
Abstract
Voltage-dependent anion channel 1 (VDAC1) is thought to contribute to the progression of tumor development. However, whether VDAC1 contributes to bone cancer pain remains unknown. In this study, we found that the expression of VDAC1 was upregulated in the L2–5 segments of the spinal dorsal horn at 2 and 3 weeks after injection of tumor cells into the tibial cavity. Intrathecal injection of a VDAC1 inhibitor significantly reversed the pain hypersensitivity and reduced the over-expression of Toll-like receptor 4 (TLR4). Intrathecal injection of minocycline, an inhibitor of microglia, also attenuated the pain hypersensitivity of rat models of bone cancer pain. These results suggest that VDAC1 plays a significant role in the development of complicated cancer pain, possibly by regulating the expression of TLR4.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer bone metastases always produce an excruciating pain, current treatments of which may be inadequate or have unacceptable side effects [1, 2]. Bone cancer pain (BCP) is a unique pain that includes features of nociceptive, neuropathic, and inflammatory pain [3]. It has been suggested that the development of BCP is associated with destruction of bone and the pathological reconstruction of nervous system [4,5,6]. However, the existing treatment strategies for BCP are not radical, but palliative or supportive. Therefore, it is urgent to explore the mechanisms of bone metastasis and develop novel therapeutic targets for the relief of BCP.
Voltage-dependent anion channels (VDACs), expressed in the mitochondrial outer membrane, function as the gatekeeper for the entry and exit of mitochondrial metabolites, thereby controlling cross-talk between mitochondria and other components of the cell. VDACs include three isoforms: VDAC1, VDAC2, and VDAC3 [7, 8]. VDAC1 is a pore-forming channel that regulates the passage of many molecules and ions [8]. Previous studies have shown that VDAC1 is expressed in many tissues including the central nervous system [9, 10]. VDAC1 is often over-expressed in many cancer types. Knockout of VDAC1 can inhibit tumor development [11]. Whether VDAC1 contributes to cancer-induced pain hypersensitivity remains unknown. It has been shown that VDAC1 is involved in neuropathic pain [12], indicating that VDAC1 activation in the spinal dorsal horn may play an important role in the development of pain hypersensitivity of bone cancer.
Toll-like receptor 4 (TLR4) is a transmembrane protein. It has both extracellular leucine-rich repeat domains and a cytoplasmic signaling domain [13]. In the central nervous system, TLR4 is predominantly expressed in microglia [14]. It has been reported that TLR4 plays an important role in the initiation and transmission of neuropathic pain signals [15]. Damage of sensory neurons results in the stimulation of microglial TLR4 in the spinal cord, followed by activation of the microglia [16]. The significant increase of TLR4 expression in a rat model of bone cancer leads to tactile allodynia and spontaneous pain, while injection of an antagonist of TLR4 leads to the reversal of both mechanical allodynia and thermal hyperalgesia in mice with neuropathic pain [17]. Intrathecal injection of synthetic small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) targeting TLR4 reduced the nociceptive response in the rat model [18]. In our work, inhibition of VDAC1 in the spinal dorsal horn reversed the activation of microglia and reduced the expression of TLR4. Therefore, VDAC1 might be a potential target for BCP therapy.
Materials and Methods
Animals
Female Sprague-Dawley rats, weighing 160 g–180 g were housed in a temperature- (24 ± 1 °C) and light-controlled (12:12-h light-dark cycle) room with free access to food and water. All experiments were approved by the Laboratory Animal Center of Soochow University, Jiangsu Province, China.
Induction of the BCP Model
The specific details of procedures for the BCP model are documented in previous reports [19, 20]. In brief, ~1 mL Walker 256 cells (~2×107 cells) were injected into the abdominal cavity of each rat. A week later, the ascites was extracted. The cells were centrifuged for 5 min at 1200 rpm and washed 3 times with 10 mL normal saline (NS). The Walker 256 cells were counted and diluted to a final concentration of 1×108 cells per mL with NS. These cells were kept on ice until injected into rats. Four microliter of carcinoma cells (4×105) or the same volume of NS (vehicle group) was slowly injected into the tibia cavity using a 10 μL microinjection syringe with a 23-gauge needle. The syringe was left in the tibia for an additional 2 min to minimize the leakage of tumor cells. Before full recovery from anesthesia, all rats were placed on a warm pad for recovery from the surgery.
Mechanical Allodynia and Thermal Hyperalgesia Testing
Mechanical allodynia was assessed using the up-down method in the hind paw withdrawal response to von Frey fillament (VFF) stimulation. The 50% paw withdrawal threshold (PWT) was determined as previously described [19, 21, 22]. Each rat was placed in a transparent box and allowed to acclimate to the surroundings for at least 30 min. The experiments were performed in a double-blinded manner. Standard VFFs (0.4, 0.6, 1.0, 1.4, 2.0, 4.0, 6.0, 8.0, 10.0, and 15.0 g) were vertically advanced to the plantar surface of the hind paw using sufficient force until the filament bent. The trial began with the 2.0 g filament. Withdrawal of the hind paw was regarded as a positive response, and this was followed by the next greater force. If there was no response, the next lower force was applied until the trial was repeated 3 times and the final VFF was determined. The cutoff force was set at 15.0 g. Each stimulation was spaced at ~10-min intervals and the mean value was regarded as the strength to produce a withdrawal response. The paw withdrawal latency (PWL) to thermal hyperalgesia was measured as previously reported [19, 21]. Rats were placed on a clear glass plate and allowed to acclimate to the new surroundings for 30 min. A radiant heat source was focused onto the plantar surface of a hind paw. The PWL was recorded by a timer which was started by activation of the heat source and stopped by the paw withdrawal as recorded with a photo detector. To prevent tissue damage, a maximal cutoff time of 20 s was set. The average of 3 PWL measurements was taken as the result of each test session for each rat. The interval between measurements was > 5 min.
Western Blotting Analysis
Expressions of VDAC1, TLR4, p65, P2X7, and CBS (cystathionine-beta-synthase) in L2–L5 of the spinal dorsal horn from control rats and BCP rats injected with 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) or Minocycline (Mino) were measured using western blotting. Rats were rapidly sacrificed by cervical dislocation. The L2–L5 spinal dorsal horn or dorsal root ganglia (DRGs) were quickly dissected out and lysed, and the lysates were allowed to cool for 2 h, then they were centrifuged at 12,000 rpm for 20 min at 4 °C. The supernatant was collected and the total protein concentration was measured using a BCA Protein Quantitation Kit (Thermo Scientific, MA). Twenty micrograms of proteins were fractionated on 10% polyacrylamide gels (Bio-Rad, CA). Then the proteins were transferred to polyvinylidenedifluoride membranes (Bio-Rad) at 200 mA for 2 h. The membranes were blocked using a 5% dilution of fat-free milk powder in TBS (50 mmol/L Tris-HCl, 133 mmol/L NaCl, pH 7.4) for 2 h at room temperature, and incubated at 4 °C with primary antibody (anti-VDAC1 at 1:1000, Proteintech, Chicago, IL; anti-TLR4 at 1:1000, Abcam, Cambridge, UK; anti-NF-κB p65 at 1:200, Santa Cruz Biotechnology, CA; anti-P2X7 at 1:1000, Neuromics, USA; anti-CBS at 1:1000, Abcam, Cambridge, UK) in TBS containing 1% fat-free milk. The membranes were washed 3 times in TBST (0.5% Tween-20), then incubated with peroxidase–conjugated secondary antibodies (1:5000, Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h at room temperature. The antibodies were dissolved in TBS containing 1% fat-free milk. Bands were developed using an ECL kit (Amersham, Buckinghamshire, UK). The densities of protein bands were analyzed using Image J (National Institutes of Health, Betheseda, MD).
Real-Time Quantitative Polymerase Chain Reaction for mRNA
Total RNA was extracted from the L2–L5 spinal dorsal horn from control and BCP rats using Trizol (Invitrogen, CA). cDNA was synthesized from total RNA using an Omniscript RT kit 50 (Qiagen, Valencia, CA) following the manufacturer’s instructions. The mRNAs of VDAC1 and GAPDH (internal control) were measured in the quantitative polymerase chain reaction using the following primers: VDAC1 forward primer 5′GCTCTGGTGCTTGGCTATGA3′, reverse primer 5′GGGTCGACCTGATACTTGGC3′; GAPDH forward primer 5′AAGGTGGTGAAGCAGGCGGC3′, reverse primer 5′GAGCAATGCCAGCCCCAGCA3′. The VDAC1 gene amplified within the linear region of the reaction amplification curves is shown as the Ct value. The expression level of VDAC1 was normalized by the Ct value of GAPDH using the 2−ΔΔCt relative quantification method.
Immunofluorescence
Two weeks after induction of the BCP model, rats were perfused transcardially with 300 mL phosphate-buffered saline (PBS) followed by 300 mL ice-cold 4% paraformaldehyde (PFA) in PBS. The L2–L5 spinal dorsal horn was extracted and post-fixed for 4 h in PFA. The dorsal horn was cryoprotected overnight in 20% sucrose in PBS. For double-labeling, 10 μm sections of the dorsal horn were incubated with TLR4 (1:200, Abcam), VDAC1 (1:200, Proteintech), NeuN (1:200, Santa Cruz Biotechnology), CD11b (1:200, Santa Cruz Biotechnology), or GFAP (1:200, Santa Cruz Biotechnology) antibody overnight at 4 °C. They were then incubated with secondary antibody with Alexa Fluor 488 and 355 for 2 h at room temperature. Negative controls were performed omitting primary antibodies. As previously described, images were captured and analyzed using Metaview software (Universal Imaging Corporation, Downingtown, PA).
Drug Application
4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS, Sigma, MI, inhibitor of VDACs) and Mino (Sigma, inhibitor of microglia) were used. One week after tumor cell injection, 24 rats were divided into 4 groups and received an intrathecal injection of NS (n = 5) or DIDS at 0.5 (n = 6), 2 (n = 6), or 8 (n = 7) mg/kg body weight. PWT and PWL were recorded 1, 3, 6 and 24 h after DIDS treatment. To assess the effect of multiple injections of DIDS, rats received a DIDS injection once a day for 7 consecutive days. After this, PWT and PWL were measured at 1, 6, 12, 18, and 24 h. For Mino treatment, 28 rats were divided into 4 groups and received an intrathecal injection of NS (n = 7) or Mino at 50 μL (n = 7), 100 μL (n = 7), or 200 μL (n = 7). PWT and PWL were recorded 0.5, 1, 2, and 4 h after Mino injection. To assess the effect of multiple injections of Mino, the rats also received a daily injection for 7 consecutive days. PWT and PWL were determined at 1, 3, 6, 9, and 12 h after the last Mino treatment. In order to eliminate the effect of DIDS or Mino on motor function, we used the Rota-rod test after intrathecal injection of DIDS or Mino into normal rats (n = 6).
Data Analysis
Data are expressed as mean ± SEM for molecular and behavior assays. All data were analyzed using SPSS 17.0 and OriginPro 8 (OriginLab, Northampton, MA) software. The significance of differences between 2 groups was determined using Student’s t or the χ2 test. Two-way repeated-measures ANOVA followed by Tukey’s post hoc test or the Mann–Whitney test following Friedman ANOVA was performed as appropriate. A P value < 0.05 was regarded as statistically significant.
Results
Reduced PWT and PWL with Upregulation of VDAC1 Expression in Spinal Dorsal Horn of BCP Rats
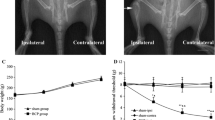
Our previous studies have shown that rats treated with Walker 256 tumor cells display a gradual increase in sensitivity to VFF and radiant heat stimuli 2 weeks after tumor cell injection [19, 21]. Therefore, we began our current studies at week 2 after tumor cell injection. The PWT to VFF in the tumor-bearing hind paw decreased from the baseline level of 14.22 ± 1.43 g to 4.34 ± 0.45 g at 2 weeks after injection (Fig. 1A, P = 0.0000454; n = 8 rats per group, Mann–Whitney test following Friedman ANOVA). Similarly, the PWL to radiant heat stimulation decreased at week 2 from 14.40 ± 0.13 s to 7.80 ± 0.21 s (Fig. 1B, P = 0.000534; n = 8 rats per group, Mann–Whitney test following Friedman ANOVA).

Upregulation of VDAC1 expression in the spinal dorsal horn of BCP rats 2 weeks after tumor cell injection. A PWT was significantly reduced 2 weeks after tumor cell injection. B PWL was significantly reduced 2 weeks after tumor cell injection. C Expression of VDAC1 at the protein level in L2–L5 spinal dorsal horn of tumor-injected rats was greatly enhanced at 2 weeks after tumor cell inoculation. P = 0.035 versus control group, Student’s t test, n = 4 rats per group. D Expression of VDAC1 at the mRNA level in L2–L5 spinal dorsal horn of tumor-injected rats was greatly enhanced at 2 weeks after tumor cell inoculation. P = 0.043 on week 2 vs control group, Student’s t test, n = 5 rats per group. E In L2–L5 DRGs, the protein level expression of VDAC1 was not altered at week 2 in BCP rats vs control group.
To determine whether the expression of VDAC1 was changed in the L2–L5 dorsal horn after tumor cell injection, we performed quantitative RT-PCR and western blotting assays 2 weeks after injection. The protein level of VDAC1 was significantly higher than in control rats (Fig. 1C, P = 0.035 at week 2 vs CON, Student’s t test, n = 4 rats per group). The mRNA level of VDAC1 expression was also higher at week 2 (Fig. 1D, P = 0.043 vs CON, Student’s t test, n = 5 rats per group). We also measured the protein level of VDAC1 in L2–L5 DRGs, but it was not altered at week 2 after tumor cell injection (Fig. 1E, P > 0.05, Student’s t test, n = 4 rats per group). These data suggested that tumor cell injection significantly enhances VDAC1 expression both at the protein and mRNA levels in the spinal dorsal horn at week 2 after tumor cell injection, and this is correlated with the changes in pain behavior in BCP rats.
To determine the time-course of pain behaviors and the expression of VDAC1, we performed experiments at week 3 after tumor cell injection. The injection resulted in continued mechanical allodynia and thermal hyperalgesia at week 3. The PWT to VFF in the tumor-bearing hind paw decreased from the baseline level of 13.65 ± 1.63 g to 3.71 ± 0.45 g at week 3 (Fig. 2A, P = 0.00001; n = 8 rats per group, Mann–Whitney test following Friedman ANOVA). The PWL to radiant heat stimulation decreased at week 3 from 14.32 ± 0.13 s to 6.32 ± 0.13 s (Fig. 2B, P = 0.00001; n = 8 rats per group, Mann–Whitney test following Friedman ANOVA).

Upregulation of VDAC1 expression in the spinal dorsal horn of BCP rats 3 weeks after tumor cell injection. A PWT was significantly reduced at week 3. B PWL was significantly reduced at week 3. C Expression of VDAC1 protein in the L2–L5 spinal dorsal horn of tumor-injected rats was greatly enhanced at 3 weeks after tumor cell inoculation. P = 0.021 vs control group, Student’s t test, n = 4 rats per group. D Expression of VDAC1 mRNA in the L2–L5 spinal dorsal horn of tumor-injected rats was greatly enhanced at 3 weeks after inoculation. P = 0.047 on week 3 vs control group, Student’s t test, n = 5 rats per group. E In L2–L5 DRGs, the protein level expression of VDAC1 was not altered at week 2 in BCP rats vs control group.
The protein level of VDAC1 in the L2–L5 spinal dorsal horn was significantly increased (Fig. 2C, P = 0.021 vs CON, Student’s t test, n = 4 rats per group). The mRNA level of VDAC1 expression in the L2–L5 spinal dorsal horn was also higher at week 3 than that in the control group (Fig. 2D, P = 0.047 vs CON, Student t test, n = 5 rats for each group). Similarly, we measured the protein level of VDAC1 in L2–L5 DRGs, but it was also unchanged at week 3 after tumor cell injection (Fig. 2E, P > 0.05, n = 4 rats per group, Student’s t test).
Distribution of VDAC1 in Spinal Dorsal Horn
Next, we examined the distribution of VDAC1 in the spinal dorsal horn. Immunofluorescence assays showed that VDAC1 was co-expressed with CD11b, a marker for microglia, and GFAP, a marker for astrocytes, but not with NeuN, a marker for neurons (Fig. 3).

Immunohistochemical staining of CD11b, GFAP, and NeuN with VDAC1 in the spinal dorsal horn. VDAC1 co-localized with most of the CD11b-positive microglia (top) and GFAP-positive astrocytes (middle) but not with NeuN-positive neurons (bottom). Scale bar, 50 μm.
Microglia Are Activated in L2–L5 Spinal Dorsal Horn After Tumor Cell Injection
Immunofluorescence results showed that microglia were activated in the L2–L5 spinal dorsal horn in BCP rats compared with control rats (Fig. 4A). The mean fluorescence intensity of microglia activated in the BCP group was stronger (0.068 ± 0.005) than that in the control group (0.051 ± 0.004) (Fig. 4B, P = 0.038, n = 4 rats per group, Student’s t test).

Activation of spinal microglia in rats with cancer-induced pain. A Immunofluorescence images showing CD11b-positive cells in control (CON) and bone cancer pain (BCP) rats; higher magnification in the upper left. B Bar graph showing that the mean fluorescence intensity of microglia activated in the BCP group was stronger than that in the control group. P = 0.038, n = 4 rats per group, Student’s t test.
Reversal of Hyperalgesia by Inhibition of VDAC1 in BCP Rats
To determine whether VDAC1 is involved in the cancer-induced hyperalgesia, we next injected the VDAC1-specific inhibitor DIDS or NS as control into BCP rats. Intrathecal injection of DIDS produced a strong amelioration in pain behaviors of BCP rats when compared with NS injection. The 50% PWT to VFF in the hind paw of rats with DIDS injection increased from the baseline level of 4.28 ± 1.23 g to 12.50 ± 2.96 g and 12.57 ± 2.59 g for 2 and 8 mg/kg 1 h after injection, and from the baseline level of 4.80 ± 1.20 g to 11.83 ± 1.44 g and 13.00 ± 2.27 g for 2 and 8 mg/kg 3 h after injection (Fig. 5A). Similarly, the PWL to radiant heat stimulation began to increase at 1 h and was maintained for 3 h after injection of DIDS. The PWL increased from the baseline level of 9.56 ± 0.87 s to 13.46 ± 1.18 s and 13.48 ± 1.02 s for 2 and 8 mg/kg 1 h after injection; and from the baseline level of 9.52 ± 0.84 s to 13.56 ± 0.46 s and 12.97 ± 0.74 s for 2 and 8 mg/kg 3 h after injection (Fig. 5B). After 7 days of administration of DIDS at 2 mg/kg body weight, a long-term effect of DIDS on PWT and PWL was found when compared with the NS-injected group. The PWT in the hind paw of rats with DIDS injection increased from the average level of 4.00 ± 0.89 g to 9.50 ± 1.25 g, 4.68 ± 1.27 g to 9.16 ± 1.27 g, 5.08 ± 1.11 g to 9.50 ± 1.25 g for 1 h, 6 h and 12 h after injection, respectively (Fig. 5C). Similarly, The PWL in the hind paw of rats with DIDS injection increased from the average level of 8.96 ± 0.54 s to 13.29 ± 0.97 s, 9.77 ± 0.35 s to 13.20 ± 0.96 s, and 9.53 ± 0.88 s to 13.30 ± 0.86 s for 1, 6, and 12 h after injection (Fig. 5D). To eliminate the effect of DIDS on motor function, the Rota-rod test was performed before and after intrathecal injection of DIDS (2 mg/kg body weight) into normal rats (Fig. 5E), and no alteration of motor function was found. These results suggest that DIDS ameliorates bone cancer-related pain behaviors.

Involvement of VDAC1 in the spinal dorsal horn of cancer pain rats. A Intrathecal application of DIDS dramatically ameliorated the PWT in dose- and time-dependent manners in BCP rats (P = 0.031 at 2 mg/kg, P = 0.020 at 8 mg/kg in the 1 h group; P = 0.005 at 2 mg/kg, P = 0.018 at 8 mg/kg in the 3 h group vs PRE; P = 0.025 at 1 h, P = 0.001 at 3 h in the 2 mg/kg group; P = 0.012 at 1 h, P = 0.004 at 3h in the 8 mg/kg group vs the corresponding NS group, 2-way repeated-measures ANOVA followed by Tukey’s post hoc test, n = 5, 6, 6, and 7 rats for NS, DIDS 0.5, 2 and 8 mg/kg, respectively). B Intrathecal application of DIDS also reversed the PWL in dose- and time-dependent manners in BCP rats (P = 0.041 at 2 mg/kg, P = 0.030 at 8 mg/kg in the 1 h group; P = 0.001 at 2 mg/kg, P = 0.012 at 8 mg/kg in the 3 h group vs PRE; P = 0.006 at 1 h, P = 0.009 at 3 h in the 2 mg/kg group; P = 0.003 at 1 h, P = 0.002 at 3 h in the 8 mg/kg group vs the corresponding NS group). C After 7 consecutive days of administration of 2 mg/kg DIDS, a long-term effect on mechanical PWT was found vs the NS-injected group (P = 0.007 at 1 h, P = 0.035 at 6 h, P = 0.030 at 12 h vs the corresponding NS group, n = 5 for NS and 6 for 2 mg/kg DIDS). D After 7 consecutive days of intrathecal injection of DIDS at 2 mg/kg, PWL tests were performed (P = 0.005 at 1 h, P = 0.013 at 6 h, P = 0.014 at 12 h vs the corresponding NS group, n = 5 for NS and 6 for 2 mg/kg DIDS). E Administration of DIDS did not have any effect on time on the Rota-rod test in healthy control rats.
Suppression of Toll-Like Receptor 4 Expression by Inhibition of VDAC1 in Bone Cancer Pain Rats
Since a previous study had shown that the protein level of TLR4 is markedly increased in the L2–L5 spinal dorsal horn of BCP rats at 2 weeks [23], we then determined whether inhibition of VDAC1 reduces TLR4 expression. The results showed that the inhibition of VDAC1 suppressed the expression of TLR4 (Fig. 6A). In contrast, DIDS treatment did not alter the expression of p65, P2X7 receptors and CBS in the dorsal horn (Fig. 6B–D).

Inhibition of VDAC1 reduced the expression of TLR4 in the L2–L5 spinal dorsal horn 14 days after tumor cell injection. A Inhibition of VDAC1 reversed the upregulation of TLR4 expression (P = 0.034 vs NS group, Student’s t test, n = 4 per group). B–D Inhibition of VDAC1 did not affect the expression of p65, P2X7 receptors and CBS (P = 0.965, 0.848 and 0.539 for p65, P2X7 receptors, and CBS vs NS, Student t test, n = 4 rats per group).
Effects of Minocycline on PWT and PWL in BCP Rats
Since upregulation of VDAC1 was found in microglia, we next aimed to determine whether microglia are involved in the maintenance of BCP. More activated microglia were expressed in the spinal dorsal horn in BCP rats than in control rats (Fig. 4A). We hypothesized that the inhibition of microglial activity may reverse the BCP behaviors. Mino, a selective inhibitor of microglia, was intrathecally injected into BCP rats. The same volume of NS was injected into the control group. Consistent with our hypothesis, intrathecal injection of Mino clearly ameliorated the BCP. The 50% PWT to VFF in the hind paw of rats with Mino-injection increased from the baseline level of 5.05 ± 1.01 g to 11.00 ± 1.60 g and 11.85 ± 1.14 g for 100 and 200 μL 0.5 h after injection, and from the baseline level of 5.25 ± 1.05 g to 9.85 ± 0.93 g and 11.14±1.03 g for 100 and 200 μL 1 h after injection (Fig. 7A). Similarly, we also tested the PWL to radiant heat stimulation. The PWL increased from the baseline level of 10.08 ± 0.58 s to 13.15 ± 1.00 s and 12.13 ± 0.59 s for 100 and 200 μL 0.5 h after injection (Fig. 7B).

Inhibition of spinal microglia attenuated bone cancer pain. A Intrathecal application of minocycline (Mino) dramatically ameliorated the PWT in dose- and time-dependent manners in BCP rats (P = 0.027 at 100 μL, P = 0.005 at 200 μL in the 0.5 h group; P = 0.034 at 100 μL, P = 0.002 at 200 μL in the 1 h group; P = 0.002 at 0.5 h, P = 0.023 at 1 h in the 100 μL group; P = 0.015 at 0.5 h, P = 0.024 at 1 h in the 200 μL group, 2-way repeated-measures ANOVA followed by Tukey’s post hoc test, n = 7 rats per group). B Intrathecal application of Mino also reversed the PWL in a time-dependent manner in BCP rats (P = 0.021 at 100 μL, P = 0.030 at 200 μL in the 0.5 h group, 2-way repeated-measures ANOVA followed by Tukey’s post hoc test, n = 7 rats per group). C After 7 consecutive days of administration of 100 μL Mino, a long-term effect on PWT was found compared with the NS-injected group (P = 0.001 at 1 h, P = 0.027 at 3 h, P = 0.045 at 6 h, 2-way repeated-measures ANOVA followed by Tukey’s post hoc test, n = 6 for NS and 7 for 100 μL Mino). D Seven consecutive days of administration of 100 μL Mino also reversed the PWL compared with the NS-injected group (P = 0.006 at 1 h, P = 0.028 at 3 h, 2-way repeated-measures ANOVA followed by Tukey’s post hoc test, n = 6 for NS and 7 for 100 μL Mino).
After 7 consecutive days of administration of 100 μL Mino, a long-term effect was determined. The PWT in the hind paw of rats with Mino injection increased from the average level of 3.56 ± 0.84 g to 10.28 ± 1.32 g, 5.00 ± 0.85 g to 9.00 ± 1.25 g, and 5.33 ± 0.98 g to 9.71 ± 1.58 g at 1, 3 and 6 h after injection (Fig. 7C). Similarly, PWL in the hind paw of rats with Mino injection increased from the average level of 9.66 ± 0.43 s to 13.18 ± 0.90 s, 11.11 ± 0.73 s to 13.91 ± 0.80 s at 1 and 3 h (Fig. 7D). These findings indicated that microglia participate in the development of pain hypersensitivity in a rat model of bone cancer.
Discussion
In the present study, we demonstrated for the first time that VDAC1 in the spinal dorsal horn is involved in the development of cancer-induced pain hypersensitivity. This conclusion was supported by the following results. First, injection of Walker 256 tumor cells into rat tibia resulted in the over-expression of VDAC1 in the spinal dorsal horn accompanied by prolonged mechanical allodynia and thermal hyperalgesia. Second, inhibition of VDAC1 by DIDS significantly attenuated the mechanical hyperalgesia in rats with BCP. Further investigation showed that VDAC1 was located in microglia, which were aberrantly activated in rats with bone cancer. Last, inhibition of microglial excitability by Mino or inhibition of VDAC1 relieved pain behaviors in rats with BCP. These findings suggested that VDAC1 plays an important role in cancer-induced pain hypersensitivity.
Located in the outer mitochondrial membrane, the VDAC functions in cellular Ca2+ homeostasis by mediating the transport of Ca2+ in and out of mitochondria. Since the intracellular Ca2+ concentration regulates a number of cellular and intercellular events, such as the cell cycle, proliferation, gene transcription, and cell death pathways, as well as processes like neuronal processing and transmission [24], the VDAC might be important in the process of chronic pain hypersensitivity. The VDAC family consists of 3 gene products (VDAC 1, 2, and 3). Of the three known isoforms, VDAC1 is the most abundantly expressed, and is thought to be closely involved in the mitochondrial response to cell stress. Besides, VDAC1 is closely associated with various cancer hallmarks, including proliferation, migration, angiogenesis, invasion, and resistance to cell death [25]. Therefore, it is necessary to study the roles of VDAC1 in complex diseases, especially cancer-induced pain. Previous studies have shown that VDAC1 is expressed in astrocytes in the central nervous system [26]. However, the expression level of VDAC1 varies under different pathophysiological conditions. VDAC1 expression is increased in the cerebellum of patients with Down’s syndrome [27]. In Alzheimer’s disease, however, the VDAC1 expression level is significantly decreased in frontal cortex [27]. In the present study, we showed that VDAC1 was significantly upregulated at both the protein and mRNA levels starting from 2 weeks after the inoculation of tumor cells. Surprisingly, VDAC1 was not expressed in neurons in the spinal dorsal horn. Further investigation demonstrated that VDAC1 was expressed not only in GFAP-labeled astrocytes but also in CD11-labeled microglia. This distinct distribution might represent a unique mechanism under cancer-induced pain conditions. Importantly, the inhibition of microglial activation by Mino or inhibition of VDAC1 relieved pain behaviors in rats with BCP, indicating that VDAC1 regulates the activation of microglia in the spinal dorsal horn. Astrocytes, together with microglia, are the primary mediators of “reactive gliosis”, the reactive response of glia to different CNS insults [28]. In the present study, VDAC1 was also expressed in astrocytes of the spinal dorsal horn. It is not clear whether spinal astrocytes are involved in the development of BCP. The role of VDAC1 in spinal astrocytes needs to be further investigated under cancer pain conditions. Thus, further understanding of the roles of VDAC1 in the central nervous system might provide new opportunities to discover pathophysiological mechanisms for cancer pain hypersensitivity for future therapeutic strategies.
Accumulating evidence suggests that VDAC1 interacts with PARK2 and is involved in its recruitment and function during mitochondrial autophagy [29]. However, a functional relationship between VDAC1 and TLR4 has not been reported. In the present study, we showed that TLR4 might be one of the downstream targets for VDAC1 under cancer pain conditions. To the best of our knowledge, this is the first report demonstrating that the inhibition of VDAC1 not only attenuated pain hypersensitivity but also reduced the expression of TLR4 in the spinal dorsal horn of BCP rats. As reported previously, TLR4 is involved in the formation and development of pain hypersensitivity, such as inflammatory pain [30], visceral pain [31], and also a BCP model [21], and is important for the pathogenesis of inflammatory reactions and the promotion of pain processing in the spinal cord [32]. Therefore, it is important to investigate the molecular mechanisms underlying the signaling pathway of VDAC1-mediated suppression of TLR4 under cancer-induced pain conditions. Another important question that needs to be addressed is how VDAC1 expression is regulated. The accumulation of oxidative damage in mitochondrial proteins, membranes, and DNA under pathophysiological states is thought to lead to mitochondrial inactivation, downstream molecular impairments, and consequent decline of biological systems [33]. Previous reports have shown that VDAC-1 is a PARP-1 substrate [34]. It is of interest to further investigate the upstream mechanisms by which VDAC1 is upregulated under cancer pain conditions. Notably, it would be better to use heat-killed cancer cells rather than simply NS as a control as described previously [35, 36].
Despite the controversy about the role of VDACs in the mitochondrial permeability transition, the importance of VDACs for therapeutic strategies in human cancer, neurodegeneration, and diabetes has recently increased. This and future studies might increase the interest on VDAC1 as an important molecular target for the treatment of the mechanical allodynia of cancer pain.
References
De Felice M, Lambert D, Holen I, Escott KJ, Andrew D. Effects of Src-kinase inhibition in cancer-induced bone pain. Mol Pain 2016, 12.
Slosky LM, BassiriRad NM, Symons AM, Thompson M, Doyle T, Forte BL, et al. The cystine/glutamate antiporter system xc- drives breast tumor cell glutamate release and cancer-induced bone pain. Pain 2016, 157: 2605–2616.
Shi FD. Neuroinflammation. Neurosci Bull 2015, 31: 714–716.
Zhu YF, Ungard R, Seidlitz E, Zacal N, Huizinga J, Henry JL, et al. Differences in electrophysiological properties of functionally identified nociceptive sensory neurons in an animal model of cancer-induced bone pain. Mol Pain 2016, 12.
Colvin L, Fallon M. Challenges in cancer pain management–bone pain. Eur J Cancer 2008, 44: 1083–1090.
Clohisy DR, Mantyh PW. Bone cancer pain. Clin Orthop Relat Res 2003: S279–288.
Brahimi-Horn MC, Giuliano S, Saland E, Lacas-Gervais S, Sheiko T, Pelletier J, et al. Knockout of Vdac1 activates hypoxia-inducible factor through reactive oxygen species generation and induces tumor growth by promoting metabolic reprogramming and inflammation. Cancer Metab 2015, 3: 8.
Shoshan-Barmatz V, Mizrachi D. VDAC1: from structure to cancer therapy. Front Oncol 2012, 2: 164.
Messina A, Reina S, Guarino F, De Pinto V. VDAC isoforms in mammals. Biochim Biophys Acta 2012, 1818: 1466–1476.
Shoshan-Barmatz V, Krelin Y, Shteinfer-Kuzmine A, Arif T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front Oncol 2017, 7: 154.
Brahimi-Horn MC, Giuliano S, Saland E, Lacas-Gervais S, Sheiko T, Pelletier J, et al. Knockout of Vdac1 activates hypoxia-inducible factor through reactive oxygen species generation and induces tumor growth by promoting metabolic reprogramming and inflammation. Cancer Metab 2015, 3: 1–18.
Janes K, Doyle T, Bryant L, Esposito E, Cuzzocrea S, Ryerse J, et al. Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy-induced neuropathic pain resulting from peroxynitrite-mediated post-translational nitration of mitochondrial superoxide dismutase. Pain 2013, 154: 2432–2440.
Li C, Yan Y, Cheng J, Gang X, Gu J, Zhang L, et al. Toll-like receptor 4 deficiency causes reduced exploratory behavior in mice under approach-avoidance conflict. Neurosci Bull 2016, 32: 127.
Lan LS, Ping YJ, Na WL, Miao J, Cheng QQ, Ni MZ, et al. Down-regulation of Toll-like receptor 4 gene expression by short interfering RNA attenuates bone cancer pain in a rat model. Mol Pain 2010, 6: 2.
De Leo JA, Tawfik VL, LaCroix-Fralish ML. The tetrapartite synapse: path to CNS sensitization and chronic pain. Pain 2006, 122: 17–21.
Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int 2004, 45: 397–407.
Bettoni I, Comelli F, Rossini C, Granucci F, Giagnoni G, Peri F, et al. Glial TLR4 receptor as new target to treat neuropathic pain: efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia 2008, 56: 1312–1319.
Pan R, Di H, Zhang J, Huang Z, Sun Y, Yu W, et al. Inducible lentivirus-mediated siRNA against TLR4 reduces nociception in a rat model of bone cancer pain. Mediators Inflamm 2015, 2015: 523896.
Zhou YL, Jiang GQ, Wei J, Zhang HH, Chen W, Zhu H, et al. Enhanced binding capability of nuclear factor-kappaB with demethylated P2X3 receptor gene contributes to cancer pain in rats. Pain 2015, 156: 1892–1905.
Fan HB, Zhang T, Sun K, Song SP, Cao SB, Zhang HL, et al. Corticotropin-releasing factor mediates bone cancer induced pain through neuronal activation in rat spinal cord. Tumour Biol 2015, 36: 9559–9565.
Wei J, Li M, Wang D, Zhu H, Kong X, Wang S, et al. Overexpression of suppressor of cytokine signaling 3 in dorsal root ganglion attenuates cancer-induced pain in rats. Mol Pain 2017, 13: 1744806916688901.
Pan HL, Liu BL, Lin W, Zhang YQ. Modulation of Nav1.8 by lysophosphatidic acid in the induction of bone cancer pain. Neurosci Bull 2016, 32: 445–454.
Jin XH, Wang LN, Zuo JL, Yang JP, Liu SL. P2X4 receptor in the dorsal horn partially contributes to brain-derived neurotrophic factor oversecretion and toll-like receptor-4 receptor activation associated with bone cancer pain. J Neurosci Res 2014, 92: 1690–1702.
Shoshan-Barmatz V, De S, Meir A. The mitochondrial voltage-dependent anion channel 1, Ca2+ transport, apoptosis, and their regulation. Front Oncol 2017, 7: 60.
Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 2008, 8: 361–375.
Benesova J, Rusnakova V, Honsa P, Pivonkova H, Dzamba D, Kubista M, et al. Distinct expression/function of potassium and chloride channels contributes to the diverse volume regulation in cortical astrocytes of GFAP/EGFP mice. PLoS One 2012, 7: e29725.
Rosa JC, De CCM. Role of hexokinase and VDAC in neurological disorders. Current Mol Pharmacol 2016, 9: 320.
Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett 2014, 565: 30.
Gatliff J, East D, Crosby J, Abeti R, Harvey R, Craigen W, et al. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10: 2279–2296.
Barcelos RP, Bresciani G, Cuevas MJ, Martinez-Florez S, Soares FA, Gonzalez-Gallego J. Diclofenac pretreatment modulates exercise-induced inflammation in skeletal muscle of rats through the TLR4/NF-kappaB pathway. Appl Physiol Nutr Metab 2017, 42: 757–764.
Tramullas M, Finger BC, Dinan TG, Cryan JF. Obesity takes its toll on visceral pain: high-fat diet induces toll-like receptor 4-dependent visceral hypersensitivity. PLoS One 2016, 11: e0155367.
Li XQ, Zhang ZL, Tan WF, Sun XJ, Ma H. Down-regulation of CXCL12/CXCR4 expression alleviates ischemia-reperfusion-induced inflammatory pain via inhibiting glial TLR4 activation in the spinal cord. PLoS One 2016, 11: e0163807.
Bai L, Wang X, Li Z, Kong C, Zhao Y, Qian JL, et al. Upregulation of chemokine CXCL12 in the dorsal root ganglia and spinal cord contributes to the development and maintenance of neuropathic pain following spared nerve injury in rats. Neurosci Bull 2016, 32: 27–40.
Baek SH, Bae ON, Kim EK, Yu SW. Induction of mitochondrial dysfunction by poly(ADP-ribose) polymer: implication for neuronal cell death. Mol Cells 2013, 36: 258–266.
Tong Z, Luo W, Wang Y, Yang F, Han Y, Li H, et al. Tumor tissue-derived formaldehyde and acidic microenvironment synergistically induce bone cancer pain. PLoS One 2010, 5: e10234.
Li Y, Cai J, Han Y, Xiao X, Meng XL, Su L, et al. Enhanced function of TRPV1 via up-regulation by insulin-like growth factor-1 in a rat model of bone cancer pain. European J Pain 2014, 18: 774–784.
Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (2016YFC1302200), the National Natural Science Foundation of China (31730040, 81070884, and 81471137), the Suzhou Health Planning Commission’s Key Clinical Diagnosis and Treatment Program (LCZX201606) and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China. This project is subject to the Preponderant Clinic Discipline Group Project funding from the Second Affiliated Hospital of Soochow University (XKQ2015008).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Kong, X., Wei, J., Wang, D. et al. Upregulation of Spinal Voltage-Dependent Anion Channel 1 Contributes to Bone Cancer Pain Hypersensitivity in Rats. Neurosci. Bull. 33, 711–721 (2017). https://doi.org/10.1007/s12264-017-0195-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-017-0195-1