
Abstract
Factor XII deficiency is a rare inherited disorder caused by clotting factor XII (FXII, F12) deficiency. It is often asymptomatic but can have both thrombotic and haemorrhagic symptoms. The aim of this study was to describe the spectrum of F12 gene mutations in a Russian population and learn more about the relationship between F12 variants and clinical phenotypes. We obtained and analysed genetic and clinical data from 33 apparently unrelated patients with FXII plasma levels below 60% and genetic data from 26 healthy controls with no history of FXII deficiency. Forty mutant alleles and six different deleterious substitutions were identified. Of these substitutions, three were major in the Russian population (c.-62C > T, c.-57G > C and c.1532-1G > A, total frequency 92.5%) and the three others (p.615 del C, c.1180_1181delCA, and CD218 TAT- > CAT p.Tyr218His) were rare and novel in the world population. Eight patients with mild FXII deficiency were found to be homozygous for a hypomorphic variant of functional polymorphism C46T and have no other deleterious substitutions in the F12 gene. Contrary to data in the literature, our study showed that mild haemorrhagic manifestations are common among patients with FXII deficiency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hageman’s disease is a rare inherited disorder caused by clotting factor XII (Hageman factor, FXII) deficiency. Descriptions of clinical manifestations of FXII deficiency in the literature are extremely controversial. Even though FXII takes part in the intrinsic coagulation cascade, it is not usually associated with a clinical bleeding predisposition [1]. In contrast, since the initial publication of the paper by Ratnoff et al. [2], there have been many reports suggesting that FXII deficiency may actually predispose to thrombosis [3]. Nonetheless, analysis of the literature indicates that most patients with venous thrombosis and FXII deficiency have some additional risk factors of venous thrombosis [4]. The research into the clinical manifestations of FXII deficiency has become especially important in the context of experiments on mouse models. They indicate that in mice, a factor XII knock-out or inhibition protects from vascular occlusive events while having only a minimal impact on haemostasis [5]. These experiments have aroused wider interest in factor XII as a novel target for antithrombotic therapy. Nonetheless, the clinical manifestations may be different between mice and humans [6]. This observation makes clinical studies on patients with hereditary deficiencies of FXII relevant and important.
To date, 60 pathogenic variants in the F12 gene have been described in the HGMD (Human Gene Mutation Database). Nonetheless, in addition to pathogenic variants, functional polymorphisms are of interest because in contrast to rare genetic variants, they determine FXII activity variation in the general population. For the F12 gene, the only known functional polymorphism that causes a moderate FXII deficiency in the homozygous state is c.-4 C > T. The frequency of the T allele (hypomorphic variant) is significantly higher in the Asian population than in the European population, thereby explaining significantly lower levels of plasma coagulant activity of the Hageman factor in the healthy Asian population compared to Europeans [7].
The aim of this study was to describe the F12 gene mutation spectrum in a Russian population and learn more about the relations between genetic variants of the F12 gene and clinical phenotypes.
Materials and methods
We obtained and analysed genetic and clinical data from 33 apparently unrelated patients with FXII plasma levels below 60%. Patients participating in the study were referred to the National Medical Research Center for Hematology (Moscow, Russia) in three different ways: patients have sought medical advice regarding blood coagulation issues (either haemorrhagic or thrombotic symptoms) (n = 11), patients have been discovered by accident through a routine pre-surgical coagulation test (n = 19), and asymptomatic patients with a family history of FXII deficiency (n = 3). To estimate T allele frequency of functional polymorphism c.-4 C > T in a healthy population, we also used genetic data from 26 conditionally healthy controls without a history of FXII deficiency who contacted the Center for other health problems (e.g. acute hepatic porphyria, acute lymphoblastic leukaemia or haemophilia).
All the patients provided written informed consent. The present work was carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) and with guidelines of a local ethics committee for experiments involving humans.
Total DNA was isolated from the whole-blood samples by standard proteinase K–SDS digestion and phenol–chloroform extraction.
We analysed all functionally important regions of the factor XII gene, i.e. the promoter region, all exons and exon–intron junctions, by Sanger sequencing. For amplification of target fragments, we used primers designed in our laboratory (Table 1); all primer pairs have an annealing temperature of 62 °C. PCR was carried out using the MasterMix (ThermoScientific, Waltham, MA, USA), following the manufacturer’s protocol, in a 25 µl reaction containing 50–100 ng of the DNA template. PCR products were analysed by polyacrylamide gel electrophoresis and then purified using the Wizard PCR Preps DNA purification system (Promega, Madison, WI, USA). Sequencing was performed by means of ABI PRISM® BigDye™ Terminator v.3.1 on an Applied Biosystems 3730 DNA Analyzer (Applied Biosystems, Waltham, MA, USA). The sequencing data were analysed in SeqScape (Applied Biosystems) and aligned manually; as a reference, we used the nucleotide sequence of the human F12 gene, GenBank accession No. NG_007568. For amino acid nomenclature, we used cDNA sequence NM_000505.3.
We analysed a single patient using the TruSight One sequencing panel (Illumina) following the manufacturer’s protocol.
The pathogenicity of novel substitutions was interpreted using the standards, guidelines and nomenclature of HGVS and the following software packages: SIFT v.6.2.1, PROVEAN v.1.1.5, PolyPhen-2 v.2.2.2 and MutationTaster.
For activated partial thromboplastin time (APTT) and FXII activity measurements, we employed a CA-660 automated coagulation analyser (Sysmex, Milton Keynes, UK), the Pathromtin® SL aPTT reagent and coagulation-factor-deficient plasma FXII (Siemens Healthcare GmbH, Erlangen, Germany).
For the measurement of Hageman factor (FXIIa)-dependent fibrinolysis, we used reagents from RENAM (Moscow, Russia). The protocol included the following steps. A mixture of 8 ml of distilled H2O, 0.2 ml of 1% acetic acid, 0.5 ml of blood plasma and 0.5% of kaolin was prepared. The mixture was carefully stirred and incubated at 37 °C for 30 min and then centrifuged for 6 min at 1500 rpm. The precipitate was dissolved in 0.5 ml of 50 mM Tris–HCl (pH 7.4) containing 130 mM NaCl. Clot formation was induced by the addition of an equal volume of 0.025 M calcium chloride. After clot formation, the time of complete clot lysis was determined [8].
Local reference ranges for the coagulation variables were as follows: APTT, 29–38 s; FXII, 70–150%; and XIIa-dependent fibrinolysis, 5–12 min.
Statistical significance of differences in polymorphism frequencies was assessed by the χ2 test in StatSoft 10.
Results and discussion
Six different pathogenic variants were identified in 25 individuals (16 patients with two pathogenic variants, 9 heterozygous patients, 8 patients without any deleterious substitutions). Three of them are known and widespread in the world population. Three variants are novel and have not been previously documented in the HGMD.
Microdeletion c.615delC in exon 7 [Nchrom = 1 (2.4%)] was found for the first time. This mutation produces a frameshift followed by a stop codon after 44 alternate codons downstream: p.(Gly206GlufsTer45). This frameshift mutation leads to the loss of the kringle domain and the entirety of the light chain of FXII.
The c.1180_1181delCA microdeletion in exon 10 [Nchrom = 1 (2.4%)] has not been previously reported either. This frameshift mutation results in a stop codon after 38 alternate codons: p.(His394GlnfsTer39).
We also detected a novel missense substitution, c.652T > C p.(Tyr218His) [Nchrom = 1 (2.4%)], in exon 9 (in the kringle domain). This variant was predicted to be deleterious by three out of four in silico analyses (Table 2) and can be interpreted as likely pathogenic based on ACMG criteria (PM2, PP3) [9].
Major mutation c.1532-1G > A [Nchrom = 19 (46.3%)] has been previously found in European populations [10]. The mutation is located at the 3′ splice acceptor site of exon 14. It creates a new acceptor site one nucleotide downstream of the natural one, which as a consequence shifts the reading frame in exon 14 one nucleotide downstream. Accordingly, the protein lacks the amino acid residues encoded by exon 14, including the functionally important translational stop codon, resulting in a detectable transcript but an unstable protein [11].
Substitutions c.-62C > T [Nchrom = 7 (17%)] and c.-57G > C [Nchrom = 12 (29.3%)] have also been described in the European population. We found them in six and eight patients, respectively. Both mutations are located in the promoter region of F12 at a putative binding site for HNF4α. Both cause significant underexpression of F12 [12].
For comparison of the patients’ phenotypes, we used only one coagulation parameter: FXII activity. APTT and XIIa-dependent fibrinolysis are strongly related to the FXII activity level (Fig. 1), indicating that these metrics are not independent.

APTT (a) and XIIa-dependent fibrinolysis (b) in groups with different FXII activity
F12 activity vs. the genotype
The results of the F12 gene sequencing allowed us to subdivide our patients into three groups: patients with two mutations in this gene (homozygous or compound heterozygous mutations, n = 16), patients heterozygous for F12 mutations (n = 9) and patients without any deleterious substitutions in this gene (n = 8). It has been demonstrated that homozygous and compound heterozygous patients exhibit almost no factor XII activity, whereas heterozygotes have intermediate values (Table 3). Our data regarding FXII activity are in agreement with the expected tendency.
The patients without any deleterious substitutions show an average FXII activity of 44.5%. All of them, except one, were homozygous for the T allele at the c.-4 position. This may be the reason for a mild FXII deficiency in this group of patients. This hypothesis is supported by the finding that in the group of patients with reduced FXII activity, the frequency of allele C at c.-4 is 0.197 (Nchrom = 66), which is significantly lower than that in the control group: 0.615 (Nchrom = 52; χ2 = 21.583, p < 0.000, df = 1). In our study, the frequency of the C allele (0.615) in the control group was intermediate between European and in Asian populations (0.75 and 0.39, respectively, according to gnomAD).
Thus, the reduction in FXII activity is caused by two kinds of factors: the presence of pathogenic variants in the F12 gene and the homozygosity for a hypomorphic variant of functional polymorphism c.-4C > T.
In this work, we identified only one patient with FXII deficiency (56%) and no abnormalities in the F12 gene. For this patient, we performed an additional analysis and tested 5000 genes by clinical exome next-generation sequencing. We did not find any genetic variations that could cause the FXII deficiency. These data indicate that the regulation of the FXII activity is a complex process that can include nonobvious mechanisms, both hereditary and epigenetic.
Clinical picture vs. mutations
We performed analysis of relationships between genetic variants of F12 gene and clinical phenotypes for 30 patients. We excluded three patients from the analysis due to them having accompanying FXII deficiency with either a mutation in the F8 gene, or with a mutation in the FGA.
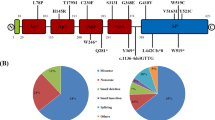
All clinical manifestations of coagulation impairment observed in our patients were classified into two categories. The first group included haemorrhage complications: bleeding gums, epistaxis, easy bruising, single abnormal bleeding, bleeding in a postoperative period or menorrhagia. The second group included thrombotic events: thrombophlebitis. Detailed information for each patient are described in Supplementary Table 1.
In a large proportion of patients (n = 20), mild or moderate haemorrhagic manifestations of FXII deficiency were observed during their lifetime (Table 4). Six of them have sought medical advice regarding haemorrhagic manifestations. In one case, haemorrhagic symptoms were partly related to non-steroidal anti-inflammatory drugs (NSAIDs), whereas in the other six patients who occasionally took an NSAID, either there was no bleeding at all (n = 4) or the bleeding episodes were not connected with the use of NSAIDs (n = 2). Two patients had cases of thrombosis, one of which was associated with an additional provoking factor (thrombophlebitis after installation of a catheter). It is also noteworthy that bleeding and thrombosis symptoms in the patients that have sought medical advice regarding blood coagulation issues are only a little more clinically prominent than the symptoms in the patients who have been discovered by accident through a routine pre-surgical coagulation test (Supplementary Table 1).
Our sample does not allow us to unambiguously investigate relations between specific pathogenic variants and clinical manifestations, because genetic variants are mostly present in combinations with each other. Only a few trends can be discerned.
Patients homozygous for the T allele at the c.-4 position who have no mutations in the F12 gene demonstrate a wide variety of clinical manifestations and a decrease in FXII activity to 26–58% (Table 4).
All three common mutations (c.-62C > T, c.-57G > C and c.1532-1G > A) in the homozygous state result in very low or no FXII activity. When these mutations are heterozygous and are in a compound state with each other or with different mutations in the F12 gene, FXII activity is low too. The only exception is one patient, who is a compound heterozygote of mutations c.-62C > T and c.615delC and has 24% FXII activity. It is possible that in this case, both substitutions are located in the same allele; however, no pedigree data are available for this patient, and therefore, no segregation analysis can be performed. In a heterozygous state, mutations c.-62C > T, c.-57G > C and c.1532-1G > A induce mild FXII deficiency, which can become moderate when they are combined with the T allele at c.-4. Only in one heterozygous patient (genotype c.1532-1G/A, c.-4T/T) did we see severe FXII deficiency (0.7%), which could be caused by an additional mutation, which could not be identified by the methods utilised in this work (for example, an extended deletion or a deep intronic mutation that disrupts normal splicing).
In addition, we note that out of 22 women in the study population, 5 had a miscarriage. Association between factor 12 deficiency and miscarriage have been noted in literature [13].
We showed an influence of pathogenic and hypomorphic variants in the F12 gene and of their combinations on the level of FXII activity. Nonetheless, we failed to identify any relation between clinical manifestations and pathogenic variants. Nevertheless, it is noteworthy that in contrast to literature data, in our study, mild haemorrhagic manifestations are common among patients with FXII deficiency.
Data availability
All data associated with this study are available in the main text, its supplementary materials and through the corresponding author upon request.
References
Simão F, Feener EP. The effects of the contact activation system on hemorrhage. Front Med. 2017;4:1–10.
Ratnoff OD, Busse RJ, Sheon RP. The demise of John Hageman. N Engl J Med. 1968;279:760–1. https://www.nejm.org/doi/pdf/10.1056/NEJM196810032791407
Goodnough LT, Saito H, Ratnoff OD. Thrombosis or myocardial infarction in congenital clotting factor abnormalities and chronic thrombocytopenias: a report of 21 patients and a review of 50 previously reported cases. Medicine (United States). 1983;62:248–55.
Girolami A, Randi ML, Gavasso S, Lombardi AM, Spiezia F. The occasional venous thromboses seen in patients with severe (Homozygous) FXII deficiency are probably due to associated risk factors: a study of prevalence in 21 patients and review of the literature. J Thromb Thrombolysis. 2004;17:139–43.
Renné T, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202(2):271–81.
Geerts H. Of mice and men. CNS Drugs. 2009;23:915–26.
Gordon EM, Donaldson VH, Saito H, Su E, Ratnoff OD. Reduced titers of hageman factor (factor XII) in orientals. Ann Intern Med. 1981;95:697–700.
Kozlov AA et al. (2013) Manual for laboratory doctors. Working with coagulometers "Renam" reagents. M: Print
Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;175(17):405–23.
Lombardi AM, et al. Genetic study in patients with factor XII deficiency: a report of three new mutations exon 13 (Q501STOP), exon 14 (P547L) and -13C>T promoter region in three compound heterozygotes. Blood Coagul Fibrinolysis. 2008;19:639–43.
Manfred S, et al. The novel acceptor splice site mutation 11396(G–>A) in the factor XII gene causes a truncated transcript in cross-reacting material negative patients. Hum Mol Genet. 1995;4:1235–7.
Sabater-Lleal M, et al. Combined cis-regulator elements as important mechanism affecting FXII plasma levels. Thromb Res. 2010;125:e55–e60.
Mordillo C, et al. "Molecular analysis of multiple genetic variants in Spanish FXII-deficient families. Haematologica. 2007;92(11):1569–72.
Acknowledgements
The study was funded by state assignment RK No. AAAA-A18-118032290163-2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
About this article

Cite this article
Demidova, E., Salomashkina, V., Pshenichnikova, O. et al. Factor XII deficiency: a clinical and molecular genetic study. Int J Hematol 117, 678–683 (2023). https://doi.org/10.1007/s12185-023-03535-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-023-03535-9