
Abstract
Coagulation factor XII deficiency is a rare autosomal recessive disorder, which could be found in a consanguineous family. We studied a Chinese family in which the activated partial thromboplastin time (APTT) of the proband had clearly prolonged up to 101.7 s, associated with low FXII activity of 3% and FXII antigen < 1%. To analyze the gene mutation in this FXII-deficient patient, we performed FXII mutation screening, and analyzed the DNA sequence of the F12 gene. A ClustalX-2.1-win and four online bioinformatics software services were used to study the conservatism and effects of the mutation. A transient in vitro expression study was performed to elucidate the possible pathological mechanism. Sequence analysis revealed a homozygous c.1681 G > A point mutation in exon 14, causing a novel Gly542Ser mutation in the catalytic domain. The results of the conservatism and bioinformatics analyses both indicated that the mutation likely affects the function of the protein. Additional expression studies in COS-7 cells showed that the antigen level of mutant FXII (FXII-Gly542Ser) was lower than wild type in culture medium, whereas the corresponding level of FXII antigen in cell lysates was equivalent. These results suggest that the Gly542Ser mutation causes FXII deficiency through intracellular degradation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coagulation factor XII (FXII), a single-chain glyco-protein, produced by hepatocytes and then secreted into plasma as an inactive serine protease precursor [1]. The molecular weight is 80 kDa, composed of 596 amino acid residues [2]. FXII is the initiator of intrinsic coagulation system. It is converted to activated FXII (FXIIa) by proteolytic cleavage at the Arg353-Val354 polypeptide bond, Then, FXIIa converted factor XI to activated factor XI (FXIa), initiating the rapid intrinsic coagulation pathway. In addition, it also plays a vital role in fibrinolysis regulation, complement activation, bradykinin production and inflammation [3,4,5].
Congenital FXII deficiency is an autosomal recessive disease due to mutations in the FXII gene (F12). It can be classified as three types: a cross-reacting material (CRM)-negative group (FXII:Ag cannot be detected), a CRM-positive group (FXII:Ag is normal), and a CRM-reduced group (FXII:Ag reduced). Most patients with FXII deficiency demonstrate no significant clinical symptoms, while several cases present with bleeding, thrombosis or recurrent abortions [6, 7]. And now several clinical investigations manifested which was a risk factor for thrombosis [8]. They are usually detected by chance for a prolonged activated partial thromboplastin time (APTT) on health checkup or pre-operative screening, which always presents with a decreased FXII activity (FXII:C) [9]. The molecular mechanism for FXII deficiency has been described in only a few cases so far [10, 11].
In this paper, we detected a patient with FXII deficiency from a consanguineous marriage family, then studied the phenotype and genotype of the proband and his families. We also analyzed the conservative and harm of the mutation by bioinformatics software and performed a transient in vitro expression study to explore possible mechanisms responsible for this deficient phenotype.
Materials and methods
Patients
The proband was a 38-year-old Chinese man, who went to the First Affiliated Hospital of Wenzhou Medical University (China) for a bronchoscope examination. He had never experienced bleeding or thrombosis in the past. Pre-operative coagulation screening revealed activated partial thromboplastin time (APTT) was extremely prolonged at 101.7 s. Further assessment showed FXII:C decreased at 3%(normal: 72–113%) and FXII:Ag level was less than 1% (normal: 72–113%). Except the fibrinogen (FIB) and d-Dimer (D-D) were slightly high, other indexes, such as prothrombin time (PT), thrombin time (TT), lupus anticoagulant (LAC), factor VIII activity (FVIII:C), factor IX activity (FIX:C), and factor XI activity (FXI:C), were all within normal ranges. No evidence showed abnormalities in liver and kidney functions.
Family study showed that there was a consanguineous relationship between the proband’s parents, as his paternal grandmother and maternal grandmother were sister-German (Fig. 1). A total of another six family members from three generations of his family participated in our research.

The family tree of the inherited coagulation FXII deficiency
Our study was approved by the Ethics Committee of the First Affiliated Hospital of Wenzhou Medical University (China). Peripheral blood samples from the probands and their family members were collected after obtaining written informed consent.
Coagulations tests
Peripheral blood samples from the probands and their family members were collected into standard 0.129 mmol/l trisodium citrate tubes. Routine coagulation tests, including APTT, PT, TT and FIB, were tested by STAR analyzer (Stago, France). d-Dimer was detected by immune turbidimetry method. Lupus anticoagulant was detected by clotting method. Coagulation activities of FXII, FVIII, FIX, and FXI were measured with a one-step clotting method. And the FXII:Ag was measured using enzyme-linked immunosorbent assay (ELISA). The reference ranges of these indexes in our laboratory were established with 100 healthy subjects.
Genetic analysis
The genomic DNA of all the participants was isolated from peripheral blood using the QIAamp DNA Blood Mini kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s protocol. All the 14 exons along with their intron–exon boundaries and 5′,3′-untranslated regions (reference sequence, GenBank: AF538691.1) were amplified by polymerase chain reaction (PCR) in a 25 μl reaction volume on a thermal cycler (Eppendorf, Hamburg, Germany). PCR products were then subjected to direct sequencing on an ABI PRISM 3700 (Applied Biosystems, Foster City, USA). The primers are as shown in Table 1.
When mutational sites were detected, they were identified by reverse sequencing. The corresponding mutational exons of the family members were amplified and sequenced to confirm whether they carry the same mutation.
Conservation analysis
The multiple sequence alignment software ClustalX-2.1-win (Science Foundation Ireland, Dublin, Ireland) was used to analyze the conservative property of Gly542 in homo sapiens and its seven homologous species (Mus musculus, Rattus norvegicus, Pan troglodytes, Macaca mulatta, Canis lupus familiaris, Bos Taurus, and Xenopus tropicalis) (HomoloGene, http://www.ncbi.nlm.nih.gov/homologene).
Bioinformatics prediction
We adopted four online bioinformatics software to predict whether the amino acid substitution could affect protein function. They were (1) Polymorphism Phenotyping v2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2) (J. Craig Venter Institute, Tauranga, New Zealand), (2) Protein Variation Effect Analyzer (PROVEAN, http://provean.jcvi.org/seq_submit.php) (J. Craig Venter Institute, Rockville, USA), (3) Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org) (the Genome Institute of Singapore, Singapore, Singapore), (4) Mutation Taster (http://http://www.mutationtaster.org) (Charité-Universitätsmedizin Berlin, Berlin, Germany). The reference protein ID of FXII was ‘P00748’ and the Ensembl transcript ID was ‘ENST00000253496’.
Transient in vitro expression study
The c.1681G > A nucleotide substitution was introduced into the pIRES2-EGFP expression vector (the wild type plasmid, containing the full-length FXII cDNA, provided by Ruijin Hospital, Shanghai Jiaotong University School of Medicine) using a QuickChange Site-Directed Mutagenesis Kit (Stratagene, USA). The resulting mutant plasmid was checked by sequencing subsequently.
The wild type and mutant plasmids were transfected into African green monkey kidney cells (COS-7) using the Lipofectamine 2000 reagent (Invitrogen, USA). Both culture medium and cells were collected following 48 h culture of the transfected cells in serum-free DMEM.
Culture supernatants were concentrated approximately 10 times using Centricon Plus-20 centrifugal filters (Millipore, USA). After centrifugated at 12,000 rpm for 10 min, the supernatants were subjected to measure the activities of FXII with a one-step clotting assay and the antigens of FXII by ELISA. The cells were washed twice with phosphate-buffered saline (PBS) and then lysed in 120 μl M-ER mammalian protein extraction reagent (Pierce Biotechnology Inc., USA) with 20 μl protease inhibitor (Roche Diagnostics GmbH, Germany), ELISA was used to evaluate FXII antigens of cell lysates.
Results
Routine coagulation indices analysis
The phenotypic detection of family members showed that his elder sister had decreased FXII:C of 52% and FXII:Ag of 50.6%, and his father, mother, elder brother, son and daughter all had decreased FXII:C and FXII:Ag to approximately 30%. We also tested the APTT, PT, TT, FIB, D-D and LAC of the six family members and found that APTT of his son was slightly high, FIB of his father and mother were slightly high, D-D of his elder brother and sister were slightly high, other indices were no abnormal results (Table 2).
DNA sequencing analysis
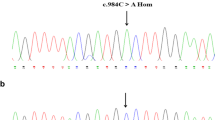
DNA sequencing analysis revealed that the proband carried a homozygous c.1681G > A point mutation in exon 14 of FXII resulting in a substitution of glycine 542 by serine (Gly341Ser) (Fig. 2①, ②). His father, mother, elder brother, elder sister, son and daughter all took a heterozygous Gly542Ser (Fig. 2③). Genotype analysis of 100 healthy subjects excluded the possibility of Gly542Ser as a polymorphism.

The sequence diagram of Gly542Ser. ① is the forward sequencing of homozygous c.1681G > A; ② is the backward sequencing of homozygous c.1681C > T; ③ is the forward sequencing of heterozygous c.1681G and ④ is the forward sequencing of wild type. The position of mutational base is indicated with an arrow
Gly542 conservation analysis
Homologous sequence alignment results showed that the Gly542 was highly conserved among the homologous species: Mus musculus, Rattus norvegicus, Pan troglodytes, Macaca mulatta, Canis lupus familiaris, Bos taurus, and Xenopus tropicalis (Fig. 3).

The conservative analysis diagrams of Gly542Ser. The targeted amino acid was indicated with an arrow, this line are all G (Gly) which indicated that the G (Gly) are conserved in the homologous species
Bioinformatics prediction of Gly542Ser
The forecasting results of the four bioinformatics softwares were ‘Probably damaging’, ‘Deleterious’, ‘Affect protein function’, and ‘Disease causing’ corresponding to ‘PolyPhen-2’, ‘PROVEAN’, ‘SIFT’, and ‘Mutation Taster’ with the scores ‘1.000’, ‘− 4.975’, ‘0.00’, and ‘1.000’, respectively (Table 3).
Transient in vitro expression results
As shown in Table 4 and Fig. 4, the FXII:Ag level in culture medium independently expressing the pIRES2-EGFP/FXII-Gly542Ser was reduced to 26.5% of the wild type recombinant protein (set as 100%), whereas the corresponding level of FXII:Ag in cell lysates was 86.8% of the wild type FXII protein. The mean values of wild-type FXII were set as 100%.

Transient expression of wild-type and mutant FXII (pIRES2-EGFP/FXII-Gly542Ser) in COS-7 cells
Discussion
In this study, DNA sequencing analysis and mutational screening revealed one genetic variation in the FXII gene of the proband, who took the homozygous Gly542Ser mutation. This homozygous mutation was found in another male patient in this area in the previous study by our research group [12]. Family study of the proband indicated that the proband had FXII:C and FXII:Ag level approximately reduced to 3% and less than 1%,and took the homozygous Gly542Ser mutation. And the FXII:C and FXII:Ag of his father, mother, elder brother, son and daughter were all decreased to around 30%, who carried the heterozygous Gly542Ser mutation. Because the proband’s parents were consanguineous marriage, we could speculate that the homozygous Gly542Ser alleles of the proband were derived from his father and mother, and the mode of its inheritance is in accordance with that of autosomal recessive trait. And we could see this mutation belongs to the CRM− group because the FXII:C and FXII:Ag level was reduced near to zero.
To further research the biological characteristics of Gly542Ser mutation, we analyzed its conservatism and possible effects on the protein. We found that Gly542 was a highly conservative site in the eight homologous species, which indicated that it was an important functional site of FXII, and played a key role in the normal function of the protein. The four bioinformatics prediction softwares all manifested the baneful effect of Gly542Ser, which was a deleterious mutation and could affect the FXII protein function and cause disease.
Gly542 lies in the catalytic domain of FXII. When the homozygous c.1681G > A mutation of F12 occurred, which led to a novel Gly542Ser mutation in FXII. It would confer a change in the catalytic domain and alter its local interactions. There were some other mutations which had been reported near or in this region: Leu519Arg [13], Asp538Asn [14] and Cys571Ser [15]. Miyata T et al. once demonstrated that the change of Cys571Ser can break the disulfide bond of Cys540–Cys571, which results in the change of serine active site or enzyme action substrate-binding site, and lose the enzyme activity [15]. Because there was no X-ray 3D structure file of the catalytic domain of FXII, we were not able to do the spatial structure analysis of the mutant protein. But, we knew that Gly542 is a nonpolar hydrophobic amino acid, at the same time, it has acidic and basic functional groups, while Ser is a polar non-charged hydrophilic amino acids. Hence, when the Gly542 mutant into Ser542, located between Cys540 and Cys571, it probably induces partial structural changes. As a consequence, the variant might be unstable and prone to intracellular proteolysis. To investigate the pathogenicity of this Gly542Ser missense mutation, we conducted the transient in vitro expression in African green monkey kidney (COS-7) cells. The results demonstrated accumulation of mutant FXII-Gly542Ser in the cell similar with wild type, while the mutant FXII-Gly542Ser showed lower levels than wild type in culture medium. From these results, we conclude that Gly542Ser substitution is susceptible to intracellular degradation,which was similar to Asp538Asn and many other reported FXII mutations [2, 14, 16, 17].
Moreover, the catalytic region was the key position of FXII for enzymatic function, which contains the activation domain and serine protease domain, and the activity center of serine protease consisted of His393, Asp442 and Ser544 residues [18]. There were only 2 amino acids range from Gly542 to Ser544, the Gly542Ser mutation may directly affect the activation of the Ser544 site. It may affect the FXII normal biological function because the catalytic domain of FXII protein could not be fully activated. Wang XF et al. had reported the Gly542Ser heterozygous mutation [19].
In summary, we reported a Gly542Ser mutation causing CRM-negative FXII deficiency in a Chinese patient family, which was a deleterious mutation analyzed through bioinformatics prediction softwares. And we also discussed its possible pathogenic mechanism that the mutation was the important reason for the decrease of FXII:C by intracellular degradation.
References
Citarella F, Ravon DM, Pascucci B, Felici A, Fantoni A, Hack CE. Structure/function analysis of human factor XII using recombinant deletion mutants. Eur J Biochem. 1996;238:240–9.
Ishii K, Oguchi S, Moriki T, Yatabe Y, Takeshita E, Murata M, et al. Genetic analyses and expression studies identified a novel mutation (W486C) as a molecular basis of congenital coagulation factor XII deficiency. Blood Coagul Fibrin. 2004;15:367–73.
Kaplan AP, Silverberg M. The coagulation-kinin pathway of human plasma. Blood. 1987;70:1–15.
McMullen BA, Fujikawa K. Amino acid sequence of the heavy chain of human a-factor XIIa (activated hageman factor). J Biol Chem. 1985;260:5328–41.
Iijima K, Arakawa Y, Sugahara Y, Matsushita M, Moriguchi Y, Shimohiro H, et al. Factor XII Osaka: abnormal factor XII with partially defective prekallikrein cleavage activity. Thromb Haemost. 2011;105:473–8.
Pauer HU, Burfeind P, Köstering H, Emons G, Hinney B. Factor XII deficiency is strongly associated with primary recurrent abortions. Fertil Steril. 2003;80:590–4.
Mordillo C, Martinez-Marchαn E, Fontcuberta J, Soria JM. Molecular analysis of multiple genetic variants in Spanish FXII-deficient families. Haematologica. 2007;92:1569–72.
Renné T, Schmaier AH, Nickel KF, Blombäck M, Maas C. In vivo roles of factor XII. Blood. 2012;120(22):4296–303.
Singhamatr P, Kanjanapongkul S, Rojnuckarin P. Molecular analysis of factor XII gene in Thai patients with factor XII deficiency. Blood Coagul Fibrinolysis. 2013;24:599–604.
Ishii K, Oguchi S, Moriki T, Yatabe Y, Takeshita E, Murata M, et al. Genetic analyses and expression studies identified a novel mutation (W486C) as a molecular basis of congenital coagulation factor XII deficiency. Blood Coagul Fibrinolysis. 2004;15:367–73.
Suzuki K, Murai K, Suwabe A, Ishida Y. Factor XII Ofunato: Lys346Asn mutation associated with blood coagulation factor XII deficiency causes impaired secretion through a proteasome-mediated degradation. Thromb Res. 2010;125:438–43.
Yang LH, Wang YY, Zhou JP, Cheng XL, Hao XP, Xie HX, et al. Identification of genetic defects underlying FXII deficiency in four unrelated Chinese patients. Acta Haematol. 2016;135:238–40.
Dai LY, Zhang DT, Lin J, Wang YY, Tong Y, Li J, et al. A pedigree analysis of Hereditary coagulation factor XII deficiency caused by a novel homozygous mutation: Leu519Arg. Chin J Lab Med. 2015;38:466–9 (China).
Li M, Xie HX, Wang MS, Ding HX. Molecular characterization of a novel missense mutation (Asp538Asn) in a Chinese patient with factor XII deficiency. Clin Lab. 2015;61:1967–71.
Miyata T, Kawabata S, Iwanaga S, Takahashi I, Alving B, Saito H. Coagulation factor XII (Hageman factor) Washington DC: inactive factor XIIa results from Cys-571-Ser substitution. Proc Natl Acad Sci USA. 1989;86:8319–22.
Kondo S, Tokunaga F, Kawano S, Oono Y, Kumagai S, Koide T. Factor XII Tenri, a novel cross-reacting material negative factor XII deficiency, occurs through a proteasome-mediated degradation. Blood. 1999;93:4300–8.
Matsuki E, Miyakawa Y, Okamoto S. A novel factor XII mutation, FXII R84P, causing factor XII deficiency in a patient with hereditary spastic paraplegia. Blood Coagul Fibrinolysis. 2011;22:227–30.
Yarovaya GA, Blokhina TB, Neshkova EA. Contact system. New concepts on activation mechanisms and bioregulatory functions. Biochemistry. 2002;67:13–24.
Wang XF, Dai J, Wang MS, Ding QL, Wang HL. Analysis of the factor XII gene mutations in two Chinese pedigrees with congenital clotting factor XII deficiency. J Diagn Concepts Pract. 2005;4:447–50 (Shanghai).
Acknowledgements
We thank all the participants in this study.
Funding
This work was supported by The Project Supported by Zhejiang Provincial Natural Science Foundation of China (LY16H080005) and The Project of Wenzhou Public Welfare Science and Technology (Y20150098).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
No potential conflict of interest was reported by the authors.
About this article

Cite this article
Zou, A., Wang, M., Jin, Y. et al. Genetic analysis of a novel missense mutation (Gly542Ser) with factor XII deficiency in a Chinese patient of consanguineous marriage. Int J Hematol 107, 436–441 (2018). https://doi.org/10.1007/s12185-017-2393-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2393-z