
Abstract
Inherited factor XIII (FXIII) deficiency is an extremely rare and under-diagnosed autosomal recessive inherited coagulopathy, which is caused by genetic defects in the F13A1 or F13B gene. More than 200 genetic mutations have been identified since the first case of inherited FXIII deficiency was reported. This study aimed to identify underlying gene mutations in a patient with inherited FXIII deficiency who presented with recurrent intracerebral hemorrhage. Levels of plasma FXIII-A antigen were measured, F13A1 and F13B genes were sequenced, mutation information was analyzed, and the mutated protein structure was predicted using bioinformatics methods. Molecular genetic analysis identified four mutations of FXIII-related genes in the proband, including three previously reported mutations inherited from his parents (c.631G>A, p.Gly210Arg and c.1687G>A, p.Gly562Arg of F13A1 gene and c.344G>A, p.Arg115His of F13B gene) and a novel spontaneous mutation of F13A1 gene (c.2063C>G, p.Ser687Cys). Molecular structural modeling demonstrated that the novel Ser687Cys mutation may cause changes in the spatial structure of FXIII-A and increase its instability. In conclusion, we identified a novel and likely pathogenic mutation of the F13A1 gene, which enriched the gene mutation spectrum of inherited FXIII deficiency. The findings may provide promising targets for diagnosis and treatment of inherited FXIII deficiency.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The plasma protransglutaminase, factor XIII (FXIII), is a heterotetramer composed of two A subunits (FXIII-A2) and two B subunits (FXIII-B2). The three-dimensional structure of the A subunit was determined by X-ray crystallography. The FXIII-A subunit includes five domains: the activation domain (residues 1–37), beta-sandwich (residues 38–184), catalytic core region (residues 185–515), beta-barrel 1 (residues 516–628), and beta-barrel 2 (residues 629–731) [1]. The crystal structure of FXIII-B is not available yet [2]. FXIII-B is composed of ten Sushi domains [3]. FXIII is the final factor involved in the blood coagulation cascade. With the help of calcium ions, it can be activated by thrombin to form active configuration (FXIIIa), which then cross-links the fibrin α-, γ-chains and fibrinolytic inhibitors (α2-plasmin inhibitor) to fibrin through covalent bonds to mechanically stabilize the fibrin clot and avoid fibrinolysis [4]. The role of FXIII is not just restricted to hemostasis; it is also involved in wound healing, tissue repair, angiogenesis, and pregnancy maintenance [5,6,7,8]. In recent years, many investigations have expanded the function of FXIII in the context of osteoblast differentiation, immunology, and adipogenesis [2].
FXIII deficiency can be inherited or acquired, and can lead to pathological bleeding episodes. Acquired FXIII deficiency is frequently caused by hyper-consumption, hypo-synthesis, or an immune-mediated process. Hyper-consumption is most common in surgery, disseminated intravascular coagulation, and pulmonary embolism. Hypo-synthesis can be seen in liver diseases, leukemia, and the administration of specific drugs (isoniazid, procainamide, and tocilizumab). Both hyper-consumption and hypo-synthesis are non-immune-mediated processes. Immune-mediated acquired FXIII deficiency is characterized by the presence of FXIII autoantibodies, mainly occurring secondary to autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis. It can also be idiopathic, and is more common in elderly individuals over 60 years of age [9, 10].
Inherited FXIII deficiency is an autosomal recessive inherited coagulopathy with an estimated prevalence of one in two million people, and the incidence is much higher in consanguineous marriages [11, 12] Almost all patients with FXIII deficiency will have a lifelong bleeding tendency [13].
A total of 235 mutations have been reported to cause inherited FXIII deficiency worldwide thus far. The complete listing of all mutations in the F13A1 and F13B genes can be found in the Human Gene Mutation Database Professional (HGMD Professional; https://www.hgmd.cf.ac.uk). The distribution of the known FXIII-related gene mutations reveals no hotspot region [14]. To further enrich the gene mutation spectrum, we performed molecular genetic analysis, laboratory examinations, and clinical phenotype study on a 25-year-old man suffering from recurrent spontaneous intracerebral hemorrhage (ICH) secondary to inherited FXIII deficiency.
Materials and methods
Patients
A 25-year-old man with inherited FXIII deficiency was recruited for this study. Basic laboratory evaluation and molecular genetic analysis of the patient and his parents were performed at Qilu Hospital of Shandong University and Shandong Hemophilia Treatment Center. The patient had no tumor, liver or kidney dysfunction, or severe infections. All participants provided informed consent to participate in the study, in accordance with the Declaration of Helsinki.
FXIII antigen assay and coagulation screen test
Peripheral blood samples were collected from the patient and his parents. Platelet-poor plasma was used to detect blood coagulation factors, and the blood cells were cryopreserved at − 80 °C for genomic DNA extraction. Coagulation indicators, including prothrombin time (PT), prothrombin time activity (PTA), international normalized ratio (INR), and activated partial thromboplastin time (APTT), were detected using an ACL TOP 300 CTS automatic blood coagulation analyzer (Werfen, Spain). FXIII-A antigen levels were measured by latex-enhanced immunoturbidimetry using a HemosIL kit (Werfen, Spain).
DNA isolation and PCR amplification
Genomic DNA was extracted from peripheral blood using a DNA extraction kit (TIANGEN, China), according to the manufacturer's instructions. Primers were designed for the sequencing of exons, 5′ untranslated region (5′ UTR), 3′ UTR, and flanking sequences of the F13A1 and F13B genes using PRIME 5.0 software (primer sequences are shown in Tables 1, 2). Amplification was performed using a Touchdown PCR program. The PCR conditions are shown in Fig. 1.

The amplification condition of Touchdown PCR was denaturation at 94 °C for 3 min, followed by 20 cycles of 30-s denaturation (94 °C), 30-s annealing (the annealing temperature was reduced by 0.5 °C each cycle until 55 °C was reached) and 40-s extension (72 °C). A final 10-min extension step finished the synthesis
Genomic analysis of F13A1 and F13B genes
The PCR products were purified and sequenced using the Sanger dideoxy chain-termination method on an ABI3730XL DNA Analyzer (Applied Biosystems, USA). DNA sequencing results were compared with the F13A1 and F13B reference sequences (NM_000129.4 and NM_001994.3) using the Blaster software to identify the mutation sites and confirmed by reverse sequencing.
Bioinformatics analysis
Single nucleotide polymorphisms (SNP) were queried using the SNP database (https://www.ncbi.nlm.nih.gov/snp/). Gene conservation analysis was performed using Clustal Omega (version 1.2.2). The prediction of the potential function of de novo mutations was evaluated using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and Provean (http://provean.jcvi.org). The mutated protein was modeled using Swiss-Pdb Viewer (version 4.1). AlphaFold (http://alphafold.ebi.ac.uk/), the deep learning algorithm developed by DeepMind, was also used to model the three-dimensional structure of mutated protein. We used the ranked_0.pdb file for further analysis, which contained the prediction result with the highest confidence. The results were visualized by PyMol (version2.5) software.
Results
Clinical presentation of the patient
A 25-year-old man who presented with a persistent unrelieved headache for 2 days accompanied by nausea and vomiting was admitted to the emergency department of our hospital. Routine blood test results and coagulation indicators were all within the normal ranges, as described below. Cranial computed tomography (CT) revealed a cerebral hemorrhage in the left temporal lobe (Fig. 2A). The patient was diagnosed with ICH. Surgery was ruled out after an assessment by neurosurgeons and he was prescribed symptomatic treatments, including hemostasis, lowering of intracranial pressure, and neurotrophic therapy. However, the patient did not exhibit any significant clinical improvement. Another CT examination four days later indicated an increase in the extent of bleeding (Fig. 2B). The patient's detailed medical history was acquired to reassess his condition. He had a history of repeated spontaneous bleeding and hematoma. The patient had undergone two craniotomies for hematoma removal due to ICH and one exploratory laparotomy for abdominal hematoma after abdominal trauma. He came from a non-consanguineous family and had no family history of pathological bleeding. Given that the patient had no abnormalities in routine coagulation tests but had significant bleeding symptoms, he was suspected to have a rare coagulation factor deficiency. A FXIII antigen assay confirmed this hypothesis. The patient was diagnosed with inherited FXIII deficiency and treated with cryoprecipitate infusion. After the infusion, he achieved rapid clinical improvement. The FXIII antigen level increased to 21.5% and a repeated CT examination showed a decreased extent of bleeding (Fig. 2C).

Cranial CT showed the extent of left temporal lobe hemorrhage (red arrow): A–C are the cranial CT images at day1, day 4 and day 19, respectively
Coagulation test results
Neither the patient nor his parents had any abnormal routine coagulation parameters. However, the patient had a significantly decreased FXIII-A antigen level at 2.4% (normal value 50–150%). His father had a mildly decreased FXIII-A antigen level (41%). Coagulation test data are presented in Table 3.
Molecular genetic analysis of F13A1 and F13B genes
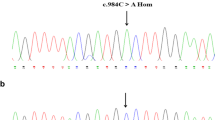
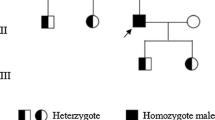
DNA sequencing revealed four missense mutations in the family: three in the F13A1 gene and one in the F13B gene (Table 4, Fig. 3 and Supplementary Table 1). The first mutation in exon 5 of the F13A1 gene, a c.631G>A replacement, resulted in a glycine to arginine substitution at position 210. This variant was present in the patient and his father in a heterozygous form. The second mutation was a G to A nucleotide exchange in exon 12 of the F13A1 gene, resulting in the substitution of glycine with arginine at position 562. This mutation originated in the patient’s mother. The third was a de novo mutation in exon 15 of the F13A1 gene, in which a C to G transition at nucleotide 2063 resulted in a serine-to-cysteine substitution at position 687. This mutation was not identified in his parents and has never been previously reported. The last mutation was located in exon 3 of the F13B gene, a G to A nucleotide change at position 344, causing an arginine to histidine substitution at amino acid position 115. The last mutation was present in a homozygous form in the patient and his parents.

Representative chromatograms from Sanger sequencing of the F13A1 and F13B genes in the pedigree. DNA sequencing of the proband revealed three heterozygous mutations in the F13A1 gene and one homozygous mutation in the F13B gene: A c.631 G>A, B c.1687 G>A, C c.2063 C>G, D c.344 G>A. The paternal and maternal sequencing maps at the corresponding locus are also shown in E–L. Red arrows indicate the mutant peaks. †When the SNP site is homozygous, it forms a single peak, and the heterozygote forms overlapping peaks. Only one base could be read at the same site. Some mutations in this figure are heterozygous; therefore, there will be inconsistent bases marked at the same site. For example, the base indicated by the red arrow in B is A instead of G
Bioinformatics analysis
As there have been no reports on the novel Ser687Cys mutation, several bioinformatic computational programs were used to analyze its pathogenicity. The frequency of the de novo mutation in the healthy population was determined using the SNP database. Results showed that the mutation was absent in the normal alleles, ruling out the possibility of a known genetic polymorphism. Clustal Omega was used to compare the homologous sequences of F13A1 genes between humans and other species. The results indicated that Ser687 of F13A1 gene was conserved among different species, including Homo sapiens, Macaca mulatta, Bos taurus, Gallus gallus, and Xenopus tropicalis (Fig. 4). Therefore, serine-to-cysteine exchange is unfavorable in terms of amino acid conservatism. Furthermore, Ser687Cys was predicted to be possibly disease-causing based on in silico analysis (Table 5). Online software assays have suggested that changes in protein function caused by the mutation are deleterious and damaging. Protein structural changes caused by the mutation were analyzed and assessed for their possible impact on protein function. The local spatial structure of the protein before and after the mutation is shown in Fig. 5.

Homologous sequence alignment of Ser687 among different species

Detailed views of the local molecular environment of the wild-type FXIII-A subunit (ACE) and the mutated FXIII-A subunit (BDF). Yellow dashed lines represent hydrogen bonds. Hydrogen bond lengths are marked with black numbers. Residues and corresponding colors are as follows: Gly210-red, Arg210-blue, Gly562-yellow, Arg562-magentas, Ser687-green, Cys687-orange
Discussion
Inherited FXIII deficiency is a rare bleeding disorder caused by mutations in the F13A1 or F13B gene. The clinical phenotype of FXIII deficiency ranges from mild to severe, with the site and severity of bleeding varying greatly among different patients [15]. Umbilical cord bleeding, muscle bleeding, and central nervous system bleeding can occur. According to bleeding location, clinical impact, and spontaneity, the European Network of the Rare Bleeding Disorders divides the severity of bleeding into four categories: asymptomatic, grade I, grade II, and grade III. Previous studies have found that patients with FXIII deficiency had the highest percentage of grade III bleeding [16]. Among all clinical manifestations, ICH is the primary cause of mortality, and develops in one-third of patients with FXIII deficiency over the disease course [17]. The brain parenchyma is the most common site of ICH (> 90%), whereas subdural and epidural hemorrhages are less common [13]. Consistent with previous studies, the patient in our case also had a parenchymal hemorrhage.
The heterogeneity of the clinical manifestations of FXIII deficiency makes its diagnosis difficult. The diagnosis of FXIII deficiency is based on clinical symptoms combined with laboratory examinations. According to the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis, the quantitative detection of FXIII activity is recommended as a first-line screening experiment. If FXIII activity decreases, subsequent tests for FXIII antigens and autoantibodies should be performed to determine the specific type of FXIII deficiency. Molecular genetic testing should also be performed if necessary [18].
Here, we presented a case of a man with ICH secondary to FXIII deficiency. The proband had experienced several severe bleeding events but had not been diagnosed with an FXIII deficiency until this time, due to the lack of understanding of the disease and limitations in diagnostic methods. Presently, there is no standardized clinical assay for FXIII activity. The most commonly used screening test is the clot solubility test, but the low sensitivity of this method leads to high false-negative rates [19]. Quantitative assays for FXIII activity can only be performed in certain specific laboratories and also have the disadvantage of low sensitivity. Therefore, when serious bleeding events of unknown reason occur but routine coagulation tests show no abnormalities, FXIII deficiency should be considered. A timely and proper diagnosis is crucial to avoid life-threatening bleeding events.
The treatment of inherited FXIII deficiency mainly depends on replacement therapy, including cryoprecipitate, fresh frozen plasma (FFP), FXIII concentrates, and recombinant FXIII. FXIII concentrates (e.g. Fibrogammin P/corifact) are currently the first choice for the treatment of the disease because of their high safety. In 2011, these drugs were approved by the FDA to treat patients with F13A1 or F13B mutations [20]. However, frequent transfusion of plasma-derived FXIII concentrates poses potential risks of allergic reactions, viral transmission, and other side effects [21]. Hence, the FDA approved the recombinant FXIII-A subunit (rFXIIIA/catridecacog) for the treatment of F13A1 mutants in 2013 [22]. Given that FXIII concentrate and rFXIIIA are not available in many countries, patients are often treated with FFP and cryoprecipitate instead. As FXIII has a long half-life (11–14 days), prophylactic treatment can prevent most serious bleeding events and greatly reduce mortality. Clinically, patients with FXIII levels as low as 2–5% can be asymptomatic, which may explain why even though the FXIII level of the patient in this case was lower than normal (21.5%) after cryoprecipitate supplement, he had no symptoms of bleeding [23, 24].
Since Duckert reported the first case in 1960, 208 FXIII-A mutations and 27 FXIII-B mutations have been identified and registered in the HGMD Professional until April 2021 [25]. The spectrum of mutations mostly consists of missense/nonsense mutations and a few small deletions/insertions and splice-site mutations. Large deletions/large duplications are very rare and only seen in the F13A1 gene. Usually, only homozygous or compound heterozygous mutations can lead to significant bleeding manifestations. In patients of non-consanguineous families, a higher frequency of compound heterozygosity mutations was observed. Homozygous or compound heterozygous mutations cause serious FXIII deficiency, while heterozygous mutations may cause mild deficiency in FXIII. This could explain the mild decrease in FXIII-A antigen in the patient's father [26, 27].
We detected three missense mutations in the F13A1 gene and one in the F13B gene in the pedigree. Their pathogenicity was analyzed as follows.
The Gly210 residue of FXIII-A subunit is highly conserved. A computerized structure-function correlation analysis of missense mutations predicted that Gly210Arg will introduce clashes with the neighboring Val210 andTyr227 on the adjacent β-strand, causing misfolding and rapid degradation of the mutated protein [28, 29].
A Gly562Arg mutation causes a conformational change in the FXIII-A subunit. Molecular modeling predicted that the Gly562Arg mutation can increase the total potential energy, making the mutated molecule less stable than the wild type. Mammalian expression studies have also confirmed that the mutated protein is normally synthesized but is rapidly degraded, which is consistent with the prediction of molecular modeling [30].
FXIII is converted into the active form by the synergistic action of thrombin and Ca2+. Thrombin cleaves the peptide bond between Arg37 and Gly38 in each FXIII-A subunit. The FXIII-B subunits dissociate from the FXIII-A2B2 tetramer in the presence of Ca2+. FXIII in the plasma is then transformed into FXIIIa with or without proteolysis. Komanasin et al. showed that the Arg115His mutation of F13B gene is only associated with an increased rate of dissociation of the FXIII tetramer, without affecting protein expression or secretion. They demonstrated that the mutation was benign, based on functional tests [1, 31, 32].
The American College of Medical Genetics and Genomics (ACMG) provides a set of classification criteria for newly discovered gene variants. Each pathogenic criterion was weighed as very strong (PVS1), strong (PS1–4), moderate (PM1–6), or supporting (PP1–5). The novel mutation from the proband, c.2063C>G (p. Ser687Cys), was not detected in his biological parents, which was classified as strong pathogenic evidence (PS2). In addition, the mutation was not found in the 1000 Genomes Project or the Exome Sequencing Project. Based on this, the level of evidence for pathogenicity was PM2. Using multiple missense mutation interpretation tools (including SIFT, Polyphen2 and PROVEAN), the mutation was predicted to be deleterious (PP3). In summary, the novel mutation was judged to be a likely pathogenic mutation according to the ACMG guideline [42].
The function of protein largely depends on its spatial conformation. We detected the protein environment surrounding the mutated residue using protein structure visualization tools. Substitution of Gly210 by a larger Arg residue would introduce clashes with the neighboring Val211 and Tyr227, which will significantly destroy the folding of catalytic domains (Fig. 5A, B). The mutant Arg562 residue generates adverse short contacts with adjacent residues (Ser368, Phe559, Gln622), and the replacement of a small residue by a large charged amino acid is expected to produce unstable misfolding structure (Fig. 5C, D). Structure–function correlation analysis results by computer modeling of the above two mutations were similar to the results of in vitro cell experiments, which confirms the feasibility of in silico analysis [28,29,30]. Therefore, we performed an in silico analysis of the novel mutation (Ser687Cys) to investigate the possible effect of the missense mutation on the structure of the FXIII-A subunit (Fig. 5E, F). Ser687 is located on the surface of the β-barrel 2 domain. When serine is replaced by a more hydrophobic cysteine, it tends to stay away from the surrounding water molecules and entrap itself within the protein, which may lead to the protein folding into an unstable spatial conformation. Ser687 shares three hydrogen bonds with neighboring residues, including Asn654, Pro685, and Arg684 (Table 5). The mutated Cys687 residue will still form a hydrogen bond with Asn654, but the distance between oxygen and nitrogen atoms will change (2.37–2.67 Å). There would not be hydrogen bonds formed by Cys687 and Pro685. Furthermore, cysteine contains a highly reactive sulfhydryl group that forms two additional hydrogen bonds with Arg684. The sulfhydryl group introduced by cysteine may offer the possibility of forming covalent disulfide bond cross-links with other cysteine-containing proteins, resulting in the disruption of the local surrounding structure. As mentioned earlier, Ser687Cys is a spontaneous mutation, it must coexist with Gly210Arg on the paternal chromosome, or with Gly562Arg on the maternal chromosome. We used AlphaFold to simulate mutant protein structure when two amino acids were substituted in one translation product. (Supplementary Fig. 1A, B) The prediction results show that Cys687 is far away from Arg210 and Arg562, and there is no direct interaction between them. Despite this, the effect of Ser687Cys on protein structure is undeniable. We also applied AlphaFold to mimic the effect of the Ser687Cys mutation alone on protein structure. The prediction result of AlphaFold (Supplementary Fig. 1C) is similar to the result of Swiss-Pdb Viewer mentioned earlier in this article (Fig. 5F). The Ser687Cys mutation caused serine to be replaced by cysteine, which alters intermolecular forces between amino acid residues and increases instability of the mutant protein. By using these two different models in our computational analyses of protein stability, we will increase the robustness of our findings.
There is no doubt that in silico prediction cannot replace biological functional experiments. Phenotypic changes caused by gene mutations may not be significant because of gene dosage effects [14]. In addition, given the high risk of grade III bleeding, most patients start alternative treatment as soon as they are diagnosed, making genotype–phenotype association analysis more difficult to implement.
Conclusion
We reported a patient of inherited FXIII deficiency with ICH, who was a compound heterozygote with two known missense mutations (Gly210Arg and Gly562Arg) and a novel missense mutation (Ser687Cys) in the F13A1 gene, and a benign mutation in the F13B gene (Arg115His). Bioinformatics analysis predicted the novel mutation to be a likely pathogenic gene. We acknowledge the limitations of the research. According to the current research results, it is premature to conclude that Ser687Cys is a disease-causing mutation, and more in vitro assays are needed to provide sufficient evidence at the mRNA or protein level. In the future, we will try to construct in vitro expression vectors containing mutant cDNA to further study the effects of Ser687Cys mutation on gene transcription, protein synthesis and protein stability.
Data availability
Research data of the study are available from the corresponding author on reasonable request.
References
Yee VC, Pedersen LC, Le Trong I, Bishop PD, Stenkamp RE, Teller DC. Three-dimensional structure of a transglutaminase: human blood coagulation factor XIII. Proc Natl Acad Sci USA. 1994;91(15):7296–300.
Schroeder V, Kohler HP. Factor XIII: structure and function. Semin Thromb Hemost. 2016;42(4):422–8.
Ichinose A, McMullen BA, Fujikawa K, Davie EW. Amino acid sequence of the b subunit of human factor XIII, a protein composed of ten repetitive segments. Biochemistry. 1986;25(16):4633–8.
Durda MA, Wolberg AS, Kerlin BA. State of the art in factor XIII laboratory assessment. Transfus Apher Sci. 2018;57(6):700–4.
Inbal A, Lubetsky A, Krapp T, Castel D, Shaish A, Dickneitte G, et al. Impaired wound healing in factor XIII deficient mice. Thromb Haemost. 2005;94(2):432–7.
Dardik R, Loscalzo J, Inbal A. Factor XIII (FXIII) and angiogenesis. J Thromb Haemost. 2006;4(1):19–25.
Inbal A, Muszbek L. Coagulation factor deficiencies and pregnancy loss. Semin Thromb Hemost. 2003;29(2):171–4.
Soendergaard C, Kvist PH, Seidelin JB, Nielsen OH. Tissue-regenerating functions of coagulation factor XIII. J Thromb Haemost. 2013;11(5):806–16.
Muszbek L, Katona É. Diagnosis and management of congenital and acquired FXIII deficiencies. Semin Thromb Hemost. 2016;42(4):429–39.
Yan MTS, Rydz N, Goodyear D, Sholzberg M. Acquired factor XIII deficiency: a review. Transfus Apher Sci. 2018;57(6):724–30.
Dorgalaleh A, Rashidpanah J. Blood coagulation factor XIII and factor XIII deficiency. Blood Rev. 2016;30(6):461–75.
Dorgalaleh A, Naderi M, Shamsizadeh M. Morbidity and mortality in a large number of Iranian patients with severe congenital factor XIII deficiency. Ann Hematol. 2016;95(3):451–5.
Naderi M, Dorgalaleh A, Alizadeh S, Tabibian S, Hosseini S, Shamsizadeh M, et al. Clinical manifestations and management of life-threatening bleeding in the largest group of patients with severe factor XIII deficiency. Int J Hematol. 2014;100(5):443–9.
Karimi M, Bereczky Z, Cohan N, Muszbek L. Factor XIII deficiency. Semin Thromb Hemost. 2009;35(4):426–38.
Lak M, Peyvandi F, Ali Sharifian A, Karimi K, Mannucci PM. Pattern of symptoms in 93 Iranian patients with severe factor XIII deficiency. J Thromb Haemost. 2003;1(8):1852–3.
Peyvandi F, Palla R, Menegatti M, Siboni SM, Halimeh S, Faeser B, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost. 2012;10(4):615–21.
Alavi SER, Jalalvand M, Assadollahi V, Tabibian S, Dorgalaleh A. Intracranial hemorrhage: a devastating outcome of congenital bleeding disorders-prevalence, diagnosis, and management, with a special focus on congenital factor XIII deficiency. Semin Thromb Hemost. 2018;44(3):267–75.
Kohler HP, Ichinose A, Seitz R, Ariens RA, Muszbek L. Diagnosis and classification of factor XIII deficiencies. J Thromb Haemost. 2011;9(7):1404–6.
Karimi M, Peyvandi F, Naderi M, Shapiro A. Factor XIII deficiency diagnosis: challenges and tools. Int J Lab Hematol. 2018;40(1):3–11.
Nugent D. Corifact™/Fibrogammin® P in the prophylactic treatment of hereditary factor XIII deficiency: results of a prospective, multicenter, open-label study. Thromb Res. 2012;130(Suppl 2):S12–4.
Ashley C, Chang E, Davis J, Mangione A, Frame V, Nugent DJ. Efficacy and safety of prophylactic treatment with plasma-derived factor XIII concentrate (human) in patients with congenital factor XIII deficiency. Haemophilia. 2015;21(1):102–8.
Carcao M, Altisent C, Castaman G, Fukutake K, Kerlin BA, Kessler C, et al. Recombinant FXIII (rFXIII-A2) prophylaxis prevents bleeding and allows for surgery in patients with congenital FXIII A-subunit deficiency. Thromb Haemost. 2018;118(3):451–60.
Jain S, Acharya SS. Management of rare coagulation disorders in 2018. Transfus Apher Sci. 2018;57(6):705–12.
Franchini M, Marano G, Mengoli C, Piccinini V, Pupella S, Vaglio S, et al. Inhibitors in patients with congenital bleeding disorders other than hemophilia. Semin Thromb Hemost. 2018;44(6):595–603.
Duckert F, Jung E, Shmerling DH. A hitherto undescribed congenital haemorrhagic diathesis probably due to fibrin stabilizing factor deficiency. Thromb Diath Haemorrh. 1960;5:179–86.
Carcao M, Fukutake K, Inbal A, Kerlin B, Lassila R, Oldenburg J, et al. Developing the first recombinant factor XIII for congenital factor XIII deficiency: clinical challenges and successes. Semin Thromb Hemost. 2017;43(1):59–68.
Thomas A, Biswas A, Dodt J, Philippou H, Hethershaw E, Ensikat HJ, et al. Coagulation factor XIIIA subunit missense mutations affect structure and function at the various steps of factor XIII action. Hum Mutat. 2016;37(10):1030–41.
Vysokovsky A, Rosenberg N, Dardik R, Seligsohn U, Inbal A. Effect of four missense mutations in the factor XIII A-subunit gene on protein stability: studies with recombinant proteins. Blood Coagul Fibrinolysis. 2006;17(2):125–30.
Vysokovsky A, Saxena R, Landau M, Zivelin A, Eskaraev R, Rosenberg N, et al. Seven novel mutations in the factor XIII A-subunit gene causing hereditary factor XIII deficiency in 10 unrelated families. J Thromb Haemost. 2004;2(10):1790–7.
Takahashi N, Tsukamoto H, Umeyama H, Castaman G, Rodeghiero F, Ichinose A. Molecular mechanisms of type II factor XIII deficiency: novel Gly562-Arg mutation and C-terminal truncation of the A subunit cause factor XIII deficiency as characterized in a mammalian expression system. Blood. 1998;91(8):2830–8.
Komanasin N, Catto AJ, Futers TS, van Hylckama VA, Rosendaal FR, Ariëns RA. A novel polymorphism in the factor XIII B-subunit (His95Arg): relationship to subunit dissociation and venous thrombosis. J Thromb Haemost. 2005;3(11):2487–96.
Muszbek L, Bereczky Z, Bagoly Z, Komáromi I, Katona É. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev. 2011;91(3):931–72.
Anwar R, Miloszewski KJ, Markham AF. Identification of a large deletion, spanning exons 4 to 11 of the human factor XIIIA gene, in a factor XIII-deficient family. Blood. 1998;91(1):149–53.
Jiao WY, Wu JS, Ding QL, Wang XF, Xu XC, Ding KY, et al. Identification of a novel mutation of F (13) A gene in a pedigree with factor XIII deficiency. Zhonghua Xue Ye Xue Za Zhi. 2007;28(9):598–601.
Ivaskevicius V, Windyga J, Baran B, Schroeder V, Junen J, Bykowska K, et al. Phenotype-genotype correlation in eight Polish patients with inherited Factor XIII deficiency: identification of three novel mutations. Haemophilia. 2007;13(5):649–57.
Otaki M, Inaba H, Shinozawa K, Fujita S, Amano K, Fukutake K. Characterization of a large deletion that leads to congenital factor XIII deficiency. Rinsho Byori. 2008;56(3):187–94.
Ma QL, Zhou KY, Zhou P, Cai WW. Identification of a novel large deletion of factor subunit A mRNA associated with hereditary factor deficiency. Zhonghua Xue Ye Xue Za Zhi. 2012;33(4):299–302.
Thomas A, Ivaškevičius V, Zawadzki C, Goudemand J, Biswas A, Oldenburg J. Characterization of a novel large deletion caused by double-stranded breaks in 6-bp microhomologous sequences of intron 11 and 12 of the F13A1 gene. Hum Genome Var. 2016;3:15059.
Ma S, Chen C, Liang Q, Wu X, Wang X, Wu W, et al. Phenotype and genotype of FXIII deficiency in two unrelated probands: identification of a novel F13A1 large deletion mediated by complex rearrangement. Orphanet J Rare Dis. 2019;14(1):182.
Shanbhag S, Ghosh K, Shetty S. Genetic basis of severe factor XIII deficiency in a large cohort of Indian patients: identification of fourteen novel mutations. Blood Cells Mol Dis. 2016;57:81–4.
Kershaw G. Detection and measurement of factor inhibitors. In: Favaloro EJ, Lippi G, editors. Hemostasis and thrombosis: methods and protocols. New York: Springer; 2017. pp. 295–304.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Acknowledgements
Zi Sheng and Yunhai Fang designed the research; Jun Peng, Jihua Qiu, and Xinsheng Zhang conducted the research; Lijie Yan collected the clinical data; Lijie Yan and Tiantian Wang analyzed the data and drafted the paper. All authors revised and approved the final version of the manuscript. This study was funded by the National Natural Science Foundation of China (82000126, 91942306, and 81770133), Shandong Medical and Health Science and Technology Development Program (202103040601), and China Postdoctoral Science Foundation (2020M672075). The authors have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors state that they have no interests which might be perceived as posing a conflict or bias.
Ethics approval statement
Ethics approval (No. KYLL-202205-023-1) was obtained from The Medical Ethics Committee of Qilu Hospital, Shandong University.
Patient consent statement
All participants gave informed consent to our research in accordance with the Declaration of Helsinki.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The work was done in Qilu Hospital of Shandong University and Shandong Hemophilia Treatment Center.
Supplementary Information
Below is the link to the electronic supplementary material.
12185_2023_3594_MOESM1_ESM.tiff
Supplementary file1 (TIFF 26384 kb) Supplementary Fig. 1 Three-dimensional structure of mutant protein predicted by AlphaFold. A Mutant protein structure when Ser687Cys exists together with Gly210Arg in one allele. B Mutant protein structure when Ser687Cys exists together with Gly562Arg in one allele. C Local protein structure caused by Ser687Cys mutation. Yellow dashed lines represent hydrogen bonds. Hydrogen bond lengths are marked with black numbers. Residues and corresponding colors are as follows: Arg210-blue, Arg562-magentas, Cys687-orange
12185_2023_3594_MOESM2_ESM.tiff
Supplementary file2 (TIFF 26384 kb) Supplementary Fig. 1 Three-dimensional structure of mutant protein predicted by AlphaFold. A Mutant protein structure when Ser687Cys exists together with Gly210Arg in one allele. B Mutant protein structure when Ser687Cys exists together with Gly562Arg in one allele. C Local protein structure caused by Ser687Cys mutation. Yellow dashed lines represent hydrogen bonds. Hydrogen bond lengths are marked with black numbers. Residues and corresponding colors are as follows: Arg210-blue, Arg562-magentas, Cys687-orange
12185_2023_3594_MOESM3_ESM.docx
Supplementary file3 (TIFF 26384 kb) Supplementary Fig. 1 Three-dimensional structure of mutant protein predicted by AlphaFold. A Mutant protein structure when Ser687Cys exists together with Gly210Arg in one allele. B Mutant protein structure when Ser687Cys exists together with Gly562Arg in one allele. C Local protein structure caused by Ser687Cys mutation. Yellow dashed lines represent hydrogen bonds. Hydrogen bond lengths are marked with black numbers. Residues and corresponding colors are as follows: Arg210-blue, Arg562-magentas, Cys687-orange
12185_2023_3594_MOESM4_ESM.tiff
Supplementary file4 (DOCX 15 kb) Supplementary Table 1 Sequencing results of F13A1 and F13B genes in the patient'sparents
About this article

Cite this article
Yan, L., Wang, T., Qiu, J. et al. Identification of a novel mutation in the factor XIII A subunit in a patient with inherited factor XIII deficiency. Int J Hematol 118, 26–35 (2023). https://doi.org/10.1007/s12185-023-03594-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-023-03594-y