
Abstract
The aim of this study was to assess the safety and optimal dose of deferasirox for the treatment of iron overload after allogeneic hematopoietic cell transplantation (HCT). The primary endpoint was the maximum tolerated dose of deferasirox that was determined by the intrapatient dose escalation methods. A total of 16 patients with post-HCT iron overload were enrolled in the study. After excluding one case of early relapse, 15 remained evaluable. Their median age was 42 years (range 22–68). Median time from HCT to deferasirox administration was 9 months (range 6–84). Deferasirox was started at a dose of 5 mg/kg, and the dose was increased to 7.5 and 10 mg/kg every 4 weeks unless there were no grade ≥ 2 of adverse events. Achievement rates of planned medication were 80% in 5 mg/kg (12 of 15), 73% in 7.5 mg/kg (11 of 15), and 60% in 10 mg/kg (9 of 15), respectively. The reasons for discontinuation of the drug were grade 2 of adverse events (n = 4), late relapse (n = 1), and self-cessation (n = 1). None of the patients developed grade ≥ 3 of adverse events or exacerbation of GVHD. Among 11 evaluable cases, mean value of ferritin decreased from 1560 ng/ml pre-treatment to 1285 ng/ml post-treatment. These data suggested that 10 mg/kg of deferasirox may be maximum tolerated dose when given after HCT. Our dose escalating method of deferasirox is useful to identify the optimal dosage of the drug in each patient.
Trial Registration
UMIN000011251
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Iron overload after allogeneic hematopoietic cell transplantation (HCT) remains as a major clinical issue. Iron overload is frequently observed prior to HCT and associated with increased morbidity and mortality after HCT. Several studies have also demonstrated the prognostic impact of post-transplant hyperferritinemia and its association with iron overload [1, 2]. Therefore, interventions to reduce excess body iron may be beneficial both before and after HCT. To this end, many studies have investigated iron chelation therapy after HCT [3,4,5,6,7,8]. Deferasirox is a well-known oral iron chelator and used in iron chelation therapy after HCT. However, the efficacy of deferasirox in this setting remains to be elucidated, and its toxicities prevent its use in transplant patients [3,4,5]. This could be explained in part by the lack of sufficient data on the optimal dosage of deferasirox, duration of therapy. Therefore, we designed a multicenter phase I study of deferasirox to evaluate the safety in patients with iron overload after HCT.
Methods
Study design
KSGCT1302 (UMIN000011251) was a multicenter, single-arm, open-label phase I study. The objectives were to evaluate the safety and optimal dosage of deferasirox in patients with iron overload after allo-HCT.
Eligibility criteria included time from HCT to enrollment ≥ 6 month after HCT, serum ferritin level ≥ 1,000 ng/ml, red blood cell (RBC) transfusions ≥ 20 units, disease remission, creatinine (Cr) within upper limit of normal, within grade 1 of aspartate aminotransferase (AST) and alanine aminotransferase (ALT), performance status of ≤ 1, and unfit for phlebotomy. The exclusion criteria were history of iron chelating therapy after HCT, presence of moderate or severe GVHD, uncontrollable complications such as infection, and history of hepatitis B or C virus infection. Safety of deferasirox was assessed by intrapatient dose escalation methods (Fig. 1). Following study registration, deferasirox was administered at a dose of 5 mg/kg for 4 weeks. Then, the dose was increased to 7.5 and 10 mg/kg every 4 weeks. For safety evaluation, patients were followed-up every 2 weeks. Assessment of liver iron content (LIC) was performed at selected sites and patients were examined on the basis of gradient echo techniques reported by Gandon et al. [9]. Treatment failure criteria were defined by the presence of any grade 2–4 adverse events and/or disease relapse. Namely, grade 1 adverse events within 4 weeks of treatment were defined as tolerable and successful medication for the planned dose. Security of adverse event severity was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0). Primary endpoint was the maximum tolerated dose of deferasirox. Secondary endpoints contained the change of biomarkers and the profile of adverse events.

Intrapatient dose escalation method. DFX indicates deferasirox
This study was conducted in accordance with the Declaration of Helsinki. The protocol was approved by the institutional review board of each attended site. All patients provided written informed consent prior to enrollment.
Statistical analysis
Analyses were performed with EZR version 1.24 statistical software, which is a graphical user interface for R version 2.13.0. [10]. Biomarkers were examined with repeated-measures ANOVA. Mauchly’s test was used for evaluating the sphericity of biomarkers. Maximum tolerated dose was defined from the intrapatient maximum dose, in which 50–80% of the eligible patients experienced successful treatment. From a one-sided significant level of α = 0.05 and 80% statistical power, the number of patients required was calculated to be 15. The registration target was set at 20 patients, including 5 ineligible patients.
Results
A total of 16 patients were enrolled to the present study between March 2013 and January 2016. One patient was excluded from the analysis due to early relapse and 15 were deemed eligible. Patient characteristics are shown in Table 1. The median age was 42 years (range 22–68). Diseases included acute myeloid leukemia (n = 6), acute lymphoblastic leukemia (n = 2), aplastic anemia (n = 2), non-Hodgkin’s lymphoma (n = 2), and others (n = 3). The median duration from HCT to deferasirox administration was 9 months (range 6–84). The median RBC transfusion volume was 54 units (range 20–156). The median serum ferritin value was 1537 ng/ml (range 1027–7655). Only two cases (case 1, 7) were RBC transfusion dependent and the others were independent during the study. All the patients had concomitant drugs with deferasirox including immunosuppressant or preventative medication for infection.
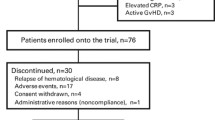
Clinical course of all cases is shown in Fig. 2. One patient (case 16) relapsed 2 weeks after starting deferasirox and was excluded from further analysis. In the remaining 15 patients, 9 patients succeeded in reaching to the final 10 mg/kg dose. Six patients were withdrawn during the treatment due to: grade 2 of elevation of AST/ALT/total bilirubin after 5 mg/kg (n = 3), late relapse after 7.5 mg/kg (n = 1), self-cessation, and grade 2 of nausea/vomiting after 10 mg/kg (n = 2). Thus, achievement rates of receiving planned dose of deferasirox were 80% after 5 mg/kg (12/15), 73% after 7.5 mg/kg (11/15), and 60% after 10 mg/kg (9/15), respectively.

Clinical courses of DFX treatment. DFX indicates deferasirox; G2 grade 2 of adverse events, T-bil total bilirubin, AST aspartate aminotransferase, ALT alanine aminotransferase, RLPS relapse
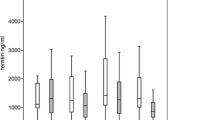
Changes in the mean value of biomarkers for evaluating iron overload were evaluated among 11 patients (Fig. 3). The mean value of serum ferritin decreased from 1560 ng/ml pre-treatment to 1,285 ng/ml post-treatment (P = 0.007). Changes in the other iron-related markers were as follows: unsaturated iron binding capacity from 90 to 142 μg/dl, hemoglobin from 12.1 to 12.6 g/dl, and transferrin from 167 to 225 mg/ml. Changes in the markers for organ function were as follows: Cr from 0.80 to 1.05 mg/dl, and ALT from 26 to 28 IU/l. LIC between pre- and post- therapy was only measured in 3 cases, and the mean LICs before and after the treatment were 200 μmol/g, and did not differ significantly.

Changes in biomarker levels during the treatment (n = 11): a serum ferritin (SF). b Unsaturated iron binding capacity (UIBC). c Hemoglobin (Hb). d Transferrin. e Creatinine. (Cr) d Alanine aminotransferase (ALT)
All adverse events observed in all eligible patients are listed in Table 2. Besides the grade 2 of adverse events described above, grade 1 of adverse events was observed in 12 cases, including elevated Cr (60%), AST (40%), ALT (40%), and alkaline phosphatase (33%), diarrhea (27%), abdominal pain (20%), skin rash (20%), constipation (20%), and nausea (13%). None of the patients developed exacerbation of GVHD or grade ≥ 3 of adverse events.
Discussion
To the best of our knowledge, this the first report evaluating the maximum tolerated dose of deferasirox for transplant patients with iron overload in a multicenter prospective study. Using an intrapatient dose escalation method, we observed that the rate of successful medication decreased with the increasing the dose of deferasirox, and we concluded the maximum tolerated dose of deferasirox as 10 mg/kg after HCT. In the previous studies, the average dose of deferasirox administrated was under 10 mg/kg, and some patients discontinued due to adverse events [3, 4]. Thus, our results seem consistent with those previous findings.
One of the characteristics of this protocol was the strict criteria for selecting patients with iron overload. Hyperferritinemia related to HCT is known to be influenced by several factors, including liver dysfunction, inflammatory disease, and GVHD [11]. Referring to the Japanese guideline for iron overload, the iron overload was defined according to both elevated serum ferritin (≥ 1,000 ng/ml) and a severe history of RBC transfusion (≥ 20 units) [12]. The evaluation of LIC is an ideal tool to define the status of iron overload, but it is not practical, because the technique is unavailable or limited to selected sites during post-transplant clinical care. We also excluded patients with moderate or severe chronic GVHD, non-remission disease, renal dysfunction, or grade 2 of liver dysfunction for precise evaluation. To examine the safety of deferasirox, the initiating time of iron chelating therapy has not established yet. The balance between early intervention and safety was discussed during the study planning. Finally, the starting time was set at 6 months after HCT. Along with the short interval of clinical care and the cost burden, it takes 3 years to enroll 16 patients in this study. Beginning at a low dose of deferasirox and performing intrapatient dose escalation in the present study was designed to ensure safety and continuity to avoid early discontinuation. Although the clinical significance remains unresolved, we think that the study could provide important data on the timing, eligibility, and optimal dosage for iron chelating therapy in patients with iron overload after HCT.
Because of the study design, it may be inappropriate to evaluate the efficacy of deferasirox in the present study, as the duration of chelation is too short. Nevertheless, the mean value of ferritin decreased during the study period. In addition, related iron biomarkers, such as transferrin or unsaturated iron binding capacity, also improved. These findings are similar to those reported in the previous studies [4, 5]. These findings suggest that deferasirox may be effective as a treatment for iron overload. However, accurate interpretation of the present results is difficult, as naturally excreting iron or an improvement in inflammation status may have contributed to the decrease in ferritin. To appropriately evaluate these findings, matched comparison with a non-chelation control group would be required.
Regarding the safety of deferasirox, despite the initial 12 weeks of administration of deferasirox and the strict selection criteria, several grade 1 or 2 adverse events occurred. Those adverse events included gastrointestinal events, such as diarrhea, nausea or vomiting, skin rash, and liver and renal dysfunction. All the adverse events were also reported in the previous studies. Continuous elevation of Cr levels was observed in the late phase of the study, suggesting that the dose of deferasirox should not be increased over 10 mg/kg. Renal impairment has also been reported as the most frequent adverse events in the previous studies and this may be the highest barrier to maintaining deferasirox therapy [4, 5]. Indeed, all the patients had concomitant drugs with renal damage including immunosuppressant or preventative medication. These findings may explain the difference of adverse events among transplant and non-transplant patients. Nevertheless, no grade 3 adverse events were found in our study, and suggesting that the intrapatient dose escalation method deemed to be reasonable approach for durable treatment with deferasirox.
There were several limitations to the present study. First, the patients were all Japanese and their characteristics were heterogeneous. Second, the dose and the duration of deferasirox were limited to 10 mg/kg and 12 weeks. Thus, higher doses and longer duration were not validated. From our pilot study, deferasirox over 10 mg/kg was estimated to be difficult to use for transplant patients due to adverse events. Moreover, patients after 6 month were almost transfusion independent and the effectiveness was sufficiently preserved even in 10 mg/kg of deferasirox. Third, the present evaluation of iron overload was insufficient. Other parameters to measure iron deposition, such as MRI, should be included during future investigations. Finally, although not included as endpoints, the evaluation of outcomes, including survival, relapse, non-relapse mortality, or late complications, was lacking. A benefit has not been established for the therapeutic intervention of iron overload. Therefore, it is important to elucidate the purpose of the treatment for iron overload, be it controlling late complications, preserving organ function or outcome-based, including survival, disease relapse, or non-relapse mortality.
Conclusion
With the intrapatient dose escalation method, we concluded that the maximum tolerated dose is likely to be 10 mg/kg of deferasirox when given after HCT. Further studies are warranted to assess the efficacy of iron chelation therapy with the optimal dose of deferasirox defined in each patient by the method used in this study.
References
Meyer SC, O’Meara A, Buser AS, Tichelli A, Passweg JR, Stern M. Prognostic impact of posttransplantation iron overload after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19(3):440–4.
Tachibana T, Tanaka M, Numata A, Matsumoto K, Tomita N, Fujimaki K, et al. Clinical significance of pre- and 1-year post-transplant serum ferritin among adult transplant recipients. Leuk Lymphoma. 2014;55(6):1350–6.
Jaekel N, Lieder K, Albrecht S, Leismann O, Hubert K, Bug G, et al. Efficacy and safety of deferasirox in non-thalassemic patients with elevated ferritin levels after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2016;51(1):89–95.
Vallejo C, Batlle M, Vázquez L, Solano C, Sampol A, Duarte R, et al. Phase IV open-label study of the efficacy and safety of deferasirox after allogeneic stem cell transplantation. Haematologica. 2014;99(10):1632–7.
Sivgin S, Eser B, Bahcebasi S, Kaynar L, Kurnaz F, Uzer E, et al. Efficacy and safety of oral deferasirox treatment in the posttransplant period for patients who have undergone allogeneic hematopoietic stem cell transplantation (alloHSCT). Ann Hematol. 2012;91(5):743–9.
Majhail NS, Lazarus HM, Burns LJ. A prospective study of iron overload management in allogeneic hematopoietic cell transplantation survivors. Biol Blood Marrow Transplant. 2010;16(6):832–7.
Sivgin S, Baldane S, Akyol G, Keklik M, Kaynar L, Kurnaz F, et al. The oral iron chelator deferasirox might improve survival in allogeneic hematopoietic cell transplant (alloHSCT) recipients with transfusional iron overload. Transfus Apher Sci. 2013;49(2):295–301.
Michallet M, Sobh M, Labussière H, Lombard C, Barraco F, El-Hamri M, et al. Potential anti-leukemic activity of iron chelation after allogeneic hematopoietic stem cell transplantation in patients with acute myeloid leukemia. Leuk Lymphoma. 2017;58(1):237–40.
Gandon Y, Olivié D, Guyader D, Aubé C, Oberti F, Sebille V, et al. Non-invasive assessment of hepatic iron stores by MRI. Lancet. 2004;363(9406):357–62.
Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48(3):452–8.
Brissot E, Savani BN, Mohty M. Management of high ferritin in long-term survivors after hematopoietic stem cell transplantation. Semin Hematol. 2012;49(1):35–42.
Suzuki T. Iron overload and iron chelation therapy. Rinsho Ketsueki. 2012;53(1):25–35.
Acknowledgements
We would like to thank all of the members of KSGCT, the data center, and the Safety and Efficacy Committee.
Author information
Authors and Affiliations
Consortia
Contributions
TT is the primary investigator and designed the protocol, managed the clinical trial, and wrote the manuscript. JK, SMachida, MTanaka, and MTakeuchi contributed to the protocol design and patient enrollment. TS, YN, SK, TM, and EY contributed to case registration. SMorita contributed to biomedical statistics. YK and HK contributed to study design and management. SO supervised the study.
Corresponding author
Ethics declarations
Conflict of interest
All authors except the two below have no conflict of interest to declare: Yuho Najima: personal financial interest on speakers’ bureaus from Novartis; Satoshi Morita: personal financial interest on speakers’ bureaus from Novartis.
About this article

Cite this article
Tachibana, T., Kanda, J., Machida, S. et al. Deferasirox for the treatment of iron overload after allogeneic hematopoietic cell transplantation: multicenter phase I study (KSGCT1302). Int J Hematol 107, 578–585 (2018). https://doi.org/10.1007/s12185-017-2396-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2396-9