity up to 36 kb, significantly lowers immune responses, and prolongs transgene expression (Alba et al. 2005). Nonimmunogenic transgenes delivered with gutless AdVs in mouse demonstrated a lifetime expression, although it is usually confined to several months in case of AdVs of earlier generations (Chen et al. 1997; Morral et al. 1998; Moorhead et al. 1999).
Susceptibility of inner ear cells to AdV transduction is determined by the presence of adenovirus receptors—coxsackievirus and adenovirus receptor along with ανβ3/ανβ5 integrin co-receptors (Zhang and Bergelson 2005; Husseman and Raphael 2009). In rat cochlea, these receptors are expressed in inner ear cells transduced by Ad5 vectors in vivo. Upon perilymphatic perfusion, only those cells lining the perilymphatic spaces expressed transgene, but no expression occurred in cells of the organ of Corti, stria vascularis, or spiral ganglion. Upon endolymphatic perfusion, transduction was found in the cells lining scala media, Hensen’s, Deiters’, pillar and phalangeal cells, and the satellite cells surrounding the spiral ganglion neurons. Transduction of sensory hair cells was not generally seen, although they express the required receptors. Some IHCs and OHCs were transduced after syringe injection into basal turn of the scala tympani (Venail et al. 2006). Likewise, direct injection of first-generation AdVs into the basal turn of the guinea pig cochlea in vivo transduced spiral ganglion cells, but not hair cells (Li Duan et al. 2002). Transduction of neonatal rat cochlear cells by AdVs was demonstrated after 72 h with the highest transgene expression level in IHCs (Dazert et al. 1997). Thus, AdVs may be broadly useful for gene transfer to the inner ear, targeting within confined fluid-filled spaces. In organotypic cultures of human vestibular epithelia, first- and second-generation AdVs type 5 efficiently targeted both hair cells and supporting cells, which suggests that successful implementation of AdVs for human inner ear gene therapy in vivo may be possible (Kesser et al. 2007).
Examples of Ad-mediated transgene delivery into the inner ear are numerous and have been thoroughly reviewed elsewhere (Husseman and Raphael 2009; Sacheli et al. 2013; Chien et al. 2015a, b). More recently, AdVs have been used to mediate delivery of Math1 (Atoh1) transcription factor to transdifferentiate cells in the lateral epithelial ridge to hair cell-like cells (Yang et al. 2013b); Ad-mediated overexpression of heat shock protein 70 resulted in significant preservation of hair cells after ototoxic shock (Takada et al. 2015).
In summary, AdVs are very efficient gene delivery vectors. Although there is room for improvement of their transduction and targeting properties, the ability of AdVs to target multiple inner ear cell types and their large cargo capacity suggests that AdVs hold promise for gene therapy use in human inner ear disorders.
Adeno-Associated Virus
Adeno-associated viruses (AAV) were discovered more than 50 years ago (Atchison et al. 1965) and are now widely regarded as promising vectors for a number of viral gene delivery applications. AAVs are small replication-deficient adenovirus-dependent viruses from the Parvoviridae family (Table 1). They have an icosaedrical capsid of 20–25 nm in diameter and a genome of 4.8 kb flanked by two inverted terminal repeats (ITRs). After uncoating in a host cell, the AAV genome can persist in a stable episome state by forming high molecular weight head-to-tail circular concatamers, or can integrate into the host cell genome (Yang et al. 1999). Both scenarios provide long-term and high-level transgene expression.
AAVs are endemic in human populations, although multiple preclinical studies have not identified any associated diseases (Calcedo and Wilson 2013; Mingozzi and High 2013). At the same time, since AAV infection is common in childhood, up to 50 % of some populations have preexisting immunity to AAV. It is of note that such people are currently excluded from AAV clinical trials, which may skew the results of general AAV efficiency in humans (Zinn et al. 2015).
Twelve natural occurring serotypes of human AAV have been characterized to date. These serotypes have different inherent tropisms and transduction efficiencies in muscles, lung, liver, brain, retina, and vasculature. Multiple attempts of AAV pseudotyping and capsid engineering resulted in considerable improvement of tropism and efficiency of transduction (Rabinowitz and Samulski 2000; Zincarelli et al. 2008; Yu et al. 2009; Geguchadze et al. 2012; Aschauer et al. 2013; Sallach et al. 2014; Zinn et al. 2015; Castle et al. 2016; Mao et al. 2016; Marsic and Zolotukhin 2016; Payne et al. 2016; Sayroo et al. 2016; De Silva et al. 2016). As for cells of the inner ear, AAV1–4, 7, and 8 were shown to infect spiral limbus, spiral ligament, and spiral ganglion cells in vivo. Infection of IHCs was also shown for AAV1–3, 5, 6, and 8. AAV1 was the most effective and occasionally infected OHCs and supporting cells (Akil et al. 2012; Askew et al. 2015). Also, AAV5 was shown to be efficient for Claudius cells, spiral ganglion, and inner sulcus cells (Jero et al. 2001; Li Duan et al. 2002; Liu et al. 2005; Kilpatrick et al. 2011; Chien et al. 2015a, b). Among pseudotyped vectors, AAV2/1 was found to efficiently transduce progenitor cells giving rise to IHCs and OHCs in mouse cochlea (Bedrosian et al. 2006), and AAV2/2 was optimal for IHCs of guinea pig cochlea (Konishi et al. 2008a, b).
One of the major disadvantages of AAV is small cargo capacity. Compared to adenoviruses, which are able to package up to 36 kb, AAV with an insert size more than 4.8 kb is unstable. However, several approaches have been established to circumvent the small packaging capacity. The first approach is based on the ability of AAV genomes to undergo intermolecular ITR-mediated head-to-tail concatamerization (Trapani et al. 2013). Placing a splicing donor at the 3′-end of the 5′-half of a transgene and a splicing acceptor at the 5′-end of the 3′-half of a transgene can yield transplicing of two halves of the concatemer and formation of mature messenger RNA (mRNA) and synthesis of full-length protein. The trans-splicing-mediated dual gene delivery approach was successfully implemented in several animal models (Cao et al. 2012; Trapani et al. 2013; Colella et al. 2014; Dyka et al. 2014; Tal-Goldberg et al. 2014; Wang et al. 2014; Fine et al. 2015). The development of this method, placing ITRs from different AAV serotypes on the ends of trans-splicing gene fragments, allows direct intermolecular recombination and enhanced efficiency of dual AAV vector trans-splicing (Yan et al. 2007). With this approach, an efficient trans-splicing of three fragments of complementary DNA (cDNA) from the DMD coding sequence (ORF size 11.1 kb) was demonstrated in muscle tissues of dystrophic mdx mice (Koo et al. 2014). While promising for gene sequences larger than 4.8 kb, trans-splicing has not been used for inner ear gene therapy.
Another possible method for overcoming the limited AAV cargo capacity relies on intein-mediated protein splicing (Topilina and Mills 2014). With this approach, N- and C-terminal fragments of a protein are encoded fused with N- and C-terminal fragments of trans-acting split-inteins. After dual delivery of the two transgenes into a single cell, expression of the two intein fusion proteins will form a functional intein assembly and splice itself out with accurate ligation of the two fragments of the target protein. To date, split-genes of Becker-form dystrophin (size 6.3 kb) was successfully delivered via AAV1 vectors and expressed a functional protein in muscle of a Duchenne muscular dystrophy mouse model (Li et al. 2008). In another example, intein splicing was demonstrated in a cell culture with delivery of split Cas9 in two recombinant AAV vectors (Truong et al. 2015).
Anc80L65
Anc80L65 is an effective representative of combinatorial library of AAV evolutionary ancestors (Anc80Lib) obtained by methods of maximum likelihood phylogeny from capsid sequences of 75 naturally occurring and clinically relevant AAVs (Zinn et al. 2015). In terms of production, yield of Anc80L65 is generally better than AAV2 but significantly depressed compared to the AAV8 (Zinn et al. 2015). Thus, production and purification of the vector may be a limiting factor for application in humans.
Anc80L65 demonstrated exceptional transduction efficiency in murine muscle, retina, and liver tissues, outperforming the most robust wild-type AAV8 in some of them. In addition, Anc80L65 showed outstanding efficiency of transduction of inner ear hair cells (Landegger et al. 2017). Delivery into rodent cochleas at early postnatal stages through the round window resulted in ~90 % transduction of both IHC and OHC with doses of genome-containing particles 2–3 times lower than required for other AAV serotypes. Delivery via canalostomy at adult stages leads to nearly 100 % IHC transduction, but fewer OHCs (Suzuki et al. 2017). Despite this caveat, this is the highest efficiency for AAV transduction of inner ear hair cells reported to date. Moreover, previous data showed that other conventional AAV serotypes do not target OHCs with high efficiency when delivered via the round window membrane which has limited recovery of auditory function (Askew et al. 2015). Along with IHCs and OHCs, Anc80L65 also transduced vestibular sensory organs, including type I and type II hair cells of the utricle and semicircular canals, raising the possibility for treatment of vestibular dysfunction as well. Another advantage of Anc80L65 for use in the treatment of hereditary hearing loss is its safety. In mouse, there were no detrimental effects on sensory cell function, ABRs, and DPOAEs or vestibular function. These advantages make Anc80L65 an attractive vector for inner ear gene delivery, not only for IHCs and OHCs, but also other inner ear cell types (Suzuki et al. 2017; Landegger et al. 2017).
Further investigation will be required to validate Anc80L65 as a safe and effective vector for larger animal models that provides selective expression in targeted tissues of the inner ear without unwanted biodistribution. Nonetheless, it is clear that Anc80L65 and other synthetic AAV serotypes may be useful alternatives for inner ear gene therapy.
Exo-AAV (Exosome-Associated AAV)
In a recent paper, György et al. (2017) described a new way to increase transduction efficiency of conventional AAVs by associating the virus with exosomes (exosome-associated AAV or exo-AAV). Exosomes are cell-secreted membrane vesicles of nanometer size that are believed to be an important component of intercellular communication (Colombo et al. 2014).
Delivery of exo-AAV-GFP in cochlear explant cultures or into mouse inner ears by RWM or cochleostomy injection showed significant transduction efficiency in cochlear hair cells: up to 65 % for IHCs and 50 % for OHCs in vitro and more than 95 % for IHCs and around 50 % for OHCs in vivo. GFP-positive hair cells were also evident in the utricle and the semicircular canals of the vestibular system. Exo-AAV1 gene therapy partially rescued hearing and improved balance-related abnormal movement in a mouse model of hereditary deafness (György et al. 2017).
While the exosomes are potent carriers of AAVs for delivery to inner ear cells, concerns associated with possible side effects of their usage should be taken into account. Exosomes are known to carry multiple proteins and nucleic acids, including RNAs. The repertoire of biologically active contents of exosomes has not been fully characterized but may include molecules important for intercellular communication that can influence target cell function, transcriptome alteration, and abnormal activation of signaling pathways and possibly subsequent activation of pathological states, including cancer (Bakhshandeh et al. 2017; Fevrier 2004; Guo et al. 2015; Melo 2014). For this reason, producing exo-AAV in immortalized or cancer cell lines may be suboptimal. Alternatively, packaging of AAVs into artificial exosomes could be an option for production of safe exo-AAVs (De La Peña et al. 2009; Kooijmans, 2012).
Nonviral Delivery
Several nonviral delivery vectors that have been implemented for gene therapy of hearing disorders include cationic liposomes, dendrimers, and nonliposomic polymers. These synthetic compounds can be produced in nonbiological environments and are biologically safe and nonimmunogenic. Although nonviral delivery methods are not widely used, for some specific applications, they provide a suitable alternative and may be superior to viral vectors.
Cationic bilayer liposomes (CLs) are routinely used for effective delivery of negatively charged DNA and RNA into cells. CLs provide reliable protection of cargo from degradation and neutralization by extracellular enzymes and antibodies. In addition, due to their positive charge which opposes negatively charged lipids of cell plasma membrane, CLs effectively initiate endocytosis and facilitate escape of their contents from degradative endosomes. Many cationic lipid formulations have been developed to enhance carrying capacity and transfection efficacy of single- and double-stranded nucleic acids of different sizes with low accompanying cytotoxicity. However, a common lipophilic agent Lipofectamine 2000 affects inner hair cell viability in in vitro conditions (Ren et al. 2010). Due to the cytotoxicity, use of lipofection to deliver nucleic acids to hair cells in vivo is rare (Li et al. 2006). However, in one example of cell-gene therapy, lipofection was used with NIH3T3 cells to express recombinant brain-derived neurotrophic factor (BDNF), and the cells were transplanted into the mouse inner ear. This resulted in survival of the grafted cells in the cochlea for up to 4 weeks with sustainable production of BDNF, which protected hair cells and spiral ganglion neuron in stressed conditions (Okano et al. 2006).
Apart from DNA delivery, two CL formulations, RNAiMAX and Lipofectamine 2000, were recently used to deliver genome-editing proteins Cre recombinase and Cas9:sgRNA complexes into OHCs in vivo (Zuris et al. 2014). Both Cre recombinase and Cas9 do not have determined negative charge, so they were tagged with negatively charged protein domains to achieve stable association with CL. Lipofectamine 2000 was the most efficient and resulted in 90 % Cre-mediated recombination and 20 % Cas9-mediated genome modification in the OHCs, although some cell loss was observed. It is of note that since Cas9 can repeatedly edit the same genomic region and may have off target effects, it may be less desirable to have constitutive long-term expression. This caveat suggests that delivery of gene editing proteins may be a niche for which protein delivery by nonviral vectors is more attractive than constitutive expression of DNA sequences encoding these proteins.
Several additional nonviral gene delivery vectors of different compositions, such as dendrimers and polyamine nonliposomal transfection agents, have been tested for their ability to carry DNA into inner ear cells. Dendrimers are nanocomposite materials with branched structure formed by monomeric subunits diverging from a central nucleus. Combinations with different nuclei, attached monomers and surface chemical groups may allow dendrimers to have a range of properties suitable for binding nucleic acids of different structures and sizes (Shcharbin et al. 2009). Round window membrane injection of DNA in complexes with dendrimers Superfect or hyperbranched polylysine nanoparticles resulted in transfection of organ of Corti cells.
Polyamine nonliposomal transfection agents are based on complex formation between nucleic acids, nontoxic cell proteins (e.g., histones), and small amounts of polyamine polymers (Budker et al. 1997). One such reagent, Mirus TransIT, delivered transgenes into Rosenthal’s canal and in the area of IHCs. At the same time, combination of nonliposomal agents and DNA condensing component Effectene was not effective. However, all the agents provided transgene expression in the spiral ganglia (Praetorius et al. 2008). The potential of linear polyethylenimine nanoparticles (Zhou et al. 2015) as well as poly(lactic-co-glycolic) acid nanoparticles (Tamura et al. 2005) was also tested but demonstrated cytotoxicity for several types of inner ear cells.
In addition to carrier-mediated gene delivery, electrical field-mediated DNA transfer (electroporation) is gaining popularity. Upon electroporation, pulses of high-voltage electric field generated by an electrode inserted into the cochlea can temporarily break cell membranes and allow the formation of pores that permit extracellular DNA to enter cells. Electroporation is advantageous for its high efficacy and absence of immune response. The main disadvantages are possible tissue damage caused by electrode placement within the cochlea or inappropriate parameters for electrical stimulation. In recent years, in vivo electroporation was used in several studies to induce transgene expression in inner ear cells. In particular, electroporation was effective for ectopic expression of transcriptional factors Neurog1, NeuroD1, and Sox2 to induce a neuronal fate in nonsensory regions of the cochlea during the early postnatal period (Puligilla et al. 2010). Cochlear implant electrode arrays have also been used for close-field electroporation to deliver naked DNA encoding BDNF into mesenchymal cells lining the cochlear perilymphatic canals of the guinea pig to stimulate regeneration of atrophied spiral ganglion neurites (Pinyon et al. 2014).
TARGETS
Genetic hearing loss arises from mutations that affect the function of many different inner ear cell types. A thorough understanding of the biology of each cell type is required for the development of appropriate therapeutic interventions that target the various cell types. In addition to understanding cellular function and a given cell type’s contributions to auditory function, a detailed understanding of gene expression is required. For example, an understanding of the precise spatial and temporal pattern of expression of a wild-type gene is needed to define therapeutic windows of opportunity for intervention. In addition to vectors that transduce the targeted cell type, promoters that drive expression at the appropriate level are also needed. In the following sections, we discuss several inner ear cell types and their ability to be targeted by emerging gene therapy approaches.
Hair Cells
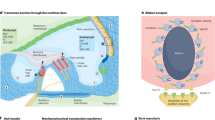
Sensory hair cells, more often than the rest of the inner ear, have been a focus of gene therapy studies. This is not surprising, given that more than 50 % of gene mutations that cause hereditary deafness are expressed in hair cells (Walsh et al. 2002; Wang et al. 1998; Kurima et al. 2002a, b; Yasunaga et al. 1999; Naz et al. 2004). The most fragile and most commonly affected part of the hair cell is the hair bundle—a structure that plays a pivotal role in the auditory mechanoelectrical transduction process. The hair bundle is located at the apex of hair cells and consists of tufts of modified microvilli, known as stereocilia, which are organized in a staircase pattern and maintained by different types of links (such as the tip links and top connectors). Near the tips of the stereocilia, mechanotransduction channels are localized (Beurg et al. 2009), which convert mechanical signals into electrochemical activity. Numerous mutations in genes encoding components of the mechanotransduction machinery in hair bundles disrupt the process of converting mechanical information into electrical signals. For a number of animal models with mutations in genes involved in mechanotransduction, successful therapy by viral and nonviral methods has been described. Among those are genes of transmembrane channel-like proteins 1 and 2 (TMC1 and TMC2; Askew et al. 2015), whirlin (Chien et al. 2016; Isgrig et al. 2017), harmonin (Pan et al. 2017), and TMHS (György et al. 2017). Because hair cells are nondividing, postmitotic cells arranged in a compact epithelial layer, they are challenging targets for gene delivery. However, recent progress has revealed a number of suitable vectors that target both inner and outer hair cells along the length of the cochlea at both neonatal and mature stages. As such, vectors are no longer a limiting factor and there are multiple options for the design of gene therapeutic interventions. Rather, the biological limitations, including control of proper expression level and delivery during therapeutically amenable windows, are important challenges that remain and may be unique to each form of genetic deafness. For example, genes expressed early in development or those required for morphogenesis of the hair bundle may have a limited window of opportunity (Frolenkov et al. 2004). Likewise, gene mutations that lead to death of hair cells will limit the potential for therapeutic intervention.
Stria Vascularis
The stria vascularis (SV) is a highly vascularized epithelium which lines the lateral wall of the scala media. It is composed of three cell types: marginal, intermediate, and basal cells, together with capillary endothelial cells (Wangemann and Schacht 1996). It is responsible for the production and maintenance of both the high endolymphatic K+ concentration for the scala media and the endocochlear potential (EP), +80–120 mV relative to perilymph, both essential for normal inner ear function. The SV plays a critical role in sound detection by hair cells: as a result of hair bundle deflections, K+ in the endolymph enters hair cells to depolarize the cells. Eventually, the K+ exits passively through potassium channels localized along the hair cell basolateral membrane and then is transported into supporting cells and fibrocytes via a network of gap junction channels. From there, K+ moves to the stria vascularis, which completes the loop by secreting K+ back into the endolymph (Steel and Barkway, 1989). The process allows for potassium homeostasis in the endolymph and shifts the energy burden from hair cells to the marginal cells of the stria vascularis.
It is not surprising that defects in genes that mediate the K+ recycling in the inner ear cause inherited deafness. For example, autosomal recessive Jervell and Lange-Nielsen syndrome is caused by mutations in KCNQ1 and KCNE1 genes which are expressed in marginal cells of the SV (Lee et al. 2000). The proteins produced by these two genes work together to form potassium channels that transport potassium ions out of the SV and into the potassium-rich endolymph and help generate the endocochlear potential.
Chang et al. (2015) injected AAV1 gene therapy vectors into the scala media of a mouse model of JLN syndrome, in which the mice were deficient in KCNQ1. Using exogenous expression of KCNQ1, they demonstrated successful gene therapy treatment and partial recovery of ABR thresholds. The study was a first for a gene specifically affecting the function of the stria vascularis, suggesting that this inner ear structure may be amenable to clinical intervention in humans (Chang et al. 2015).
Supporting Cells
The development, function, and maintenance of inner ear sensory epithelia are heavily dependent upon supporting cells, which are nonsensory cells that reside between and around hair cells (Wan et al. 2013). The supporting cells provide mechanical support to the epithelium and the hair cells. They also help to maintain an environment in the epithelium that enables hair cells to function. Supporting cells are coupled to each other by large numbers of gap junctions (Souter, 2008; Kikuchi et al. 1995). The apical surfaces of supporting cells contact the endolymph (located in the scala media), and its basolateral surfaces contact with perilymph—Na+-rich fluid located in the scala vestibuli and the scala tympani that bathes the basolateral surface of hair cells and supporting cells. Due to their negative resting potential, supporting cells can absorb Na+ from the scala media through apical sodium channels (ENaCs), effectively reducing endolymphic Na+ concentration, which is important for the generation and maintenance of the endocochlear potential. Supporting cells also help to remove K+ ions from the intercellular spaces of the sensory epithelium after it exits hair cells and thereby maintain the low K+ environment around the hair cell body, which is necessary for transduction and auditory sensitivity. As discussed above, the gap junction network in supporting cells provides a means to shuttle the K+ ions away from hair cells preventing local accumulation, depolarization, and death (Gulley and Reese 1976; Wangemann 2006).
Gap junctions are clusters of intercellular channels that allow direct diffusion of ions and small molecules between adjacent cells (Goodenough and Paul 2009). The protein subunits that form gap junction channels are members of the connexin protein family (Souter, 2008). Connexin 26 (Cx26) and Connexin 30 (Cx30) are the primary members of the connexin family that are expressed in the inner ear. Cx26 mutations are among the most common causes of genetic hearing loss in humans (Denoyelle et al. 1997; Kelsell et al. 1997; Estivill et al. 1998). Mutations in Gjb2 which encodes Cx26 can cause autosomal-dominant or autosomal-recessive deafness. Recently, it was shown that hearing loss in Gjb2 knockout mice might be restored by delivering normal copies of the Gjb2 into the inner ear using AAV delivering approach. Yu et al. (2014) managed to reestablish intercellular gap junction networks in mutant mouse cochleas by Gjb2 gene therapy.
Spiral Ganglion Neurons
The auditory afferent neurons, also known as spiral ganglion neurons (SGNs), receive synaptic input from auditory hair cells, encode the information into trains of action potentials, and transmit auditory information via the eighth cranial nerve to the cochlear nucleus in the brainstem. Two populations of SGNs have been described. Type I SGNs account about 90 % of the entire population and innervate exclusively IHCs. Type II SGNs account for 5–10 % of the population and innervate numerous OHCs (Spoendlin, 1981; Ota and Kimora 1980). For normal function, SGNs require both neurotrophin 3 (NT-3) and BDNF (Ernfors et al. 1995; Pirvola et al. 1997). BDNF and NT-3 are expressed by the sensory hair cells in the developing organ of Corti and by supporting cells. Loss of SGNs can occur directly through damage to the afferent synapses/spiral ganglion cell bodies or as a secondary consequence through lack of adequate trophic support.
In the case when death of neurons occurs because of the lack of key trophic factors, these factors can be delivered into inner ear to promote SGN survival. Hence, several animal studies showed that in vivo direct delivery (viral and nonviral) of both BDNF and NT-3 into the cochlea (scala tympani and supporting cells) rescued auditory neurons from target deprivation-induced neuronal death (Lalwani et al. 2002; Miller et al. 1997). Moreover, not only neurotrophic factors expressed in the inner ear may protect SGN’s survival. Delivery of glial cell-derived neurotrophic factor (GDNF) into the inner ear of guinea pigs (ototoxic model) promoted the survival of SGNs (Kuang et al. 1999; Yagi et al. 2000; Kanzaki et al. 2002).
Preserving SGNs from degeneration is of paramount importance for cochlear implant patients, since the implants require SGNs to convey the auditory information from the periphery to brainstem nuclei (Pfingst et al. 2011). Furthermore, the use of growth factors that promote survival, protection, or regrowth of IHC-SGN synapses may be of benefit for patients with hidden hearing loss due to synaptopathy (Kobel et al. 2016; Hickox et al. 2017; Liberman and Kujawa 2017).
GENE THERAPY STRATEGIES
Gene Replacement
The basic principle of gene replacement is to supplement a nonfunctional mutant gene by delivering the correct coding sequence of functional cDNA for a gene of interest into targeted cell types. Ideal candidates for this form of gene therapy are inherited disorders caused by loss-of-functional mutations, such as recessive diseases. The absence of factors that would negatively affect a delivered cDNA in the target cell is critical for gene replacement, for example dominant-negative proteins, or even protein fragments that might outcompete the wild-type therapeutic sequence. The time of transgene delivery is also critical. If gene expression begins during prenatal development, the absence of a functional gene may lead to significant developmental consequences which may not be recoverable by delivery of the functional sequence at later stages. In other words, for gene replacement to be successful, there must be a viable clinical window available for intervention. In addition to an available window, if the target gene is expressed into adulthood, expression of the exogenous sequence must be maintained for extended periods of time, perhaps a lifetime. Alternatively, although not yet attempted, it may be possible to reintroduce the therapeutic sequence at multiple time points. Gene replacement strategies are less likely to be successful for overcoming the influences of dominant mutations. For these forms of genetic deafness, alternate approaches may be better suited.
Gene Silencing
Gene silencing is a form of gene regulation that may be used to “switch off” expression of a mutant gene. The approach may be useful for dominant mutations in heterozygous animals. In this scenario, silencing of the mutant allele while allowing expression of the wild-type allele may be sufficient to overcome the consequences of the dominant hearing loss. Gene silencing can be accomplished at either the genomic, transcriptional, or post-transcriptional levels.
At the transcriptional level, it may be achieved by inducing modification in histone protein or by changing the environment for binding of transcriptional complexes such as RNA polymerase, transcription factors, etc. In such cases, mRNA is not transcribed. The most popular tools to implement this approach are catalytically inactive Cas9 endonuclease and engineered zinc finger transcription factors, as well as their fusion complexes with effector enzymes that introduce required epigenetic modifications. The simplest example is the gRNA-dCas9 DNA recognition complex that spatially interferes with transcriptional elongation, RNA polymerase binding, or transcription factor binding (Qi et al. 2013). Fusion complexes of KRAB (Kruppel-associated box repressor) with dCas9 and zinc finger domains draw a diverse group of histone modifiers that cooperate to form heterochromatin. Targeting of the dCas9-KRAB and ZF-KRAB to distal regulatory elements effectively represses them and leads to silencing of the controlled genes (Thakore et al. 2015; Chen et al. 2014b, Luo et al. 2017). A complex of dCas9 with methyltransferase 3A efficiently methylates promoter regions of targeted genes, decreasing their transcription (McDonald et al. 2016). Zinc finger domains fused to the catalytic domain of histone methyltransferases affect local methylation of H3K9 and consequently repress gene expression (Snowden et al. 2002).
For post-transcriptional gene silencing, turning off gene expression occurs at the mRNA transcript level by RNA interference (RNAi) (Zamore et al. 2000; Elbashir et al. 2001). Gene silencing by RNAi was first described in 1998 by Andrew Fire and Craig Mello (Fire et al. 1998). Since that time, this endogenous mechanism has found wide application in biology, including gene therapy. Conceptually, RNA interference is a natural biological process in which noncoding RNA molecules regulate gene expression by preventing mRNA translation. The central role in RNAi is played by two types of short complementary small RNA—microRNA (miRNA) or small interfering RNA (siRNA). These two small RNAs can be designed to bind specific mRNA sequences and either increase or decrease their activity, for example by preventing an mRNA from producing a protein.
A main advantage of this method is that RNAi is a natural target-specific method for gene regulation. Its sequence specificity makes it particularly adept for silencing dominant mutations (Maeda et al. 2005; Shibata et al. 2016), without affecting wild-type sequences or off target sequences.
Gene Editing
Precise direct manipulation of genome sequences in the inner ear (Zou et al. 2015) is becoming an emerging reality with the technologies of gene editing. The main instruments of gene editing are engineered nuclease-based enzymes able to find a target genome sequence and to introduce single- or double-strand DNA, which stimulate innate DNA repairing machinery. The repair proceeds via two major pathways—nonhomologous end-joining (NHEJ) or homology-directed repair (HDR). NHEJ results in nonspecific breakage healing with stochastic small insertions or deletions (indels), which may lead to shifts of reading frame, premature stop sequences, or truncated proteins. HDR relies on the presence of homologous template (like sister chromatid or exogeneously supplied template) and leads to specific repair of DNA breakage. This pathway can be exploited to precisely manipulate a native gene sequence. Moreover, recent technologies allow directed mutagenesis without DNA breakage (Komor et al. 2016).
There are several main instruments for gene editing developed up to date. Zinc finger nucleases represent chimeric nucleases constructed from FokI restriction endonuclease cleavage domain and tandems of DNA-binding domains from zinc finger transcriptional factors, mainly murine transcriptional factor Zif268 (Klug 2010). TALENs are derivatives from the TALE proteins from Xantomonas proteobaceria that have conservative amino acid repeats binding a single DNA base pair (Chen and Gao 2013). Meganucleases are re-engineered homing endonucleases of the LAGLIDADG family able to target custom sequences (Silva et al. 2011). Chimeric proteins containing domains from zinc fingers, TALENs, and meganucleases are designed to take advantage of all these tools to enhance binding affinity and cleavage specificity (Kleinstiver et al. 2012; Wolfs et al. 2014). Nucleases from CRISPR/Cas and /Cpf1 endonucleases were adapted from prokaryotic immune systems for resistance to phages and plasmids. It is considered as the most pervasive and easy-to-use system with multiple applications. Cas9 and Cpf1 can be programmed to find a desired DNA target by a single guide RNA molecule which is partially complementary to the target sequence. Both Cpf1 and Cas9 require the presence of a protospacer adjacent motif (PAM) immediately following the DNA target sequence, although the PAM sequences matching Cpf1 and Cas9 are different. To date, much effort has been directed toward the design of CRIPSR nucleases with altered PAM specificities and diminished off target activities allowing CRISPR/Cas approaches with even more precision (Kleinstiver et al. 2015a, b; Hirano et al. 2016).
PRELIMINARY RESULTS WITH GENETIC HEARING LOSS
Genetic forms of sensorineural hearing loss account for almost half of all cases of congenital deafness with more than 300 genetic loci implicated [hereditaryhearingloss.org]. HL due to genetic factors can have different modes of inheritance and may be delayed in onset with different progression rates. Many “deafness” genes have key roles in inner ear development, structure, or function, and some of them may be suitable targets for gene therapy. Although a number of deafness genes will likely not be good targets for gene therapy, given their temporal expression pattern, requirement for cell survival, or key role during a critical window of development, several have already been the target of proof-of-principle studies in animal models. These cutting-edge gene therapy studies have targeted Vglut3, Tmc1, Whirlin, Ush1-c, Clarin-1, and GJB2 and will be reviewed in the following sections.
Vglut3 (SLC17A8)
Mutations in Vglut3 (SLC17A8) causing autosomal dominant hearing loss occur in approximately 1.6 % of hearing loss patients in a diverse representative sample (Sloan-Heggen et al. 2016). AAV-mediated delivery of the Vglut3 cDNA into Vglut3 knock-out (KO) mouse cochleas was the first example of successful gene therapy in mammalian hair cells (Akil et al. 2012). Vglut3 encodes a vesicular glutamate transporter (VGLUT3) which is essential for transporting the neurotransmitter glutamate into synaptic vesicles. In humans, missense mutations in this gene lead to progressive high-frequency autosomal dominant hearing loss (DFNA25) (Ruel et al. 2008). Vglut3-KO mice are profoundly deaf with early degeneration of some cochlear ganglion neurons (Seal et al. 2008). Hearing loss in these rodents is a consequence of lack of glutamate release by inner hair cells, which results in the absence of auditory neuron depolarization in response to sound. Akil et al. (2012) used AAV2/1 to deliver Vglut3 cDNA into neonatal (postnatal days 1 to 12) KO mouse cochleas. Based on data from auditory brainstem response (ABR) and acoustic startle reflexes, they demonstrated that auditory function in injected ears recovered within 2 weeks. It should be noted that in spite of using vectors that contain a promoter with broad activity, chicken b-actin, the Vglut3 transgene was only expressed in the targeted auditory inner hair cells. Partial recovery of the morphological features of inner hair cell-associated afferent ribbon synapses was also reported in this study (Akil et al. 2012). The data revealed the potential for gene therapy to reconstitute molecular and cellular components required for the normal function of the auditory system. The success of Vglut3 gene therapy motivated the search for other gene therapy targets for more common forms of genetic deafness.
Tmc1
TMC1 encodes the TMC1 protein, a component of the hair bundle transduction machinery (Kawashima et al. 2011; Pan et al. 2013) which is located at the apical pole of hair cells (Kurima et al. 2015). Mutations in TMC1 causing hereditary hearing loss occur in approximately 2.3 % of all hearing loss patients in a diverse representative sample, including autosomal dominant (3.2 %) and autosomal recessive (2.2 %) forms (Sloan-Heggen et al. 2016) Mutations in human TMC1 can account for 4 to 8 % of genetic deafness in some populations (Kalay et al. 2005; Santos et al. 2005; Kitajiri et al. 2007; Hilgert et al. 2008; Sırmacı et al. 2009). It has been implicated in the pathogenesis of both dominant and recessive nonsyndromic HL, DFNA36 and DFNB7/11, respectively (Kurima et al. 2002a, b).
In a recent report, Askew et al. (2015) described AAV2/1-mediated gene transfer of Tmc1 coding sequence into the mouse inner ear. Two strains of Tmc1 mutant mice were used: Tmc1-KO mice which served as a recessive loss-of-function model for human DFNB7/11, and Beethoven (Bth) mice, a model of human DFNA36, which carry a dominant point mutation. Both animal models show several physiological deficits in hair cell mechanosensory transduction. In vivo injection of the AAV2/1-Tmc1 vectors at P0–P2 restored mechanotransduction in inner hair cells but not outer hair cells of Tmc1-KO mice. In spite of the fact that AAV2/1 was capable of driving exogenous gene expression in vitro, OHCs were not efficiently targeted in vivo. The small number of OHCs transduced was not sufficient to restore distortion product otoacoustic emissions (DPOAEs). However, because IHCs were transduced with 80–90 % efficiency, there was recovery of ABR responses with elevated thresholds. Furthermore, the recovery was sufficient to drive acoustic startle responses, a behavioral measure of auditory function. In addition, AAV2/1-Tmc2 vectors were capable of preserving auditory function and IHCs survival in Bth mice, which supports the hypothesis that Tmc1 and Tmc2 perform somewhat redundant functions and can partially substitute for each other, at least in IHCs. However, Bth mice AAV2/1-Tmc2 vectors did not recover startle responses, suggesting that alternate approaches for overcoming the influence of dominant mutations may be required.
Using an alternate approach, Shibata et al. (2016) developed artificial miRNA designed to inhibit the progressive hearing loss phenotype in Bth mice. The mouse carries the semidominant Tmc1 c.1235T>A (p.M412 K) allele, which is orthologous to a human TMC1 mutation, c.1253T>A (p.M418 K) (Zhao et al. 2014). Moreover, natural progression of hearing loss in humans with p.M418K closely mimics the phenotype of the Bth-heterozygous mice. Artificial miRNA carried in an AAV vector was delivered into inner ears of Bth mice by single intracochlear injection at early developmental stages. Treated mice showed improved hair cell survival compared to untreated mice. This procedure was able to delay the onset of hearing loss for up to 35 weeks. This was substantially longer than the recovery of function demonstrated by Vglut3 gene delivery in postnatal mice, which deteriorates by 7 weeks (Akil et al. 2012), as well as in utero injection of rAAV2/1-MsrB3 to rescue MsrB3−/− mice which deteriorates by 4 weeks (Kim et al. 2016).
WHRN (Whirlin)
WHRN is one of the causative genes of Usher syndrome type 2, the most common clinical type of the disease, characterized by congenital moderate to severe hearing loss (Yang et al. 2012). Mutations in WHRN causing autosomal recessive hearing loss occur in approximately 0.5 % of hearing loss patients in a diverse representative sample (Sloan-Heggen et al. 2016). The gene is expressed in hair cells and encodes the cytoskeletal PDZ-scaffold protein whirlin. Two major isoforms of whirlin (long and short) have distinct locations in stereocilia and also across hair cell types. Lack of both isoforms causes abnormally short stereocilia and profound deafness (Ebrahim et al. 2016). Whirlin appears to operate as a quaternary complex with other Usher 2 proteins, USH2A, GPR98, and PDZD7, and is localized at the ankle link region of the hair bundle (Chen et al. 2014a). Abnormal expression or mutations of any of these genes leads to disorganization and gradual degeneration of hair bundles in mice (Chen et al. 2014a). In addition, interaction of whirlin with myosin-XVa is critical for proper whirlin delivery to the tips of stereocilia (Belyantseva et al. 2005).
Recovery of hearing with gene therapy for whirlin mutations has thus far met with mixed success. Whirlin−/− mice treated with AAV8-Whirlin cDNA recovered stereocilia morphology but did not recover normal auditory function, perhaps due to insufficient length and number of regenerated stereocilia, low virus infectivity rate, or isolated isoform delivery (Chien et al. 2016). However, in a follow-up study, some recovery of hearing and balance function was demonstrated following AAV8-whirlin injection via the posterior semicircular canal (Isgrig et al. 2017). Higher transduction efficiency or delivery of both whirlin isoforms may further enhance recovery.
Ush1c (Harmonin)
Type I Usher syndrome is characterized by congenital onset deafness, vestibular dysfunction, and retinitis pigmentosa. The severity of the phenotype depends on the specific Ush1c mutation, which includes point mutations, expansions of exons, and multiexon continuous deletions (Ouyang et al. 2005; Lentz et al. 2013; Bitner-Glindzicz et al. 2000; Verpy et al. 2000). Mutations in Ush1c causing autosomal recessive hearing loss occur in approximately 0.3 % of hearing loss patients in a diverse representative sample (Sloan-Heggen et al. 2016). In the mouse inner ear, Ush1c was found to be expressed in hair cells only. The Ush1c product, harmonin, is a multi-PDZ domain-containing protein synthesized in three isoforms. All the isoforms contain an N-terminal helical domain, two PDZ domains, and a coiled-coil domain. Harmonin-b is localized in stereocilia and forms a stable tertiary complex with unconventional myosin VIIa through the adaptor protein Sans. Besides, harmonin-b also interacts with cadherin-23 and protcadherin-15 (Yan et al. 2010). These Ush 1 proteins form a scaffold which facilitates development and maintenance of stereocilia into highly organized hair bundles. Therefore, mutations affecting structure or production of harmonin lead to hair bundle disorganization resulting in deafness (Boëda et al. 2002). Harmonin-a is localized at hair cell synapses where it is thought to be required for calcium channel localization and proper synaptic transmission (Gregory et al. 2011, 2013).
For one of the Ush1c mutations, c.216G>A which leads to formation of defective mRNA, correction of splicing was achieved by using antisense oligonucleotides (Lentz et al. 2013). Due to their high sequence specificity, antisense oligonucleotides have tremendous therapeutic potential for gene expression modification related to alternative splicing defects (Kole and Sazani 2001). Delivery requires efficient localization to pre-mRNA splicing sites in cell nuclei and overcoming cellular barriers, including the vascular endothelial barrier, reticuloendothelial system, renal clearance system, and blood-brain barrier (Juliano 2016). Traversing the blood-brain barrier using systemic administration has been met with limited success; however, antisense oligonucleotides can reach the inner ear via the reticuloendothelial system and vasculoendothelial system using chemical modifications and cellular processes. The addition of phosphorothioate backbones, neutral backbones, 2′ modifications, neutral siRNAs, and polymer RNAs to antisense oligonucleotides increases their cellular affinity in endocytosis (Juliano 2016). Most of the currently used modified antisense oligonucleotides in clinical trials contain 2′-deoxynucleotide-phosphorothioate backbones (Tamm et al. 2001). The primary mechanism of cellular uptake and transport of antisense oligonucleotides is via accumulation in late endosomes and multivesicular bodies (Juliano 2016). The transport to the cell nuclei is then regulated by the multiprotein complex ESCRT, recently found to be critical to antisense oligonucleotide effectiveness in disease therapy (Henne et al. 2011; Wagenaar et al. 2015). Through this mechanism, antisense oligonucleotides can modify gene expression in the inner ear. In a mouse model, a single intraperitoneal injection of antisense oligonucleotide (300 mg per kg body weight) increased correct harmonin expression and improved hair cell stereocilia organization, low-frequency hearing (8–16 kHz), and vestibular function. It is of note that improvements in vestibular function were observed for mice injected with antisense oligonucleotide on up to postembryonic day 13 (P13), whereas injection on P16 did not bring any improvement suggesting the window of clinical intervention may be limited. However, the therapeutic effects were sustained for 3 months following treatment, indicating good potential of this approach (Lentz et al. 2013). The study suggests that delivery of antisense oligonucleotides may suppress the consequences of specific point mutations and may be a viable approach for treatment of some forms of genetic hearing loss.
How antisense oligonucleotides introduced via IP injection make their way across the blood-labyrinthine barrier and into the targeted cell type has not been clarified.
In a recent study, gene replacement was successfully implemented to correct the pathological phenotype due to the c.216G>A mutation in the same mouse model of Ush1c discussed above (Pan et al. 2017). Two splice forms of harmonin (harmonin-a1 and harmonin-b1), under the control of CMV promoter, were delivered with AAV2/Anc80 vector into the inner ear via the RWM at early postnatal stages. To demonstrate successful targeting of the proteins in vitro and in vivo after the RWM injection, AAV vectors bearing fluorescently tagged harmonins were used. It was shown that the tagged harmonin-b was correctly localized to the distal end of stereocilia near the tip-link insertions (Boëda et al. 2002; Lefèvre et al. 2008; Grillet et al. 2009) and the tagged harmonin-a localized to the synapse, which was consistent with the previous localization work (Gregory et al. 2011, 2013).
RWM injection of AAV2/Anc80.CMV.harmonin-b at P0–P1 restored mechanotransduction as well as auditory and vestibular function in deaf and dizzy mice to unprecedented levels. ABR thresholds were as low as 25 dB in some Ush1c injected mice. The rescue was associated with recovery of mRNA expression encoding for wild-type harmonin. Scanning electron microscopy revealed decreased hair cell loss and preserved hair bundles with normal staircase morphology in rescued ears (Pan et al. 2017).
ABR and DPOAE thresholds in mice co-injected with harmonin-a1 and harmonin-b1 vectors were similar to those injected with harmonin-b1 alone. Thus, co-injection did not provide further improvement, suggesting that harmonin-a1 may be dispensable for auditory function. Along with rescue of audiological function, the use of viral-mediated gene delivery restored vestibular function: mice injected with AAV2/Anc80.CMV.harmonin-b1 and those co-injected with harmonin-a1 and harmonin -b1 vectors maintained balance function consistent with control mice (Pan et al. 2017).
USH3A (Clarin-1)
Mutations in USH3A are the only currently identified cause of Usher syndrome type 3, which usually has postlingual onset (mostly during the first decade) and is primarily characterized by progressive deafness, blindness, as well as variable vestibular balance disorder (Adato et al. 2002). The USH3A gene is expressed in the organ of Corti and spiral ganglion cells of the mouse ear and encodes the protein clarin-1, a transmembrane protein of the tetraspanin superfamily. Clarin-1 is an essential hair bundle protein and acts as a modulator of mechanotransduction activity and presynaptic ribbon assembly (Ogun and Zallocchi 2014; Geng et al. 2012; Gopal et al. 2015). Clarin-1 is apparently indispensable for survival of hair cells, since ribozyme-mediated decay of clarin-1 mRNA leads to pronounced apoptosis of the cells in mouse inner ear. Although the mechanism is unclear, clarin-1 deficiency-mediated hair cell death is one of the factors that causes the clinical manifestations of Usher syndrome, type 3 (Aarnisalo et al. 2007).
Ush3A gene therapy has not been reported but remains an attractive target, due to its later onset and short sequence. However, an alternate approach was recently reported to mitigate progression of Usher syndrome, type 3 invoked by the N48K mutation in clarin-1. This mutation leads to elimination of an N-linked glycosylation site and failure in clarin-1 plasma membrane expression by preventing its engagement with the endoplasmic reticulum calnexin folding cycle and/or imposing a chaperone-independent energetic folding defect of clarin-1 (Lukacs et al. 2016). High-throughput screening identified a regulatory molecule called BioFocus 844 (BF844) that stabilizes clarin-1N48K and restores its plasma membrane expression. It thereby attenuates hearing loss in Ush3A mutant mice. Although the molecular targets of BF844 have not been identified, it appears to influence the proteostasis network preventing ERAD-mediated degradation of the mutated clarin-1 (Alagramam et al. 2016).
Gjb2 (Connexin26)
Mutations in the gap junction protein beta 2 (GJB2) gene are among the most common causes of genetic HL worldwide. Mutations in Gjb2 causing autosomal recessive hearing loss occur in approximately 21.5 % of hearing loss patients in a diverse representative sample, including autosomal-dominant (1.6 %) and autosomal-recessive (25.3 %) forms (Sloan-Heggen et al. 2016). The GJB2 gene encodes the transmembrane protein Cx26 which together with other connexins form channels that allow rapid transport of ions or small molecules between cells. Cx26 is expressed in an inner ear network of supporting cells that connect the organ of Corti through the basilar membrane to the cells of the stria vascularis (Wangemann 2006). This network is critical for recycling potassium ions released from the hair-cell basolateral membrane back to the potassium-rich endolymph that bathes hair bundles. GJB2 is involved in both autosomal-dominant (DFNA3) and autosomal-recessive (DFNB1) forms of deafness (Richard et al. 1998; Maeda et al. 2005, 2007). Despite great interest in the GJB2 gene as a potential target for gene therapy, it has remained a difficult task because germline mutations in mouse Gjb2 result in death in utero. One solution to this problem was the creation of mouse models with inducible or cell-specific deletion of Gjb2.
The restoration of Cx26 function to recover hearing has been met with partial success (Ahmad et al. 2007). Maeda et al. (2007) used RNAi to suppress the expression of a dominant-negative mutation (p.R75W) in the Gjb2 gene. They transfected cochleas of adult mice in vivo with a plasmid vector expressing GJB2-R75W-eGFP and showed that this allele variant causes hearing loss by a dominant-negative effect. This pathological phenotype was corrected by delivering RNAi that specifically silenced GJB2-R75W expression. It was confirmed that siRNA was potent enough to modify the phenotype caused by the expression of GJB2-R75W-eGFP, decreasing the expected ABR threshold shift (Maeda et al. 2007).
Yu et al. (2014) reported that scala media delivery of AAV1 that carried the Gjb2 coding sequence could recover gap junction intercellular networks in Gjb2-deficient cochleas and promoted recovery of dye diffusion, as well as reduced cell death and reduced spiral ganglion neuron degeneration. Unfortunately, the approach did not lead to recovery of hearing function (Yu et al. 2014), perhaps due to limited viral transduction efficiency in the targeted cell type (<50 %), or an injection time point (P0–P1) that was beyond the developmental window for functional recovery. However, more recently, Iizuka et al. (2015) applied perinatal injection of the Gjb2 gene into the cochlea using AAV5. The approach demonstrated preservation of some cochlear structures and improved ABR thresholds in a conditional Gjb2 knock-out mouse model (Iizuka et al. 2015).
Gene therapy has also been applied to improving spiral ganglion function associated with Gjb2 mutations. Spiral ganglion cells in the organ of Corti in Cx26 knock-out mice degenerate during development to varying degrees from the cochlear base to the apex (Takada et al. 2014). The degeneration can be prevented by the application of BDNF to spiral ganglion neurons in Gjb2 knock-out mice. Delivery of BDNF using adenovirus, primarily in Rosenthal’s canal at the base of the cochlea, rescues neuronal function (Takada et al. 2014). This suggests that the potential for combining gene and neurotrophic factor delivery using viral and nonviral gene therapy methods could enhance hearing loss treatment and recovery.
TRANSLATION TO THE CLINIC
Genetic forms of hearing loss in humans present a challenging but unique opportunity for therapeutic intervention (Minoda et al. 2015). Human inner ear gene therapy has the potential to restore or prevent decay of auditory function. Translation of the proof-of-concept success stories from animal models, highlighted above, into gene therapies for humans with genetic deafness has not yet begun. However, in other systems, there are over 1800 clinical trials that have been either completed, are in progress, or approved involving gene therapy (Ginn et al. 2013). Although clinical trials for genetic hearing loss have not been initiated, several genes involved in hearing loss have also been implicated in other conditions, where gene therapy trials have begun. These include syndromic diseases such as Usher’s syndrome, a rare disease that presents with blindness, deafness, and balance disorder, and disorders such as cardiovascular, craniofacial development, and neurological conditions. There has been success using gene therapy in the retinal disease Leber congenital amaurosis, with sustained recovery of vision at 3 years, and in hemophilia B with initial improvement in symptoms (Ali et al. 2000; Cideciyan et al. 2009; Maguire et al. 2009; Simonelli et al. 2010; Jacobson et al. 2012, Manno et al. 2006). Lessons from gene therapy in these other systems may help inform the design and implementation of gene therapy development for genetic hearing loss. The emerging field of inner ear gene therapy would be well-advised to heed the lessons learned from other, more advanced gene therapy fields and avoid the pitfalls that have troubled other fields, as reviewed in the next sections.
Safety
The successes using gene therapy in mouse models of genetic hearing loss warrant careful consideration for translation into therapeutic strategies for treating genetic deafness in humans. Since hearing loss has a very low morbidity rate, it will be of utmost importance to adhere to a key principle of the Hippocratic oath: do no harm. Safety concerns must be given full consideration, including the safety of the injection technique, the gene delivery vector, and the exogenous gene sequence. As discussed in prior sections, the safety of various routes of delivery into the inner ear will need to be evaluated in humans. Damage to the fragile structures of the human ear will need to be minimized in order to optimize outcome. The physical volume delivered will also require consideration. Given the small volume of the inner ear fluids (~100 μL), the volume and rate of delivery of the injected bolus will need to be well-defined. Thorough pharmacology and toxicology studies for each gene therapy strategy and a range of doses will require full and careful analysis. A significant concern is that a single negative outcome could set back the field of inner ear gene therapy while still in its infancy. Safety studies will also need to be designed that consider the consequences of exogenous gene expression in target cells and off target cells. Are there deleterious effects of overexpression or knock-down in the target cell type or off target cell type? For gene silencing or gene editing approaches, are there potential safety concerns associated with off target effects? Are there long-term safety concerns of the exogenous construct in either cell type? What is the biodistribution of the gene therapy reagents? Will vectors injected into the inner ear remain locally within the targeted organ or can they escape, entering other bodily tissues? Is there a potential for leakage into the cerebral-spinal fluid or the brain? Might the vector capsid or the protein product of the therapeutic gene induce an immune response, and if so, to what extent? Some strategies may necessitate a second injection of the therapeutic vector. In this case, does repeated exposure evoke a stronger immune response? Answers to these and perhaps other unforeseen safety questions will need to be addressed for each investigational new gene therapy reagent. While some risks may be inherent in any potential gene therapy procedure, the potential risks and benefits will need to be well-defined and evaluated to enable optimal design of clinical studies, and in the long term, to allow patients and physicians to make the best well-informed decisions.
Efficacy
The majority of inner ear gene therapy studies have evaluated the approach in embryonic, neonatal, and young adult mice. Many reports have suggested that there may be a critical period for gene therapy to be effective in hearing preservation or recovery. In addition to timing of delivery, dosing of gene therapy will be extremely important, given the strong dose-dependent relationship between vector titers and transduction efficiency, and it will be crucial to evaluate and establish dose-response relationships prior to clinical translation. Percentages of target cells transduced and off target cells transduced will need to be quantified. Furthermore, mechanisms that allow for control over the level of gene expression will be valuable and perhaps necessary for some applications. One such approach for controlling expression level has recently been demonstrated using 4-OH-tamoxifen (Parker et al. 2013). To conditionally express Atoh1, Parker et al. (2013) developed a stand-alone genetic construct that takes advantage of the tamoxifen sensitivity of a mutated estrogen receptor (ER) ligand-binding domain. In this construct, Atoh1-ER-DsRed is translated to ATOH1-ER-DSRED, a fusion protein that remains isolated and inactive in the cytoplasm. With the administration of 4-hydroxy-tamoxifen, the fusion protein ATOH1-ER-DSRED translocates to the nucleus, wherein it binds to the Atoh1 enhancer, and activates downstream Atoh1 signaling pathways. The construct is reversible wherein the removal of tamoxifen leads to downregulation of Atoh1 signaling.
In addition to expression level, the longevity of expression will need to be determined. Maintaining the effects of gene therapy will be important for clinical translation and may require supplementation as is the case for neurotrophic growth factors, including BDNF, particularly in spiral ganglion neurons (Zanin et al. 2014). In this case, adequate BDNF production was ensured by the introduction of stably transfected fibroblasts which secreted BDNF and was able to maintain survival of spiral ganglion cells for at least 30 weeks. Obviously, longer-term studies will be needed.
Other Animal Models
In addition to mouse models, other mammalian model systems will be needed to advance inner ear gene therapy toward clinical application. Since the FDA encourages testing of any experimental new treatment in more than one animal model, additional model systems may be worth establishing for translational inner ear research. The guinea pig is a useful and arguably underutilized mammalian animal model. Konishi et al. (2008a, b) demonstrated early success with a guinea pig model of genetic deafness, demonstrating that AAV vectors could successfully be transferred to guinea pig cochlea (Konishi et al. 2008a, b). The guinea pig was also an important model in a combined cochlear implant/gene therapy study. In this case, neurotrophin gene therapy enhanced cochlear implant device performance by stimulating spiral ganglion neurite regeneration, which could be targeted and concentrated to mesenchymal cells using focused electric fields generated by different cochlear implant electrodes (Pinyon et al. 2014).
While many animal models lack the power of mouse genetics, they may nonetheless provide a platform for tests of safety, vector targeting, generation of the therapeutic molecule at the nucleic acid and protein levels, protein localization, and other key therapeutic goals. Moreover, with the advent of CRISPR/Cas9 approaches, the ability to generate genetic hearing loss models in other species is possible. Genetic hearing loss models in larger animals, such as pigs or nonhuman primates, which have ears that are anatomically more similar to those of humans, may provide a valuable platform for extending translational proof-of-principle studies.
Human Tissue In Vitro
Translating gene therapy for inner ear disorders from animal models to human patients in clinical trials could also be facilitated by studies of human inner ear tissue in vitro. Human vestibular epithelia harvested during surgical excision of eighth cranial nerve tumors have been used as a source of live human inner tissue (Kesser et al. 2007; Staecker et al. 2014; Talyor et al. 2015; Landegger et al. 2017). In some cases, the human tissue with living hair cells has been maintained in organotypic culture for up to 10 days. While a limited source, cultured human inner ear tissue may be a viable platform for testing gene therapy reagents in vitro. Indeed, Kesser et al. (2007) demonstrated that adenoviral vectors encoding GFP and the potassium channel gene KCNQ4 were able to target and transduce human vestibular hair cells in vitro. Furthermore, the KCNQ4 protein was translated and correctly targeted to the basolateral membrane. Landegger et al. (2017) used human vestibular tissue to test the synthetic AAV vector, Anc80L65, and reported transduction and GFP expression in large numbers of type I and type II hair cells. While efficacy studies for genetic inner ear disorders may not be possible using this experimental paradigm, the approach may be valuable for examining targeting, gene and protein expression, protein localization, cellular toxicity as well as other aspects of gene therapy reagents in live human tissue. Positive outcomes with human tissue in in vitro studies could boost confidence that the gene therapy reagent in question may have similar effects in human tissue in vivo. Human clinical trials are already underway using Ad-Atoh1 gene therapy. While gene therapy for genetic hearing loss has not yet entered clinical trials, recent success with animal models provides hope that gene therapy in the human inner ear gene may be feasible in the not too distant future.
CONCLUSIONS
These are exciting times for inner ear gene therapy. The prospects of translating the successes with animal models to the clinic seem palpable. Increased interest from government funding agencies, private foundations, industry, physicians, and patients is mounting. The coming years are likely to see additional advances. Despite the growing interest and enthusiasm for inner ear gene therapy, a measured and methodical approach is warranted. A large number of unanswered questions still need to be addressed. With over 100 different genes identified that cause genetic hearing loss, each unique cause will need to be characterized and evaluated for its potential to be the target of successful gene therapy intervention. Despite the challenges, there are reasons for optimism as the field of inner gene therapy advances toward the collective goal of developing novel and effective treatments for patients with genetic hearing loss.
References
Aarnisalo AA, Pietola L, Joensuu J, Isosomppi J, Aarnisalo P, Dinculescu A, Lewin AS, Flannery J, Hauswirth WW, Sankila EM, Jero J (2007) Anti-clarin-1 AAV-delivered ribozyme induced apoptosis in the mouse cochlea. Hear Res 230:9–16. doi:10.1016/j.heares.2007.03.004
Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, Olender T, Bonne-Tamir B, Ben-Asher E, Espinos C, Millán JM (2002) USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet 10:339–350. doi:10.1038/sj.ejhg.5200831
Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Söhl G, Willecke K, Chen P, Lin X (2007) Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc Natl Acad Sci 104:1337–1341. doi:10.1073/pnas.0606855104
Akil O, Seal RP, Burke K, Wang C, Alemi A, During M, Edwards RH, Lustig LR (2012) Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron 75:283–293. doi:10.1016/j.neuron.2012.05.019
Alagramam KN, Gopal SR, Geng R, Chen DH, Nemet I, Lee R, Tian G, Miyagi M, Malagu KF, Lock CJ, Esmieu WR (2016) A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat Chem Biol 12:444–451. doi:10.1038/nchembio.2069
Alba R, Bosch A, Chillon M (2005) Gutless adenovirus: last-generation adenovirus for gene therapy. Gene Ther 12:S18–S27. doi:10.1038/sj.gt.3302612
Ali RR, Sarra GM, Stephens C, de Alwis M, Bainbridge JW, Munro PM, Fauser S, Reichel MB, Kinnon C, Hunt DM, Bhattacharya SS (2000) Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet 25:306–310. doi:10.1038/77068
Aschauer D, Kreuz S, Rumpel S (2013) Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One 8:e76310. doi:10.1371/journal.pone.0076310
Askew C, Rochat C, Pan B, Asai Y, Ahmed H, Child E, Holt JR (2015) Tmc gene therapy restores auditory function in deaf mice. Science Translational Medicine 7:295ra108. doi:10.1126/scitranslmed.aab1996
Bakhshandeh B, Kamaleddin MA, Aalishah KA (2017) Curr Stem Cell Res Ther 12(1):31–36
Bedrosian J, Gratton M, Brigande J (2006) In vivo delivery of recombinant viruses to the fetal murine cochlea: transduction characteristics and long-term effects on auditory function. Mol Ther 14:328–335. doi:10.1016/j.ymthe.2006.04.003
Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, Friedman TB (2005) Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 7:148–156. doi:10.1038/ncb1219
Beurg M, Fettiplace R, Nam JH, Ricci AJ (2009) Localization of inner hair cell mechanotransducer channels using high-speed calcium imaging. Nature Neurosci 12:553–558. doi:10.1038/nn.2295
Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, Barnes PD, O'Brien RE, Farndon PA, Sowden J, Liu XZ, Scanlan MJ, Malcolm S, Dunne MJ, Aynsley-Green A, Glaser B (2000) A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the usher type 1C gene. Nat Genet 26(1):56–60.
Boëda B, El-Amraoui A, Bahloul A et al (2002) Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J 21:6689–6699. doi:10.1093/emboj/cdf689
Borkholder DA, Zhu X, Frisina RD (2014) Round window membrane intracochlear drug delivery enhanced by induced advection. J Control Release 174:171–176
Budker V, Hagstrom JE, Lapina O, Eifrig D, Fritz J, Wolff JA (1997) Protein/amphipathic polyamine complexes enable highly efficient transfection with minimal toxicity. BioTechniques 23(1):139, 142–7.
Calcedo R, Wilson JM (2013) Humoral immune response to AAV. Front Immunol 4:341. doi:10.3389/fimmu.2013.00341
Cao M, Khan JA, Kang BY, Mehta JL, Hermonat PL (2012) Dual AAV/IL-10 plus STAT3 anti-inflammatory gene delivery lowers atherosclerosis in LDLR KO mice, but without increased benefit. Int J Vasc Med 2012:524235. doi:10.1155/2012/524235
Castle MJ, Turunen HT, Vandenberghe LH, Wolfe JH (2016) Controlling AAV tropism in the nervous system with natural and engineered capsids. Methods Mol Biol 1382:133–149. doi:10.1007/978-1-4939-3271-9_10
Chang Q, Wang J, Li Q, Kim Y, Zhou B, Wang Y, Lin X (2015) Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol Med 7:1077–1086. doi:10.15252/emmm.201404929
Chen HH, Mack LM, Kelly R, Ontell M, Kochanek S, Clemens PR (1997) Persistence in muscle of an adenoviral vector that lacks all viral genes. Proc Natl Acad Sci U S A 94:1645–1650
Chen K, Gao C (2013) TALENs: Customizable molecular DNA scissors for genome engineering of plants. J Genet Genomics 40:271–279. doi:10.1016/j.jgg.2013.03.009
Chen Q, Zou J, Shen Z, Zhang W, Yang J (2014a) Whirlin and PDZ domain-containing 7 (PDZD7) proteins are both required to form the quaternary protein complex associated with Usher syndrome type 2. J Biol Chem 289(52):36070–36088. doi:10.1074/jbc.M114.610535
Chen Y, Whitehead EH, Guimaraes C (2014b) Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159:647–661. doi:10.1016/j.cell.2014.09.029
Chien WW, Isgrig K, Roy S, Belyantseva IA, Drummond MC, May LA, Cunningham LL (2016) Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Mol Ther 24:17–25. doi:10.1038/mt.2015.150
Chien WW, Monzack EL, McDougald DS, Cunningham LL (2015a Jan) Gene therapy for sensorineural hearing loss. Ear Hear 36(1):1–7
Chien WW, McDougald DS, Roy S et al (2015b) Cochlear gene transfer mediated by adeno-associated virus: comparison of two surgical approaches. Laryngoscope 125:2557–2564. doi:10.1038/srep02996
Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, Windsor EA, Conlon TJ, Sumaroka A, Roman AJ, Byrne BJ (2009) Vision 1 year after gene therapy for Leber’s congenital amaurosis. N Engl J Med 361:725–727. doi:10.1056/NEJMc0903652
Colella P, Trapani I, Cesi G et al (2014) Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther 21:450–456. doi:10.1038/gt.2014.8
Colombo M, Raposo G, Théry C (2014) Annu Rev Cell Dev Biol 30:255–289. doi:10.1146/annurev-cellbio-101512-122326
Cronin J, Zhang X-Y, Reiser J (2005) Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther 5:387–398. doi:10.2174/1566523054546224
Dazert S, Battaglia A, Ryan AF (1997) Transfection of neonatal rat cochlear cells in vitro with an adenovirus vector. Int J Dev Neurosci 15(4–5):595–600
De La Peña (2009) J Immunol Methods 344(2):121–132. doi:10.1016/j.jim.2009.03.011
De Silva SR, Charbel Issa P, Singh MS et al (2016) Single residue AAV capsid mutation improves transduction of photoreceptors in the Abca4(−/−) mouse and bipolar cells in the rd1 mouse and human retina ex vivo. Gene Ther 23:767–774. doi:10.1038/gt.2016.54
Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, Dodé C (1997) Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet 6:2173–2177
Dyka FM, Boye SL, Chiodo VA, Hauswirth WW, Boye SE (2014) Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum Gene Ther Methods 25:166–177. doi:10.1089/hgtb.2013.212
Ebrahim S, Ingham NJ, Lewis MA, Rogers MJ, Cui R, Kachar B, Steel KP (2016) Alternative splice forms influence functions of whirlin in mechanosensory hair cell stereocilia. Cell Rep 15:935–943. doi:10.1016/j.celrep.2016.03.081
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411(6836):494–498.
Ernfors P, Van De Water T, Loring J, Jaenisch R (1995) Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron 14:1153–1164
Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma L, Zelante L (1998) Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 351:394–398
Fevrier B (2004) Proc Natl Acad Sci U S A 101(26):9683–9688. doi:10.1073/pnas.0308413101
Fine EJ, Appleton CM, White DE, Brown MT, Deshmukh H, Kemp ML, Bao G (2015) Trans-spliced Cas9 allows cleavage of HBB and CCR5 genes in human cells using compact expression cassettes. Sci Rep 5:10777. doi:10.1038/srep10777
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis Elegans. Nature 391(6669):806–811.
Frolenkov GI, Belyantseva IA, Friedman TB, Griffith AJ (2004) Genetic insights into the morphogenesis of inner ear hair cells. Nat Rev Genet 5:489–498. doi:10.1038/nrg1377
Fukui H, Raphael Y (2013) Gene therapy for the inner ear. Hear Res 297:99–105
Geguchadze RN, Machen L, Zourelias L, Gallo PH, Passineau MJ (2012) An AAV2/5 vector enhances safety of gene transfer to the mouse salivary gland. J Dent Res 91:382–386. doi:10.1177/0022034512437373
Géléoc GS, Holt JR (2014) Sound strategies for hearing restoration. Science 344(6184):1241062. doi:10.1126/science.1241062
Geng R, Melki S, Chen DHC, Tian G, Furness DN, Oshima-Takago T, Holt JR (2012) The mechanosensory structure of the hair cell requires clarin-1, a protein encoded by Usher syndrome III causative gene. J Neurosci 32:9485–9498. doi:10.1523/JNEUROSCI.0311-12.2012
Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J (2013) Gene therapy clinical trials worldwide to 2012—an update. J Gene Med 15:65–77. doi:10.1002/jgm.2698
Goodenough DA, Paul DL (2009) Gap junctions. Cold Spring Harb Perspect Biol 1:a002576. doi:10.1101/cshperspect.a002576
Gopal SR, Chen DHC, Chou SW, Zang J, Neuhauss SC, Stepanyan R, Alagramam KN (2015) Zebrafish models for the mechanosensory hair cell dysfunction in Usher syndrome 3 reveal that clarin-1 is an essential hair bundle protein. J Neurosci 35:10188–10201. doi:10.1523/JNEUROSCI.1096-15.2015
Gregory FD, Bryan KE, Pangršič T, Calin-Jageman IE, Moser T et al (2011) Harmonin inhibits presynaptic Cav1.3 Ca2+ channels in mouse inner hair cells. Nat Neurosci 14:1109–1111. doi:10.1038/nn.2895
Gregory FD, Pangrsic T, Calin-Jageman IE, Moser T, Lee A (2013) Harmonin enhances voltage-794 dependent facilitation of Cav1.3 channels and synchronous exocytosis in mouse inner hair cells. J Physiol 591:3253–3269. doi:10.1113/jphysiol.2013.254367
Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T et al (2009) Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron 62:375–387. doi:10.1016/j.neuron.2009.04.006
Gulley RL, Reese TS (1976) Intercellular junctions in the reticular lamina of the organ of Corti. J Neurocytol 5:479–507
Guo BB, Bellingham SA, Hill AFJ (2015) Biol Chem 290(6):3455–3467. doi:10.1074/jbc.M114.605253
György B, Sage C, Indzhykulian AA, Scheffer DI, Brisson AR, Tan S, Wu X, Volak A, Mu D, Tamvakologos PI, Li Y, Fitzpatrick Z, Ericsson M, Breakefield XO, Corey DP, Maguire CA (2017) Rescue of Hearing by gene delivery to inner-ear hair cells using exosome-associated AAV. Mol Ther 25(2):379–391.
Han JJ, Mhatre AN, Wareing M et al (1999) Transgene expression in the guinea pig cochlea mediated by a lentivirus-derived gene transfer vector. Hum Gene Ther 10:1867–1873. doi:10.1089/10430349950017545
Harris JP, Ryan AF (1995) Fundamental immune mechanisms of the brain and inner ear. Otolaryngol Head Neck Surg 112(6):639–653.
Henne WM, Buchkovich NJ, Emr SD (2011) The ESCRT pathway. Dev Cell 21(1):77–91. doi:10.1016/j.devcel.2011.05.015
Hickox AE, Larsen E, Heinz MG, Shinobu L, Whitton JP (2017) Translational issues in cochlear synaptopathy. Hear Res. 2017 Jan 6. pii: S0378-5955(16)30444-0.
Hilgert N, Alasti F, Dieltjens N, Pawlik B, Wollnik B, Uyguner O, Delmaghani S, Weil D, Petit C, Danis E, Yang T (2008) Mutation analysis of TMC1 identifies four new mutations and suggests an additional deafness gene at loci DFNA36 and DFNB7/11. Clin Genet 74:223–232. doi:10.1111/j.1399-0004.2008.01053.x
Hirano S, Nishimasu H, Ishitani R, Nureki O (2016) Structural basis for the altered PAM specificities of engineered CRISPR-Cas9. Mol Cell 61:886–894. doi:10.1016/j.molcel.2016.02.018
Husseman J, Raphael Y (2009) Gene therapy in the inner ear using adenovirus vectors. Adv Otorhinolaryngol 66:37–51. doi:10.1159/000218206
Iizuka T, Kamiya K, Gotoh S et al (2015) Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum Mol Gen 24:3651–3661. doi:10.1093/hmg/ddv109
Imtiaz A, Maqsood A, Rehman AU, Morell RJ, Holt JR, Friedman TB, Naz S (2016) Recessive mutations of TMC1 associated with moderate to severe hearing loss. Neurogenetics 17(2):115–123.
Isgrig K, Shteamer JW, Belyantseva IA, Drummond MC, Fitzgerald TS, Vijayakumar S, Jones SM, Griffith AJ, Friedman TB, Cunningham LL, Chien WW (2017) Gene therapy restores balance and auditory functions in a mouse model of Usher syndrome. Mol Ther 25(3):780–791. doi:10.1016/j.ymthe.2017.01.007
Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, Conlon TJ (2012) Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 130:9–24. doi:10.1001/archophthalmol.2011.298
Jero J, Mhatre AN, Tseng CJ et al (2001) Cochlear gene delivery through an intact round window membrane in mouse. Hum Gene Ther 12:539–548. doi:10.1089/104303401300042465
Juliano RL (2016) The delivery of therapeutic oligonucleotides. Nucleic Acids Res 44(14):6518–6548. doi:10.1093/nar/gkw236
Kalay E, Karaguzel A, Caylan R, Heister A, Cremers FPM, Cremers CWRJ, Brunner HG, de Brouwer APM, Kremer H (2005) Four novel TMC1 (DFNB7/DFNB11) mutations in Turkish patients with congenital autosomal recessive nonsyndromic hearing loss. Hum Mutat 26:591–591. doi:10.1002/humu.9384
Kanzaki S, Stöver T, Kawamoto K, Prieskorn DM, Altschuler RA, Miller JM, Raphael Y (2002) Glial cell line-derived neurotrophic factor and chronic electrical stimulation prevent VIII cranial nerve degeneration following denervation. J Comp Neurol 454:350–360. doi:10.1002/cne.10480
Kawashima Y, Géléoc GS, Kurima K, Labay V, Lelli A, Asai Y, Griffith AJ (2011) Mechanotransduction in mouse inner ear hair cells requires transmembrane channel-like genes. J Clin Invest 121:4796–4809. doi:10.1172/JCI60405
Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Leigh IM (1997) Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387:80–83. doi:10.1038/387080a0
Kesser BW, Hashisaki GT, Fletcher K, Eppard H, Holt JR (2007) An in vitro model system to study gene therapy in the human inner ear. Gene Ther 14:1121–1131. doi:10.1038/sj.gt.3302980
Kikuchi T, Kimura RS, Paul DL, Adams JC (1995) Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat Embryol 191:101–118
Kilpatrick LA, Li Q, Yang J, Goddard JC, Fekete DM, Lang H (2011) Adeno-associated virus-mediated gene delivery into the scala media of the normal and deafened adult mouse ear. Gene Ther 18:569–578. doi:10.1038/gt.2010.175
Kim MA, Cho HJ, Bae SH, Lee B, Oh SK, Kwon TJ, Ryoo ZY, Kim HY, Cho JH, Kim UK, Lee KY (2016) Methionine sulfoxide reductase B3-targeted in utero gene therapy rescues hearing function in a mouse model of congenital sensorineural hearing loss. Antioxid Redox Signal 24:590–602. doi:10.1089/ars.2015.6442
Kitahara T, Fukushima M, Uno Y, Mishiro Y, Kubo T (2003) Up-regulation of cochlear aquaporin-3 mRNA expression after intra-endolymphatic sac application of dexamethasone. Neurol Res 25(8):865–870
Kitajiri SI, McNamara R, Makishima T et al (2007) Identities, frequencies and origins of TMC1 mutations causing DFNB7/B11 deafness in Pakistan. Clin Genet 72:546–550. doi:10.1111/j.1399-0004.2007.00895.x
Kleinstiver BP, Prew MS, Tsai SQ et al (2015a) Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33:1293–1298. doi:10.1038/nbt.3404
Kleinstiver BP, Prew MS, Tsai SQ et al (2015b) Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523:481–485. doi:10.1038/nature14592
Kleinstiver BP, Wolfs JM, Kolaczyk T et al (2012) Monomeric site-specific nucleases for genome editing. Proc Natl Acad Sci 109:8061–8066. doi:10.1073/pnas.1117984109
Klug A (2010) The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem 79:213–231
Kobel M, Le Prell CG, Liu J, Hawks JW, Bao J (2016) Noise-induced cochlear synaptopathy: past findings and future studies. Hear Res. doi:10.1016/j.heares.2016.12.008
Kole R, Sazani P (2001) Antisense effects in the cell nucleus: modification of splicing. Curr Opin Mol Ther 3(3):229–234
Komor AC, Kim YB, Packer MS et al (2016) Programmable editing of a target base in genomicDNA without double-stranded DNA cleavage. Nature 533:420–424. doi:10.1038/nature17946
Konishi M, Kawamoto K, Izumikawa M et al (2008a) Gene transfer into guinea pig cochlea using adeno-associated virus vectors. J Gene Med 10:610–618. doi:10.1002/jgm.1189
Konishi M, Kawamoto K, Izumikawa M, Kuriyama H, Yamashita T (2008b) Gene transfer into guinea pig cochlea using adeno-associated virus vectors. J Gene Med 10:610–618. doi:10.1002/jgm.1189
Koo T, Popplewell L, Athanasopoulos T, Dickson G (2014) Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum Gene Ther 25:98–108. doi:10.1089/hum.2013.164
Kooijmans SA (2012) Int J Nanomedicine 7:1525–1541. doi:10.2147/IJN.S2966
Kohrman DC, Raphael Y (2013) Gene therapy for deafness. Gene Ther 20(12):1119–1123
Kuang R, Hever G, Zajic G, Yan Q, Collins F, Louis JC, Magal E (1999) Glial cell line-derived neurotrophic factor: potential for otoprotection. Ann N Y Acad Sci 884:270–291. doi:10.1111/j.1749-6632.1999.tb08648.x
Kurima K, Ebrahim S, Pan B, Sedlacek M, Sengupta P, Millis BA, Cui R, Nakanishi H, Fujikawa T, Kawashima Y, Choi BY, Monahan K, Holt JR, Griffith AJ, Kachar B (2015) TMC1 and TMC2 localize at the site of Mechanotransduction in mammalian inner ear hair cell Stereocilia. Cell Rep 12(10):1606–1617.
Kurima K, Peters LM et al (2002a) Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet 30:277–284
Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, Ghosh M (2002b) Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet 30:277–284. doi:10.1038/ng842
Lalwani AK, Han JJ, Castelein CM, Carvalho GJ, Mhatre AN (2002) In vitro and in vivo assessment of the ability of adeno-associated virus–brain-derived neurotrophic factor to enhance spiral ganglion cell survival following ototoxic insult. Laryngoscope 112:1325–1334. doi:10.1097/00005537-200208000-00001
Landegger LD, Pan B, Askew C, Wassmer SJ, Gluck SD, Galvin A, Taylor R, Forge A, Stankovic KM, Holt JR, Vandenberghe LH (2017) A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat Biotechnol 35(3):280–284.
Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Francis H (2000) Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest 106:1447–1455. doi:10.1172/JCI10897
Lefèvre G, Michel V, Weil D, Lepelletier L, Bizard E et al (2008) A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development 135(8):1427–1437. doi:10.1242/dev.012922
Lentz JJ, Jodelka FM, Hinrich AJ, McCaffrey KE, Farris HE, Spalitta MJ, Bazan NG, Duelli DM, Rigo F, Hastings ML (2013) Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat Med 19:345–350. doi:10.1038/nm.3106
Li Duan M, Bordet T, Mezzina M, Kahn A, Ulfendahl M (2002) Adenoviral and adeno-associated viral vector mediated gene transfer in the guinea pig cochlea. Neuroreport 13:1295–1299
Li J, Sun W, Wang B, Xiao X, Liu XQ (2008) Protein trans-splicing as a means for viral vector-mediated in vivo gene therapy. Hum Gene Ther 19:958–964. doi:10.1089/hum.2008.009
Li W, Kong W, Yue J (2006) Gene transfer into the mammalian inner ear using a RNAi non-viral vector. Lin Chuang Er Bi Yan Hou Ke Za Zhi 20:1024–1026
Liberman MC, Kujawa SG (2017) Cochlear synaptopathy in acquired sensorineural hearing loss: manifestations and mechanisms. Hear Res. pii: S0378-5955(16)30250-7.
Liu Y, Okada T, Sheykholeslami K et al (2005) Specific and efficient transduction of cochlear inner hair cells with recombinant adeno-associated virus type 3 vector. Mol Ther 12:725–733. doi:10.1016/j.ymthe.2005.03.021
Logan AC, Haas DL, Kafri T, Kohn DB (2004) Integrated self-inactivating lentiviral vectors produce full-length genomic transcripts competent for encapsidation and integration. J Virol 78:8421–8436. doi:10.1128/JVI.78.16.8421-8436.2004
Luo H, Schmidt JA, Lee YS, Oltz EM, Payton JE (2017) Targeted epigenetic repression of a lymphoma oncogene by sequence-specific histone modifiers induces apoptosis in DLBCL. Leuk Lymphoma 58:445–456. doi:10.1080/10428194.2016.1190973
Maeda Y, Fukushima K, Kawasaki A, Nishizaki K, Smith RJ (2007) Cochlear expression of a dominant-negative GJB2 R75W construct delivered through the round window membrane in mice. Neurosci Res 58:250–254. doi:10.1016/j.neures.2007.03.006
Maeda Y, Fukushima K, Nishizaki K, Smith RJ (2005) In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum Mol Genet 14:1641–1650. doi:10.1093/hmg/ddi172
Maguire AM, High KA, Auricchio A, Wright JF, Pierce EA, Testa F, Mingozzi F, Bennicelli JL, Ying GS, Rossi S, Fulton A (2009) Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 374:1597–1605. doi:10.1016/S0140-6736(09)61836-5
Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M (2006) Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat Med 12:342–347. doi:10.1038/nm1358
Mao Y, Wang X, Yan R, Hu W, Li A, Wang S, Li H (2016) Single point mutation in adeno-associated viral vectors-DJ capsid leads to improvement for gene delivery in vivo. BMC Biotechnol 16:1. doi:10.1186/s12896-015-0230-0
McDonald JI, Celik H, Rois LE (2016) Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biology Open bio-019067. doi:10.1242/bio.019067
Melo S. A.(2014). Cancer cell. 5:707–721. doi:10.1016/j.ccell.2014.09.005
Miller JM, Chi DH, O'Keeffe LJ, Kruszka P, Raphael Y, Altschuler RA (1997) Neurotrophins can enhance spiral ganglion cell survival after inner hair cell loss. Int J Dev Neurosci 15:631–643
Mingozzi F, High KA (2013) Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122:23–36. doi:10.1182/blood-2013-01-306647
Minoda R, Miwa T, Ise M, Takeda H (2015 Aug) Potential treatments for genetic hearing loss in humans: current conundrums. Gene Ther 22(8):603–609
Moorhead JW, Clayton GH, Smith RL, Schaack J (1999) A replication-incompetent adenovirus vector with the preterminal protein gene deleted efficiently transduces mouse ears. J Virol 73:1046–1053
Morral N, Parks RJ, Zhou H et al (1998) High doses of a helper-dependent adenoviral vector yield supraphysiological levels of alpha1-antitrypsin with negligible toxicity. Hum Gene Ther 9:2709–2716
Naz S, Griffith AJ, Riazuddin S, Hampton LL, Battey JF, Khan SN, Friedman TB (2004) Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J Med Genet 41:591–595. doi:10.1136/jmg.2004.018523
Naz S, Imtiaz A, Mujtaba G, Maqsood A, Bashir R, Bukhari I, Khan MR, Ramzan M, Fatima A, Rehman AU, Iqbal M, Chaudhry T, Lund M, Brewer CC, Morell RJ, Friedman TB (2017) Genetic causes of moderate to severe hearing loss point to modifiers. Clin Genet 91(4):589–598.
Okano T, Nakagawa T, Kita T et al (2006) Cell-gene delivery of brain-derived neurotrophic factor to the mouse inner ear. Mol Ther 14:866–871. doi:10.1016/j.ymthe.2006.06.012
Ota CY, Kimura RS (1980) Ultrastructural study of the human spiral ganglion. Acta Otolaryngol 89:53–62
Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, Nance WE, Li AR, Angeli S, Kaiser M, Newton V, Brown SD (2005) Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population. Hum Genet 116:292–299. doi:10.1007/s00439-004-1227-2
Pan B, Askew C, Galvin A, et al (2017) Gene therapy restores auditory and vestibular function 1 in a mouse model of Usher syndrome, type 1c. Nat Biotechnol 35(3):264–272
Pan B, Géléoc GS, Asai Y, Horwitz GC, Kurima K, Ishikawa K, Kawashima Y, Griffith AJ, Holt JR (2013) TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron 79(3):504–515.
Parker MA, Cheng YF, Kinouchi H, Bieber R, Edge AS (2013) An independent construct for conditional expression of atonal homolog-1. Hum Gen Ther Methods 25:1–13. doi:10.1089/hgtb.2013.014
Payne JG, Takahashi A, Higgins MI et al (2016) Multilineage transduction of resident lung cells in vivo by AAV2/8 for α1-antitrypsin gene therapy. Mol Ther Methods Clin Dev 3:16042. doi:10.1038/mtm.2016.42
Pfingst BE, Bowling SA, Colesa DJ et al (2011) Cochlear infrastructure for electrical hearing. Hear Res 281:65–73. doi:10.1016/j.heares.2011.05.002
Pietola L, Aarnisalo AA, Joensuu J, Pellinen R, Wahlfors J, Jero J (2008) HOX-GFP and WOX-GFP lentivirus vectors for inner ear gene transfer. Acta Otolaryngol 128:613–620. doi:10.1080/00016480701663409
Pinyon JL, Tadros SF, Froud KE et al (2014) Close-field electroporation gene delivery using the cochlear implant electrode array enhances the bionic ear. Sci Transl Med 6:233ra54. doi:10.1126/scitranslmed.3008177
Pirvola U, Hallböök F, Xing-Qun L, Virkkala J, Saarma M, Ylikoski J (1997) Expression of neurotrophins and Turk receptors in the developing, adult, and regenerating avian cochlea. J Neurobiol 33:1019–1033
Praetorius M, Pfannenstiel S, Klingmann C, Baumann I, Plinkert PK, Staecker H (2008) Expression patterns of non-viral transfection with GFP in the organ of Corti in vitro and in vivo. Gene therapy of the inner ear with non-viral vectors. HNO 56:524–529. doi:10.1007/s00106-008-1738-6
Puligilla C, Dabdoub A, Brenowitz SD, Kelley MW (2010) Sox2 induces neuronal formation in the developing mammalian cochlea. J Neurosci 30:714–722. doi:10.1523/JNEUROSCI.3852-09.2010
Qi LS, Larson MH, Gilbert LA et al (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi:10.1016/j.cell.2013.02.022
Rabinowitz JE, Samulski RJ (2000) Building a better vector: the manipulation of AAV virions. Virology 278:301–308. doi:10.1006/viro.2000.0707
Räty JK, Lesch HP, Wirth T, Ylä-Herttuala S (2008) Improving safety of gene therapy. Curr Drug Saf 3:46–53. doi:10.2174/157488608783333925
Ren L-L, Wu Y, Han D et al (2010) Math1 gene transfer based on the delivery system of quaternized chitosan/Na-carboxymethyl-β-cyclodextrin nanoparticles. J Nanosci Nanotech 10:7262–7265. doi:10.1166/jnn.2010.2822
Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ (1998) Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet 103:393–399
Rivera T, Sanz L, Camarero G, Varela-Nieto I (2012) Drug delivery to the inner ear: strategies and their therapeutic implications for sensorineural hearing loss. Curr Drug Deliv 9(3):231–242
Ruel J, Emery S, Nouvian R, Bersot T, Amilhon B, Van Rybroek JM, Rebillard G, Lenoir M, Eybalin M, Delprat B, Sivakumaran TA (2008) Impairment of SLC17A8 encoding vesicular glutamate transporter-3, VGLUT3, underlies nonsyndromic deafness DFNA25 and inner hair cell dysfunction in null mice. Am J Hum Genet 83:278–292. doi:10.1016/j.ajhg.2008.07.008
Sacheli R, Delacroix L, Vandenackerveken P, Nguyen L, Malgrange B (2013) Gene transfer in inner ear cells: a challenging race. Gene Ther 20:237–247. doi:10.1038/gt.2012.51
Sallach J, Di Pasquale G, Larcher F et al (2014) Tropism-modified AAV vectors overcome barriers to successful cutaneous therapy. Mol Ther 22:929–939. doi:10.1038/mt.2014.14
Santos RLP, Wajid M, Khan MN, McArthur N, Pham TL, Bhatti A, Lee K, Irshad S, Mir A, Yan K, Chahrour MH (2005) Novel sequence variants in the TMC1 gene in Pakistani families with autosomal recessive hearing impairment. Hum Mutat 26:396–396. doi:10.1002/humu.9374
Sayroo R, Nolasco D, Yin Z, Colon-Cortes Y, Pandya M, Ling C, Aslanidi G (2016) Development of novel AAV serotype 6 based vectors with selective tropism for human cancer cells. Gene Ther 23:18–25. doi:10.1038/gt.2015.89
Seal RP, Akil O, Yi E, Weber CM, Grant L, Yoo J, Clause A, Kandler K, Noebels JL, Glowatzki E, Lustig LR (2008) Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron 57:263–275. doi:10.1016/j.neuron.2007.11.032
Shcharbin DG, Klajnert B, Bryszewska M (2009) Dendrimers in gene transfection. Biochemistry 74(10):1070–1079.
Shibata SB, Ranum PT, Moteki H, Pan B, Goodwin AT, Goodman SS, Abbas PJ, Holt JR, Smith RJ (2016) RNA interference prevents autosomal-dominant hearing loss. Am J Hum Genet 98:1101–1113. doi:10.1016/j.ajhg.2016.03.028
Shu Y, Tao Y, Li W, Shen J, Wang Z, Chen Z-Y 2016. Adenovirus vectors target several cell subtypes of mammalian inner ear in vivo. Neural Plast. 2016: 9409846. Published online 2016 Dec 28. doi: 10.1155/2016/9409846
Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Pâques F (2011) Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11:11–27. doi:10.2174/156652311794520111
Simonelli F, Maguire AM, Testa F, Pierce EA, Mingozzi F, Bennicelli JL, Rossi S, Marshall K, Banfi S, Surace EM, Sun J (2010) Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther 18:643–650. doi:10.1001/archophthalmol.2011.298
Sırmacı A, Duman D, Öztürkmen-Akay H, Erbek S, Incesulu A, Oztürk-Hişmi B, Arici ZS, Yüksel-Konuk EB, Taşir-Yilmaz S, Tokgöz-Yilmaz S, Cengiz FB, Aslan I, Yildirim M, Hasanefendioğlu-Bayrak A, Ayçiçek A, Yilmaz I, Fitoz S, Altin F, Ozdağ H, Tekin M (2009) Mutations in TMC1 contribute significantly to nonsyndromic autosomal recessive sensorineural hearing loss: a report of five novel mutations. Int J Pediatr Otorhinolaryngol 73:699–705. doi:10.1016/j.ijporl.2009.01.005
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT (2016) Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 135(4):441–450. doi:10.1007/s00439-016-1648-8
Snowden AW, Gregory PD, Case CC, Pabo CO (2002) Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol 12:2159–2166. doi:10.1016/S0960-9822(02)01391-X
Souter M (2008) Gap junctions and connexin expression in the inner ear. Gap Junction-Mediated Intercellular Signalling in Health and Disease 797:134
Spoendlin H (1981) Differentiation of cochlear afferent neurons. Acta Otolaryngol 91:451–456
Staecker H, Schlecker C, Kraft S, Praetorius M, Hsu C, Brough DE (2014) Optimizing atoh1-induced vestibular hair cell regeneration. Laryngoscope 124:S1–S12. doi:10.1002/lary.24775
Steel KP, Barkway C (1989) Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development 107:453–463
Suzuki J, Hashimoto K, Xiao R, Vandenberghe LH, Liberman MC (2017) Cochlear gene therapy with ancestral AAV in adult mice: complete transduction of inner hair cells without cochlear dysfunction. Sci Rep 7:45524
Takada Y, Beyer LA, Swiderski DL, O'Neal AL, Prieskorn DM, Shivatzki S, Avraham KB, Raphael Y (2014) Connexin 26 null mice exhibit spiral ganglion degeneration that can be blocked by BDNF gene therapy. Hear Res 309:124–135. doi:10.1016/j.heares.2013.11.00
Takada Y, Takada T, Lee MY et al (2015) Ototoxicity-induced loss of hearing and inner hair cells is attenuated by HSP70 gene transfer. Mol Ther Methods Clin Dev 2:15019. doi:10.1038/mtm.2015.19
Tal-Goldberg T, Lorain S, Mitrani-Rosenbaum S (2014) Correction of the middle eastern M712T mutation causing GNE myopathy by trans-splicing. NeuroMolecular Med 16:322–331. doi:10.1007/s12017-013-8278-2
Tamm I, Dörken B, Hartmann G (2001) Antisense therapy in oncology: new hope for an old idea? Lancet 358(9280):489–497. doi:10.1016/S0140-6736(01)05629-X
Tamura T, Kita T, Nakagawa T et al (2005) Drug delivery to the cochlea using PLGA nanoparticles. Laryngoscope 115:2000–2005. doi:10.1097/01.mlg.0000180174.81036.5a
Thakore PI, D'Ippolito AM, Song L et al (2015) Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12:1143–1149. doi:10.1038/nmeth.3630
Topilina NI, Mills KV (2014) Recent advances in in vivo applications of intein-mediated protein splicing. Mob DNA 5:5. doi:10.1186/1759-8753-5-5
Trapani I, Colella P, Sommella A et al (2013) Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med 6:194–211. doi:10.1002/emmm.201302948
Truong D-JJ, Kühner K, Kühn R, Werfel S, Engelhardt S, Wurst W, Ortiz O (2015) Development of an intein-mediated split–Cas9 system for gene therapy. Nucleic Acids Res 43:6450–6458. doi:10.1093/nar/gkv601
Venail F, Wang J, Ruel J et al (2006) Coxsackie adenovirus receptor and ανβ3/ανβ5 integrins in adenovirus gene transfer of rat cochlea. Gene Ther 14:30–37. doi:10.1038/sj.gt.3302826
Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Slim R (2000) A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet 26:51–55. doi:10.1038/79171
Wagenaar TR, Tolstykh T, Shi C, Jiang L, Zhang J, Li Z, Yu Q, Qu H, Sun F, Cao H, Pollard J (2015) Identification of the endosomal sorting complex required for transport-I (ESCRT-I) as an important modulator of anti-miR uptake by cancer cells. Nucleic Acids Res 43(2):1204–1215. doi:10.1093/nar/gku1367
Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, Avraham KB (2002) From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci 99:7518–7523. doi:10.1073/pnas.102091699
Wan G, Corfas G, Stone JS (2013) Inner ear supporting cells: rethinking the silent majority. Semin Cell Dev Biol 24:448–459. doi:10.1016/j.semcdb.2013.03.009
Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, Friedman TB (1998) Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science 280:1447–1451
Wang Q, Dong B, Firrman J et al (2014) Efficient production of dual recombinant adeno-associated viral vectors for factor VIII delivery. Hum Gene Ther Methods 25:261–268. doi:10.1089/hgtb.2014.093
Wang Y, Sun Y, Chang Q et al (2013) Early postnatal virus inoculation into the scala media achieved extensive expression of exogenous green fluorescent protein in the inner ear and preserved auditory brainstem response thresholds. J Gene Med 15:123–133. doi:10.1002/jgm.2701
Wangemann P (2006) Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol 576:11–21. doi:10.1113/jphysiol.2006.112888
Wangemann P, Schacht J (1996) Homeostatic mechanisms in the cochlea. In: The cochlea. Springer, New York, pp 130–185
Wanisch K, Yáñez-Muñoz RJ (2009) Integration-deficient lentiviral vectors: a slow coming of age. Mol Ther 17:1316–1332. doi:10.1038/mt.2009.122
Wei Y, Fu Y, Liu S, Xia G, Pan S (2013) Effect of lentiviruses carrying enhanced green fluorescent protein injected into the scala media through a cochleostomy in rats. Am J Otolaryngol 34:301–307. doi:10.1016/j.amjoto.2012.12.011
Wolfs JM, DaSilva M, Meister SE, Wang X, Schild-Poulter C, Edgell DR (2014) MegaTevs: single-chain dual nucleases for efficient gene disruption. Nucleic Acids Res 42:8816–8829. doi:10.1093/nar/gku573
Yagi M, Kanzaki S, Kawamoto K, Shin B, Shah PP, Magal E, Raphael Y (2000) Spiral ganglion neurons are protected from degeneration by GDNF gene therapy. J Assoc Res Otolaryngol 1:315–325. doi:10.1007/s101620010011
Yamasoba T, Yagi M, Roessler BJ, Miller JM, Raphael Y (1999) Inner ear transgene expression after adenoviral vector inoculation in the endolymphatic sac. Hum Gene Ther 10(5):769–774
Yan J, Pan L, Chen X, Wu L, Zhang M (2010) The structure of the harmonin/sans complex reveals an unexpected interaction mode of the two Usher syndrome proteins. Proc Natl Acad Sci 107:4040–4045. doi:10.1073/pnas.0911385107
Yan Z, Lei-Butters DCM, Zhang Y, Zak R, Engelhardt JF (2007) Hybrid adeno-associated virus bearing nonhomologous inverted terminal repeats enhances dual-vector reconstruction of minigenes in vivo. Hum Gene Ther 18:81–87. doi:10.1089/hum.2006.128
Yang G, Si-Tayeb K, Corbineau S et al (2013a) Integration-deficient lentivectors: an effective strategy to purify and differentiate human embryonic stem cell-derived hepatic progenitors. BMC Biol 11:86. doi:10.1186/1741-7007-11-86
Yang J, Cong N, Han Z, Huang Y, Chi F (2013b) Ectopic hair cell-like cell induction by Math1 mainly involves direct transdifferentiation in neonatal mammalian cochlea. Neurosci Lett 549:7–11. doi:10.1016/j.neulet.2013.04.053
Yang J, Zhou W, Zhang Y, Zidon T, Ritchie T, Engelhardt JF (1999) Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J Virol 73:9468–9477. doi:10.1128/JVI.79.11.6801-6807.2005
Yang SM, Chen W, Guo WW, Jia S, Sun JH, Liu HZ, Young WY, He DZ (2012) Regeneration of stereocilia of hair cells by forced Atoh1 expression in the adult mammalian cochlea. PLoS One 7:e46355. doi:10.1371/journal.pone.0046355
Yasunaga SI, Grati MH, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, Petit C (1999) A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet 21:363–369. doi:10.1038/7693
Yu CY, Yuan Z, Cao Z, Wang B, Qiao C, Li J, Xiao X (2009) A muscle-targeting peptide displayed on AAV2 improves muscle tropism on systemic delivery. Gene Ther 16:953–962. doi:10.1038/gt.2009.59
Yu Q, Wang Y, Chang Q, Wang J, Gong S, Li H, Lin X (2014) Virally expressed connexin 26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther 21:71–80. doi:10.1038/gt.2013.59
Zanin MP, Hellström M, Shepherd RK, Harvey AR, Gillespie LN (2014) Development of a cell-based treatment for long-term neurotrophin expression and spiral ganglion neuron survival. Neuroscience 277:690–699. doi:10.1016/j.neuroscience.2014.07.044
Zamore PD, Tuschl T, Sharp PA, Bartel DP (2000) RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101(1):25–33.
Zhang Y, Bergelson JM (2005) Adenovirus receptors. J Virol 79:12125–12131
Zhao Y, Wang D, Zong L, Zhao F, Guan L, Zhang P, Shi W, Lan L, Wang H, Li Q, Han B, Yang L, Jin X, Wang J, Wang J, Wang Q (2014) A novel DFNA36 mutation in TMC1 orthologous to the Beethoven (Bth) mouse associated with autosomal dominant hearing loss in a Chinese family. PLoS One 9(5):e97064.
Zhou H, Ma X, Liu Y et al (2015) Linear polyethylenimine-plasmid DNA nanoparticles are ototoxic to the cultured sensory epithelium of neonatal mice. Mol Med Report 11:4381–4388
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE (2008) Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16:1073–1080. doi:10.3892/mmr.2015.3306
Zinn E, Pacouret S, Khaychuk V et al (2015) In silico reconstruction of the viral evolutionary lineage yields a potent gene therapy vector. Cell Rep 12:1056–1068. doi:10.1016/j.celrep.2015.07.019
Zou B, Mittal R, Grati M, Lu Z, Shu Y, Tao Y, Feng Y, Xie D, Kong W, Yang S, Chen ZY, Liu X (2015 Sep) The application of genome editing in studying hearing loss. Hear Res 327:102–108
Zuris JA, Thompson DB, Shu Y et al (2014) Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotech 33:73–80
Author information
Authors and Affiliations
Corresponding author
Additional information
Hena Ahmed and Olga Shubina-Oleinik contributed equally to this work.
Rights and permissions
About this article

Cite this article
Ahmed, H., Shubina-Oleinik, O. & Holt, J.R. Emerging Gene Therapies for Genetic Hearing Loss. JARO 18, 649–670 (2017). https://doi.org/10.1007/s10162-017-0634-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-017-0634-8