
Abstract
Acute promyelocytic leukemia (APL) is associated with a high incidence of early death, which occurs within 30 days of diagnosis. The major cause of early death in APL is severe bleeding, particularly intracranial bleeding. Although APL is known to be associated with activation of coagulation, hyperfibrinolysis, and thrombocytopenia, the precise mechanisms that cause bleeding have not yet been elucidated. I propose that a combination of four pathways may contribute to bleeding in APL: (1) tissue factor, (2) the urokinase plasminogen activator/urokinase plasminogen activator receptor, (3) the annexin A2/S100A100/tissue plasminogen activator, and (4) the podoplanin/C-type lectin-like receptor 2. A better understanding of these pathways will identify new biomarkers to determine which APL patients are at high risk of bleeding and allow the development of new treatments for APL-associated bleeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
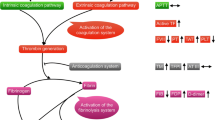
Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia (AML) with an annual incidence of 600–800 cases in the United States [1]. It is characterized by a translocation between chromosomes 15 and 17 that leads to fusion of promyelocytic leukemia (PML) gene with the retinoic acid receptor-α (RARα) gene to create a PML-RARα gene. This fusion gene is detected in approximately 90% of APL patients [2]. PML-RARα inhibits myeloid differentiation at the promyelocytic stage of myelopoiesis [3]. The development of differentiation therapies, such as all-trans retinoic acid (ATRA) and arsenic trioxide (ATO), has significantly improved prognosis of APL patients, and APL is currently considered to be a curable type of cancer. However, early death within 30 days of diagnosis remains a major problem, with rates of 4–11% in clinical trials [4,5,6,7,8,9,10,11] and 17–26% in registry studies [12,13,14,15,16,17]. Most early deaths are caused by severe bleeding, particularly intracranial bleeding [13, 15, 17]. Notably, nearly 90% of APL patients demonstrate some evidence of bleeding at presentation [18]. Consistent with the high incidence of bleeding, APL patients also exhibit higher rates of disseminated intravascular coagulation (DIC, 60–85%) [19,20,21] than other types of acute leukemia (6–22%) [22,23,24,25,26,27]. The high incidence of bleeding and DIC in APL, as compared to other hematologic malignancies, suggests that APL causes a unique disruption of the hemostatic system. However, the mechanisms that cause bleeding in APL have not been elucidated yet. APL is known to be associated with activation of coagulation, hyperfibrinolysis, and thrombocytopenia [28, 29]. The aim of this review is to summarize the current knowledge of pathways that lead to activation of coagulation, hyperfibrinolysis, and thrombocytopenia. This review will focus on tissue factor (TF) and the activation of coagulation, the urokinase plasminogen activator (uPA)/uPA receptor (uPAR) and the annexin A2 (AA2)/S100A10 (S100)/ tissue plasminogen activator (tPA) pathways and hyperfibrinolysis, and the podoplanin (PDPN)/C-type lectin-like receptor 2 (CLEC-2) pathway and thrombocytopenia (Fig. 1).

Proposed pathways to bleeding in acute promyelocytic leukemia. Acute promyelocytic leukemia (APL) cells express tissue factor (TF), urokinase plasminogen activator (uPA), uPA receptor (uPAR), annexin A2 (AA2), S100A10 (S100) and podoplanin (PDPN). APL cell-derived and monocyte cell-derived TF activate coagulation, followed by consumption of coagulation factors that leads to bleeding. The uPA/uPAR complex and the AA2/S100/tissue plasminogen activator (tPA) complex generate plasmin that cause aberrant degradation of fibrin resulting in bleeding. APL cell PDPN binds to C-type lectin-like receptor 2 (CLEC-2) on platelets, causes activation and consumption of platelets, and contributes to bleeding. The figure was created with Biorender.com
TF
TF is a transmembrane receptor for FVII/VIIa. The TF/FVIIa complex initiates the coagulation cascade [30]. TF is also released in the form of extracellular vesicles (EVs) from either cancer cells or host cells, such as activated monocytes [31].
TF in APL patients
APL patients have high levels of thrombin–antithrombin complex (TAT), which is a biomarker of activation of coagulation [32, 33]. TF has been proposed as an initiator of coagulation in APL patients. Indeed, high levels of TF were reported in mononuclear cells from bone marrow and peripheral blood of APL patients [34, 35]. Interestingly, peripheral blood mononuclear cells (PBMC) from APL/AML patients with DIC had higher levels of TF compared to PBMC from APL/AML patients without DIC [35]. These data suggest that TF is associated with DIC in APL and AML patients. Studies also showed that ATRA treatment significantly reduces TF mRNA and protein expression in bone marrow cells from APL patients [33, 34]. These data suggest that TF expression in APL cells is reduced after differentiation therapy.
We and others found high levels of EVTF activity in plasma from APL patients [36,37,38,39]. Two studies reported that APL patients with DIC had high levels of EVTF activity that were reduced after the resolution of DIC [36, 37]. These data suggest that EVTF activity is associated with DIC in APL patients. Our study included 29 APL, 253 non-APL AML and 76 acute lymphoblastic leukemia (ALL) patients. There were a total of 41 major bleeding and clinically relevant non-major bleeding cases with 7 in APL, 31 in non-APL AML, 3 in ALL patients. We found that high levels of EVTF activity were associated with bleeding in these acute leukemia patients [hazard ratio (HR): 2.32, 95% confidence interval (95% CI1.08–4.99) in an univariable model and HR: 2.33 (95% CI 1.08–5.04) in a multivariable model adjusted for age, sex, race/ethnicity] [39]. Our interpretation of this data is that TF-positive (+) EVs activate coagulation, followed by consumption of coagulation factors that leads to bleeding in these patients. Since the numbers of APL patients and bleeding cases were small in this study, we have not performed a sub-analysis for APL patients. Future studies need to investigate the association between EVTF activity and bleeding in a larger cohort of patients with APL.
TF in preclinical studies
We studied the role of TF using mouse xenograft and allograft models of APL [40]. We used a human APL cell line called NB4 to establish the xenograft model. We and others found that NB4 cells express high levels of TF [40,41,42]. To establish the allograft model, we injected splenocytes containing APL cells from a transgenic mouse that develops spontaneous APL [43] into C57BL6 mice. Both mouse models exhibited activation of coagulation, hyperfibrinolysis, thrombocytopenia and bleeding phenotype similar to APL patients.
To investigate the role of TF in coagulopathy and bleeding in APL mice, we inhibited TF in the 2 mouse models. We used an anti-human TF antibody (HTF-1) to inhibit APL cell-derived TF in the xenograft model. HTF-1 does not inhibit host-derived mouse TF. We used an anti-mouse TF antibody (1H1) to inhibit both APL cell-derived and host cell-derived TF in the allograft model. HTF-1 and 1H1 significantly reduced levels of TAT in their respective models. These data indicate that TF contributes to activation of coagulation in mouse models of APL. Strikingly, HTF-1 significantly shortened the tail bleeding time in the xenograft model. These data indicate that expression of TF by APL cells leads to bleeding in the xenograft model, presumably by inducing consumption of coagulation factors. Our study is the first to investigate the role of TF in APL using mouse models. In future studies we will determine the role of host cell-derived TF in APL using these mouse models.
The uPA/uPAR pathway
uPA is a serine protease that binds to its receptor uPAR to activate plasminogen to plasmin, which degrades fibrin [44].
The uPA/uPAR pathway in APL patients
We and others reported that APL patients have high levels of plasmin–antiplasmin complex (PAP), a biomarker of activation of fibrinolysis [39, 45]. One of the pathways considered to be involved in hyperactivation of fibrinolysis in APL is the uPA/uPAR pathway. An early study found high uPAR protein expression on PBMC from patients with APL [46]. More recently, RNAseq analysis revealed that both uPA and uPAR mRNA expression were higher in blast cells from APL patients compared to blast cells from non-APL AML patients [47]. Conflicting results on the effect of ATRA on uPA and uPAR expression in NB4 cells have been reported. Some studies showed that ATRA transiently increases uPA activity and uPAR mRNA and protein expression in NB4 cells [48, 49] whereas other studies showed that ATRA reduces uPAR mRNA expression in NB4 cells [50, 51]. The effect of ATRA on uPA and uPAR expression should be further investigated using blast cells from APL patients.
High plasma levels of uPA [33, 52] and soluble uPAR [45] were reported in APL patients. However, to date, there are no studies that investigated associations between plasma uPA or uPAR levels and DIC or bleeding in APL patients. Future studies should investigate these associations.
The AA2/S100/tPA pathway
AA2 is a calcium- and anionic phospholipid-binding protein that forms a heterotetrameric complex with S100 [53]. The AA2/S100 complex serves as a receptor for both tPA and plasminogen and facilitates the generation of plasmin [54].
The AA2/S100/tPA pathway in APL patients
The AA2/S100/tPA pathway may be a more specific fibrinolytic pathway for APL patients than the uPA/uPAR pathway. Mennel and colleagues firstly reported that AA2 protein expression in the leukocytes fraction from APL patients is higher than the leukocytes fraction from patients with other types of acute leukemia [55]. These data were later confirmed with RNAseq analysis showing that blast cells from APL patients had higher levels of AA2 mRNA expression compared to blast cells from AML patients [47]. Mennel and colleagues showed that NB4 cells, which express high levels of AA2, exhibit high plasmin generation in the presence of tPA. This high plasmin generation activity was significantly reduced when either NB4 or tPA were absent, or when an anti-AA2 antibody was added [55]. Another study also found that NB4 cells express S100 and shRNA suppression of S100 mRNA expression resulted in a reduction of plasmin generation in NB4 cells [56]. These data indicate that the AA2/S100/tPA pathway contributes to plasmin generation by NB4 cells. Interestingly, studies found that differentiation therapies significantly reduced AA2 mRNA and protein expression and S100 mRNA expression in mononuclear cells from bone marrow and peripheral blood from patients with APL [57, 58]. Similarly, differentiation therapies reduced AA2 mRNA and protein expression and S100 protein expression in NB4 cells [55, 56]. One study found that ATRA induces ubiquitin-independent proteasomal degradation of S100 protein in NB4 cells [59]. These data suggest that differentiation therapies reduce AA2 and S100 expression in APL cells. Further studies are needed to investigate if the AA2/S100/tPA pathway is associated with DIC and bleeding in APL patients.
The AA2/S100/tPA pathway in preclinical studies
One study found that inhibition of AA2 activity by treatment with L-methionine, which increases homocysteine that competes with tPA for the same binding site of AA2, decreased plasma levels of tPA in transgenic mice that spontaneously develop APL [60]. Future studies need to investigate the role of the AA2/S100/tPA pathway in hyperfibrinolysis and bleeding in mouse models of APL.
The PDPN/CLEC-2 pathway
PDPN is a transmembrane glycoprotein expressed in the kidney, liver and lymph nodes as well as cancer cells. It binds to CLEC-2 on platelets and induces aggregation [61].
The PDPN/CLEC-2 pathway in APL patients
Many papers have documented that patients with APL exhibit thrombocytopenia [62,63,64]. However, there are conflicting results regarding the association between a low platelet count and early death in APL patients. One study reported that a low platelet count was associated with early death in APL patients [14], while another study reported that a low platelet count were not associated with early death in APL patients [65]. Many studies have reported that a low platelet count was associated with bleeding in patients with APL [65,66,67,68,69]. However, three studies reported that a low platelet count was not associated with hemorrhagic death in patients with APL [70,71,72]. These data suggest that a low platelet count was associated with bleeding but not hemorrhagic death.
Bone marrow infiltration of blast cells and infection may contribute to thrombocytopenia in patients with APL [64]. A recent study also found that blast cells from patients with APL express higher levels of PDPN mRNA compared to blast cells from non-APL AML patients [47]. Interestingly, ATRA strikingly reduced PDPN mRNA expression, whereas ATO modestly reduced PDPN mRNA expression in primary APL cells from patients APL [47]. More studies are needed to determine the role of the PDPN/CLEC-2 pathway in thrombocytopenia and bleeding in APL patients.
The PDPN/CLEC-2 pathway in preclinical studies
A recent study investigated the role of PDPN in thrombocytopenia and bleeding in a xenograft mouse model [47]. The investigators overexpressed PDPN in the human AML cell line called OCI-AML5 that does not express PDPN. NSG mice bearing PDPN expressing OCI-AML5 cells exhibited a lower platelet count and prolonged tail bleeding time compared to NSG mice bearing wildtype OCI-AML5 cells [47]. These data indicate that PDPN by AML cells induces thrombocytopenia and bleeding phenotype in this model. However, this study has some limitations. First, it employed immunodeficient mice that lack interaction between platelets and immune cells. Second, AML cells were used instead of APL cells. Third, PDPN was overexpressed in a human AML cell line, which is highly artificial and may not represent the pathological level of PDPN in APL. Future studies need to investigate the role of the PDPN/CLEC-2 pathway in thrombocytopenia and bleeding in immunocompetent mice using APL cells that express PDPN.
Conclusion
I propose that the TF, the uPA/uPAR, the AA2/S100/tPA, and the PDPN/CLEC-2 pathways contribute to bleeding in APL patients. However, to date, no studies have evaluated the association between these pathways and bleeding in patients with APL. Mouse models can be used to investigate the mechanisms of coagulopathy and bleeding in APL. However, mouse models have their limitations because no single mouse model mimics all characteristics observed in patients with APL. Further studies are needed to understand the mechanisms of APL-associated coagulopathy and bleeding in patients with APL. A better understanding of these mechanisms will develop new treatments to prevent APL-associated bleeding and death in patients with APL.
Data availability
No new data were created in this article.
References
Chen Y, Kantarjian H, Wang H, Cortes J, Ravandi F. Acute promyelocytic leukemia: a population-based study on incidence and survival in the United States, 1975–2008. Cancer. 2012;118(23):5811–8.
Yilmaz M, Kantarjian H, Ravandi F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J. 2021;11(6):123.
Grignani F, Ferrucci PF, Testa U, Talamo G, Fagioli M, Alcalay M, et al. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell. 1993;74(3):423–31.
Fenaux P, Le Deley MC, Castaigne S, Archimbaud E, Chomienne C, Link H, et al. Effect of all transretinoic acid in newly diagnosed acute promyelocytic leukemia. Results of a multicenter randomized trial. European APL 91 Group. Blood. 1993;82(11):3241–9.
Asou N, Adachi K, Tamura J, Kanamaru A, Kageyama S, Hiraoka A, et al. All-trans retinoic acid therapy for newly diagnosed acute promyelocytic leukemia: comparison with intensive chemotherapy. The Japan Adult Leukemia Study Group (JALSG). Cancer Chemother Pharmacol. 1997;40:30–5.
Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337(15):1021–8.
Fenaux P, Chastang C, Chevret S, Sanz M, Dombret H, Archimbaud E, et al. A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group Blood. 1999;94(4):1192–200.
Lengfelder E, Haferlach C, Saussele S, Haferlach T, Schultheis B, Schnittger S, et al. High dose ara-C in the treatment of newly diagnosed acute promyelocytic leukemia: long-term results of the German AMLCG. Leukemia. 2009;23(12):2248–58.
Asou N, Kishimoto Y, Kiyoi H, Okada M, Kawai Y, Tsuzuki M, et al. A randomized study with or without intensified maintenance chemotherapy in patients with acute promyelocytic leukemia who have become negative for PML-RARalpha transcript after consolidation therapy: the Japan Adult Leukemia Study Group (JALSG) APL97 study. Blood. 2007;110(1):59–66.
Avvisati G, Lo-Coco F, Paoloni FP, Petti MC, Diverio D, Vignetti M, et al. AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood. 2011;117(18):4716–25.
Burnett AK, Russell NH, Hills RK, Bowen D, Kell J, Knapper S, et al. Arsenic trioxide and all-trans retinoic acid treatment for acute promyelocytic leukaemia in all risk groups (AML17): results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2015;16(13):1295–305.
Park JH, Qiao B, Panageas KS, Schymura MJ, Jurcic JG, Rosenblat TL, et al. Early death rate in acute promyelocytic leukemia remains high despite all-trans retinoic acid. Blood. 2011;118(5):1248–54.
McClellan JS, Kohrt HE, Coutre S, Gotlib JR, Majeti R, Alizadeh AA, et al. Treatment advances have not improved the early death rate in acute promyelocytic leukemia. Haematologica. 2012;97(1):133–6.
Lehmann S, Ravn A, Carlsson L, Antunovic P, Deneberg S, Mollgard L, et al. Continuing high early death rate in acute promyelocytic leukemia: a population-based report from the Swedish Adult Acute Leukemia Registry. Leukemia. 2011;25(7):1128–34.
Lehmann S, Deneberg S, Antunovic P, Rangert-Derolf A, Garelius H, Lazarevic V, et al. Early death rates remain high in high-risk APL: update from the Swedish Acute Leukemia Registry 1997–2013. Leukemia. 2017;31(6):1457–9.
Xu F, Wang C, Yin C, Jiang X, Jiang L, Wang Z, et al. Analysis of early death in newly diagnosed acute promyelocytic leukemia patients. Medicine. 2017;96(51): e9324.
Ho G, Li Q, Brunson A, Jonas BA, Wun T, Keegan THM. Complications and early mortality in patients with acute promyelocytic leukemia treated in California. Am J Hematol. 2018;93(11):E370–2.
Abedin S, Altman JK. Acute promyelocytic leukemia: preventing early complications and late toxicities. Hematology Am Soc Hematol Educ Program. 2016;2016(1):10–5.
Hoyle CF, Swirsky DM, Freedman L, Hayhoe FG. Beneficial effect of heparin in the management of patients with APL. Br J Haematol. 1988;68(3):283–9.
Stone RM, Maguire M, Goldberg MA, Antin JH, Rosenthal DS, Mayer RJ. Complete remission in acute promyelocytic leukemia despite persistence of abnormal bone marrow promyelocytes during induction therapy: experience in 34 patients. Blood. 1988;71(3):690–6.
Chang H, Kuo MC, Shih LY, Dunn P, Wang PN, Wu JH, et al. Clinical bleeding events and laboratory coagulation profiles in acute promyelocytic leukemia. Eur J Haematol. 2012;88(4):321–8.
Higuchi T, Toyama D, Hirota Y, Isoyama K, Mori H, Niikura H, et al. Disseminated intravascular coagulation complicating acute lymphoblastic leukemia: a study of childhood and adult cases. Leuk Lymphoma. 2005;46(8):1169–76.
Dixit A, Chatterjee T, Mishra P, Kannan M, Choudhry DR, Mahapatra M, et al. Disseminated intravascular coagulation in acute leukemia at presentation and during induction therapy. Clin Appl Thromb/Hemost Off J Int Acad Clin Appl Thromb/Hemost. 2007;13(3):292–8.
Uchiumi H, Matsushima T, Yamane A, Doki N, Irisawa H, Saitoh T, et al. Prevalence and clinical characteristics of acute myeloid leukemia associated with disseminated intravascular coagulation. Int J Hematol. 2007;86(2):137–42.
Libourel EJ, Klerk CPW, van Norden Y, de Maat MPM, Kruip MJ, Sonneveld P, et al. Disseminated intravascular coagulation at diagnosis is a strong predictor for thrombosis in acute myeloid leukemia. Blood. 2016;128(14):1854–61.
Sletnes KE, Godal HC, Wisloff F. Disseminated intravascular coagulation (DIC) in adult patients with acute leukaemia. Eur J Haematol. 1995;54(1):34–8.
Nur S, Anwar M, Saleem M, Ahmad PA. Disseminated intravascular coagulation in acute leukaemias at first diagnosis. Eur J Haematol. 1995;55(2):78–82.
Kwaan HC. The unique hemostatic dysfunction in acute promyelocytic leukemia. Semin Thromb Hemost. 2014;40(3):332–6.
Ikezoe T. Pathogenesis of disseminated intravascular coagulation in patients with acute promyelocytic leukemia, and its treatment using recombinant human soluble thrombomodulin. Int J Hematol. 2014;100(1):27–37.
Grover SP, Mackman N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler Thromb Vasc Biol. 2018;38(4):709–25.
Hisada Y, Sachetto ATA, Mackman N (2022) Circulating tissue factor-positive extracellular vesicles and their association with thrombosis in different diseases. Immunol Rev. 2022;312(1):61-75.
Dombret H, Scrobohaci ML, Ghorra P, Zini JM, Daniel MT, Castaigne S, et al. Coagulation disorders associated with acute promyelocytic leukemia: corrective effect of all-trans retinoic acid treatment. Leukemia. 1993;7(1):2–9.
Tallman MS, Lefebvre P, Baine RM, Shoji M, Cohen I, Green D, et al. Effects of all-trans retinoic acid or chemotherapy on the molecular regulation of systemic blood coagulation and fibrinolysis in patients with acute promyelocytic leukemia. Journal of thrombosis and haemostasis : JTH. 2004;2(8):1341–50.
Falanga A, Iacoviello L, Evangelista V, Belotti D, Consonni R, D’Orazio A, et al. Loss of blast cell procoagulant activity and improvement of hemostatic variables in patients with acute promyelocytic leukemia administered all-trans-retinoic acid. Blood. 1995;86(3):1072–81.
Dicke C, Amirkhosravi A, Spath B, Jimenez-Alcazar M, Fuchs T, Davila M, et al. Tissue factor-dependent and -independent pathways of systemic coagulation activation in acute myeloid leukemia: a single-center cohort study. Exp Hematol Oncol. 2015;4:22.
Langer F, Spath B, Haubold K, Holstein K, Marx G, Wierecky J, et al. Tissue factor procoagulant activity of plasma microparticles in patients with cancer-associated disseminated intravascular coagulation. Ann Hematol. 2008;87(6):451–7.
Thaler J, Pabinger I, Sperr WR, Ay C. Clinical evidence for a link between microparticle-associated tissue factor activity and overt disseminated intravascular coagulation in patients with acute myelocytic leukemia. Thromb Res. 2014;133(3):303–5.
Zhao H, Sun J, Yan L, Jin B, Hou W, Cao F, et al. Tissue factor-bearing microparticles are a link between acute promyelocytic leukemia cells and coagulation activation: a human subject study. Ann Hematol. 2021;100(6):1473–83.
Hisada Y, Archibald SJ, Bansal K, Chen Y, Dai C, Dwarampudi S, et al. Biomarkers of bleeding and venous thromboembolism in patients with acute leukemia. medRxiv. 2023. https://doi.org/10.1101/2023.10.18.23297216.
Hisada Y, Kawano T, Archibald SJ, Welch JS, Reeves BN, Mackman N. Tissue factor activates the coagulation cascade in mouse models of acute promyelocytic leukemia. Blood Adv. 2023;7(18):5458–69.
Falanga A, Consonni R, Marchetti M, Locatelli G, Garattini E, Passerini CG, et al. Cancer procoagulant and tissue factor are differently modulated by all-trans-retinoic acid in acute promyelocytic leukemia cells. Blood. 1998;92(1):143–51.
Marchetti M, Diani E, ten Cate H, Falanga A. Characterization of the thrombin generation potential of leukemic and solid tumor cells by calibrated automated thrombography. Haematologica. 2012;97(8):1173–80.
Welch JS, Klco JM, Varghese N, Nagarajan R, Ley TJ. Rara haploinsufficiency modestly influences the phenotype of acute promyelocytic leukemia in mice. Blood. 2011;117(8):2460–8.
Mondino A, Blasi F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004;25(8):450–5.
Wang P, Zhang Y, Yang H, Hou W, Jin B, Hou J, et al. Characteristics of fibrinolytic disorders in acute promyelocytic leukemia. Hematology. 2018;23(10):756–64.
Mustjoki S, Alitalo R, Stephens RW, Vaheri A. Blast cell-surface and plasma soluble urokinase receptor in acute leukemia patients: relationship to classification and response to therapy. Thromb Haemost. 1999;81(5):705–10.
Lavallee VP, Chagraoui J, MacRae T, Marquis M, Bonnefoy A, Krosl J, et al. Transcriptomic landscape of acute promyelocytic leukemia reveals aberrant surface expression of the platelet aggregation agonist Podoplanin. Leukemia. 2018;32(6):1349–57.
Tapiovaara H, Matikainen S, Hurme M, Vaheri A. Induction of differentiation of promyelocytic NB4 cells by retinoic acid is associated with rapid increase in urokinase activity subsequently downregulated by production of inhibitors. Blood. 1994;83(7):1883–91.
Mustjoki S, Tapiovaara H, Siren V, Vaheri A. Interferons and retinoids enhance and dexamethasone suppresses urokinase-mediated plasminogen activation in promyelocytic leukemia cells. Leukemia. 1998;12(2):164–74.
Zheng PZ, Wang KK, Zhang QY, Huang QH, Du YZ, Zhang QH, et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia. Proc Natl Acad Sci USA. 2005;102(21):7653–8.
Saumet A, Vetter G, Bouttier M, Portales-Casamar E, Wasserman WW, Maurin T, et al. Transcriptional repression of microRNA genes by PML-RARA increases expression of key cancer proteins in acute promyelocytic leukemia. Blood. 2009;113(2):412–21.
Bennett B, Booth NA, Croll A, Dawson AA. The bleeding disorder in acute promyelocytic leukaemia: fibrinolysis due to u-PA rather than defibrination. Br J Haematol. 1989;71(4):511–7.
Bharadwaj A, Bydoun M, Holloway R, Waisman D. Annexin A2 heterotetramer: structure and function. Int J Mol Sci. 2013;14(3):6259–305.
Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015;29(1):17–24.
Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, Hajjar KA. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med. 1999;340(13):994–1004.
O’Connell PA, Madureira PA, Berman JN, Liwski RS, Waisman DM. Regulation of S100A10 by the PML-RAR-alpha oncoprotein. Blood. 2011;117(15):4095–105.
Liu Y, Wang Z, Jiang M, Dai L, Zhang W, Wu D, et al. The expression of annexin II and its role in the fibrinolytic activity in acute promyelocytic leukemia. Leuk Res. 2011;35(7):879–84.
Huang D, Yang Y, Sun J, Dong X, Wang J, Liu H, et al. Annexin A2–S100A10 heterotetramer is upregulated by PML/RARalpha fusion protein and promotes plasminogen-dependent fibrinolysis and matrix invasion in acute promyelocytic leukemia. Front Med. 2017;11(3):410–22.
Holloway RW, Thomas ML, Cohen AM, Bharadwaj AG, Rahman M, Marcato P, et al. Regulation of cell surface protease receptor S100A10 by retinoic acid therapy in acute promyelocytic leukemia (APL)(☆). Cell Death Dis. 2018;9(9):920.
Jacomo RH, Santana-Lemos BA, Lima AS, Assis PA, Lange AP, Figueiredo-Pontes LL, et al. Methionine-induced hyperhomocysteinemia reverts fibrinolytic pathway activation in a murine model of acute promyelocytic leukemia. Blood. 2012;120(1):207–13.
Suzuki-Inoue K. Platelets and cancer-associated thrombosis: focusing on the platelet activation receptor CLEC-2 and podoplanin. Blood. 2019;134(22):1912–8.
Jones ME, Saleem A. Acute promyelocytic leukemia. A review of literature. Am J Med. 1978;65(4):673–7.
Avvisati G, Lo Coco F, Mandelli F. Acute promyelocytic leukemia: clinical and morphologic features and prognostic factors. Semin Hematol. 2001;38(1):4–12.
Kuchenbauer F, Buske C. Revisiting thrombocytopenia in acute promyelocytic leukemia. Leukemia. 2018;32(6):1477–8.
Altman JK, Rademaker A, Cull E, Weitner BB, Ofran Y, Rosenblat TL, et al. Administration of ATRA to newly diagnosed patients with acute promyelocytic leukemia is delayed contributing to early hemorrhagic death. Leuk Res. 2013;37(9):1004–9.
Rodeghiero F, Avvisati G, Castaman G, Barbui T, Mandelli F. Early deaths and anti-hemorrhagic treatments in acute promyelocytic leukemia. A GIMEMA retrospective study in 268 consecutive patients. Blood. 1990;75(11):2112–7.
Song YH, Peng P, Qiao C, Zhang R, Li JY, Lu H. Low platelet count is potentially the most important contributor to severe bleeding in patients newly diagnosed with acute promyelocytic leukemia. Onco Targets Ther. 2017;10:4917–24.
Minamiguchi H, Fujita H, Atsuta Y, Asou N, Sakura T, Ueda Y, et al. Predictors of early death, serious hemorrhage, and differentiation syndrome in Japanese patients with acute promyelocytic leukemia. Ann Hematol. 2020;99(12):2787–800.
Kim DY, Lee JH, Lee JH, Kim SD, Lim SN, Choi Y, et al. Significance of fibrinogen, D-dimer, and LDH levels in predicting the risk of bleeding in patients with acute promyelocytic leukemia. Leuk Res. 2011;35(2):152–8.
Mitrovic M, Suvajdzic N, Bogdanovic A, Kurtovic NK, Sretenovic A, Elezovic I, et al. International Society of Thrombosis and Hemostasis Scoring System for disseminated intravascular coagulation >/= 6: a new predictor of hemorrhagic early death in acute promyelocytic leukemia. Med Oncol. 2013;30(1):478.
de la Serna J, Montesinos P, Vellenga E, Rayon C, Parody R, Leon A, et al. Causes and prognostic factors of remission induction failure in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and idarubicin. Blood. 2008;111(7):3395–402.
Mantha S, Goldman DA, Devlin SM, Lee JW, Zannino D, Collins M, et al. Determinants of fatal bleeding during induction therapy for acute promyelocytic leukemia in the ATRA era. Blood. 2017;129(13):1763–7.
Acknowledgements
The author would like to thank Dr. Nigel Mackman and Ms. Sierra J. Archibald for their constructive comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The author declares that he has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Hisada, Y. Dysregulated hemostasis in acute promyelocytic leukemia. Int J Hematol 119, 526–531 (2024). https://doi.org/10.1007/s12185-024-03708-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-024-03708-0