
Abstract
Background
Iron overload (IOL) due to repetitive transfusions of packed red blood cells (pRBC) has a major impact on morbidity and mortality in patients with inherited bone marrow failure syndromes and hemoglobinopathies such as thalassemia and sickle cell disease. However, whether IOL influences the outcome of elderly patients with myeloid malignancies is not yet clear. Moreover, clinical trials have reported high drop-out rates during treatment with the oral iron chelator deferasirox (DFX).
Aim
Here we report the results of a 2-year prospective observational study that aimed at describing the routine use of DFX in patients with hematological malignancies with regard to safety, efficacy and handling of the drug in a routine setting.
Results
A total of 406 patients were included. 58% of the patients were male. Most of the patients had myelodysplastic syndromes (MDS) (68%) and myeloproliferative neoplasms (MPN) (14%). Median time from first transfusion to study enrollment was 1.1 years (0–25.5 years) and most patients were chelation naive (91%) at enrollment. With regard to transfusion burden, most of the patients were moderately or mildly transfusion-dependent with 53% receiving 2–4 and 27% receiving less than 2 units of pRBC per month. Serum ferritin decreased from a mean of 2305 μg/l (± 1449 μg/l) to a mean of 1910 μg/l (± 1529 μg/l) at 24 months. There was no substantial change in transfusion-dependence during the observation period. Dose adjustments were reported in 48% of the patients with dose-escalation strategies being the most frequent reason for dosage increases (49%). The median observation time was 355 days (5–1080 days). Median duration of exposure to DFX was 322 days (2–1078 days). Two-hundred and ninety (72%) patients discontinued the trial prematurely after a median time of 235 days (1–808 days). Death (29%) and adverse events (23%) were the main reasons for discontinuation. Eleven percent of the patients discontinued treatment due to sufficient decrease in serum ferritin. Most frequent adverse events were decrease in creatinine clearance (22%), increase in serum creatinine (18%) and diarrhea (16%).
Conclusion
This descriptive trial confirms the efficacy of DFX in decreasing the serum ferritin. Moreover, the high drop-out rates seen in prospective trials are recapitulated in this study, which can be attributed to adverse events in a substantial proportion of patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Repetitive transfusions of packed red blood cells (pRBC) put patients at risk for transfusional iron overload (IOL), since the organism has no means to actively eliminate excess iron from the body. Patients with inherited bone marrow failure syndromes, hemoglobinopathies and hematological malignancies in general and those with a chronic course of the disease in particular often require regular transfusions of pRBC over a long period of time. Usually, after a cumulative transfusion burden of 20 units pRBC, serum ferritin (SF) increases and IOL manifests.
Excessive body iron increase leads to an exhaustion of the iron binding capacity of transferrin resulting in the occurrence of the so called non-transferrin bound iron (NTBI). The labile plasma iron (LPI) is a sub-fraction of the NTBI, which catalyzes the generation of reactive oxygen species that can cause damage of organelles and DNA, ultimately inducing organ damage (Gattermann and Rachmilewitz 2011). Particularly in patients with inherited anemias, organ damage by IOL can manifest as cardiac failure, diabetes, liver cirrhosis and other organ failures. Treatment of IOL with deferoxamine (DFO) was the gold standard over several decades. However, use of DFO was hampered by the subcutaneous administration, which was associated with low compliance and subsequently insufficient iron chelation. Deferasirox (DFX) as an orally available iron chelator has proven better compliance and higher treatment adherence as compared to DFO. Moreover, it has been shown that DFX can lower serum ferritin, total body iron and organ iron deposits in different iron overloading states (Gattermann et al. 2010; Lee et al. 2010; List et al. 2012; Nolte et al. 2013; Kim et al. 2015; Latagliata et al. 2016).
Particularly for transfusion-dependent patients with inherited bone marrow failure syndromes and hemoglobinopathies it has been shown that thorough iron chelation can reduce morbidity and mortality in those patients (Modell et al. 2000; Borgna-Pignatti et al. 2004). However, this finding is controversial in elderly patients with MDS and other hematologic diseases. So far, only either retrospective studies or prospective non-randomized studies on iron chelation with DFX in patients with MDS and other hematological diseases have been published (Gattermann et al. 2010; Lee et al. 2010; List et al. 2012; Nolte et al. 2013; Kim et al. 2015; Latagliata et al. 2016). Although some of them suggest a beneficial effect of iron chelation on morbidity and mortality, conclusions that can be drawn from these studies are highly limited due to the study design and applied statistics (Leitch et al. 2008, 2017; Rose et al. 2010; Komrokji et al. 2011; Neukirchen et al. 2012; Delforge et al. 2014). In addition, in clinical trials with DFX such as the EPIC or the US03 trial high drop-out rates have been reported which were mainly due to gastrointestinal and renal adverse events (Gattermann et al. 2010; List et al. 2012).
Here we report on the results of a prospective observational study that investigated the safety and efficacy of DFX in patients with hematological disease in a routine setting outside clinical trials.
Methods
Study design
This was a prospective, multi-center, non-interventional, observational study on safety, tolerability and efficacy of DFX in patients with transfusion-dependent iron overload. After enrollment patients were scheduled to be followed for 24 months with 3-monthly follow-up visits during the first 12 months and 6-monthly visits thereafter. Diagnostic and medically indicated examinations were performed and documented under daily routine practice. The study was approved by the ethics committee of the medical faculty Mannheim of the University of Heidelberg as well as by the local ethics committees of the university of Göttingen and Sachsen-Anhalt.
Study population
Patients had to meet the following inclusion criteria: written informed consent of either patient him or herself or the legal representative, established indication for iron chelation (IC) according to the European label of DFX, i.e., patients with beta-thalassemia major aged 6 years or older with iron overload due to frequent blood transfusions (7 mL/kg bw per month) and patients with chronic iron overload due to blood transfusions when deferoxamine therapy is contraindicated or inadequate in the following patient groups: in pediatric patients with beta-thalassemia major with iron overload due to frequent blood transfusions (≥ 7 mL/kg/month of packed red blood cells) aged 2–5 years, in adult and pediatric patients with beta-thalassemia major with iron overload due to infrequent blood transfusions (< 7 mL/kg/month of packed red blood cells) aged 2 years and older, in adult and pediatric patients with other anemias (such as MDS or MPN and others) aged 2 years and older and in patients with chronic iron overload requiring chelation therapy when deferoxamine therapy is contraindicated or inadequate in patients with non-transfusion-dependent thalassemia syndromes aged 10 years and older.
Dosing of DFX
DFX was administered orally at doses of 10–30 mg/kg body weight (bw). The recommended starting dose was 20 mg/kg bw. DFX dose could be increased in patients showing insufficient treatment response indicated by persistently high SF levels. The maximum dose allowed to be administered according to the DFX label was 40 mg/kg bw.
Safety and efficacy assessments
Safety and tolerability were assessed by documentation of adverse events (AE), physical examinations and laboratory assessments. AE were summarized using the MedDRA (version 19.1) coding system. Serious adverse events (SAEs) were defined as AEs which were fatal or requiring an unplanned or unforeseen hospitalization or prolongation of hospitalization, or resulted in persistent or significant disability/incapacity or impairment or congenital anomalies or birth defects or other medically important events. If AEs or SAEs were considered being related to the study drug they were categorized as either non-severe adverse drug reaction (nsADR) or severe adverse drug reaction (sADR).
Efficacy was evaluated based on the change in serum ferritin levels. Moreover, data on the transfusion burden during the observation were collected.
Statistical analysis
The trial was designed as a descriptive trial testing no hypotheses. Summary statistics were used for categorical and continuous data. The main analysis sets of the study are the full analysis set (FAS) and the safety analysis set (SAS). The FAS contains all patients who were treated according to the summary of product characteristic (SmPc) (i.e., patients with a creatinine clearance of < 60 mL/min were not included), who had received at least one dose of DFX and who had at least one follow-up information documented. Patients in the FAS were stratified based on baseline SF. The SAS consisted of the FAS population and patients who did receive DFX although not fulfilling the SmPC specification of a minimum creatinine clearance of 60 mL/min. A preplanned subgroup analysis was performed on the MDS patients included in the trial. Patients were stratified by IPSS risk categories and baseline SF. Analyses were done by using the Statistical Analysis Software (SAS) (version 9.4).
Results
Patient characteristics
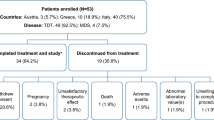
Between July 2010 and December 2014, 505 patients were included in the study database. Finally, the FAS consisted of 406 patients and the SAS consisted of 429 patients (Fig. 1). Most frequently documented primary disease was myelodysplastic syndrome (MDS) in 276 (68%) of the patients, followed by myeloproliferative neoplasm and acute myeloid leukemia. Median time from diagnosis of primary disease was 2.0 years (0–30.9 years). Four hundred and one (99%) of 406 patients were dependent on blood transfusions and median time since start of first transfusion was 1.1 year (0–25.5 years). Most of the patients had received more than 20 units of pRBC (n = 248, 61%). Of 406 patients, 369 (91%) were naive to iron chelation. See Table 1 for summary of patient characteristics in the FAS. The median observation time was 355 days (range 5–1080 days).

Analysis sets
Dosing of study drug
The reason for initiating treatment with DFX was SF > 1000 ng/mL in 349 (86%) patients and a history of > 20 transfused units of pRBC in 38 (9%) patients. The mean prescribed dosage of DFX at start of treatment was 13.6 mg/kg (SD 6.61) with 129 (32%) patients receiving < 10 mg/kg at treatment start, 83 (20%) 10 to < 15 mg/kg, 61 (15%) 15 to < 20 mg/kg, 109 (27%) 20 to < 25 mg/kg, 5 (1.2%) 25 to < 30 mg/kg, and 10 (2.5%) ≥ 30 mg/kg, respectively.
Dose adjustments
Dose adjustments were reported for 194 patients. One, two, three or more than three dose adjustments were reported for 89 (22%), 42 (10%), 30 (7.4%) and 33 (8.1%), respectively. Dose increases and decreases were reported for 145 and 103 patients, respectively. One, two, three or more than three dose increases were reported for 91 (22%), 23 (5.7%), 19 (4.7%) and 12 (3%), respectively, while dose decreases were reported for 68 (17%), 23 (5.7%), 8 (2%) and 4 (1%) of the patients.
Reasons for increases in DFX dosage were reported to be upfront intended dose-escalation strategies in 124 (49%) and too weak chelation in 63 (25%) patients. Decreases in DFX dose were reported to be due to adverse events (AEs) in 62 (39%) and patient’s decision in 25 (16%) of the patients.
Exposure to DFX and premature discontinuation
Overall, the median duration of exposure to DFX was 322 days (range 2–1078 days). Of 406 patients, 290 (72%) discontinued the treatment prematurely after a median time of 235 days (range 1–808 days). Of those, 205 (70%) discontinued treatment within the first 12 months. Discontinuation was reported to be due to death of the patient in 83 (29%), adverse events in 67 (23%), lost to follow-up in 28 (9.6%), progression of primary disease or progression to AML in 24 (8.2%), and deterioration of the general condition of the patient in 21 (7.2%) of the cases, respectively. Achievement of the target SF value as a reason for discontinuation was reported for 32 (11%) patients (Table 2).
Adverse events
Of 429 patients included in the SAS, 377 (88%) experienced an AE. Gastrointestinal disorders and a decrease in creatinine clearance were reported in 131 (31%) and 96 (22%) patients, respectively, representing the most frequent AEs in this patient cohort. Diarrhea (n = 67, 16%) and nausea (n = 45, 10%) were most prevalent gastrointestinal disturbances reported and were considered nsADR in 54 (80%) and 35 (78%) of the cases. In 13 of 96 patients (13%), the decrease in creatinine clearance was considered a nsADR and in 56 (58%) a sADR. Overall, sADRs were documented for 149 (35%) patients. Table 3 gives an overview on the most frequent AEs.
Of 1655 reported AEs, 392 (24%) and 272 (16%) were assessed to be nsADRs and sADRs. There was no obvious change in incidences of nsADRs or sADRs with regard to treatment duration (Table 4).
With regard to adverse events, overall there was no substantial difference between the different starting doses (Table 5) nor was the frequency of gastrointestinal disturbances or changes in creatinine clearance or blood creatinine levels (Table 6).
Measurement of liver iron concentration and liver function parameters
In a total of 11 patient measurement of liver iron concentration (LIC) was performed by magnetic resonance imaging (MRI). Of those, ten had baseline values available. Mean LIC was 9.5 mg Fe/g dry weight (dw) (± 5.6 mg Fe/g dw). However, no follow-up measurements were performed. There were no significant changes in liver and biliary serum parameters, i.e., alanine amino transferase (ALT), aspartate amino transferase (AST), alkaline phosphatase (AP) and bilirubin detected during follow-up as compared to baseline values.
Efficacy
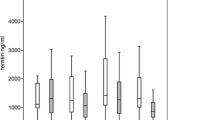
Mean serum ferritin was 2305 µg/L (± 1449 µg/L) at baseline and declined to a mean of 1910 µg/L (± 1529 µg/L) at 24 months. Regarding the changes according to the initially planned dose groups, changes in SF from baseline to 24 months were as follows: + 332 µg/L (± 2424 µg/L) in the < 10 mg/kg dose group, − 523 µg/L (± 1227 µg/L) in the 10 to <15 mg/kg dose group, − 315 µg/L (± 1021 µg/L) in the 15 to <20 mg/kg dose group, − 670 µg/L (± 1444 µg/L) in the 20 to < 25 mg/kg dose group, − 964 µg/L (± 408 µg/L) in the 25 to < 30 mg/kg dose group and − 208 µg/L (± 1823 µg/L) in the > 30 mg/kg dose group, respectively. The overall change in SF was − 254 (± 1761 µg/L).
Overall, during the observation period the median number of pRBC was 24 (0–134) and the median number of pRBC per month was 2 (0–21) at baseline and 2 (0–22) at 24 months.
Discussion
Here we report on the results of a German 2-year non-interventional trial that aimed at describing the routine use of DFX in the treatment of IOL in hematologic diseases and further characterizing the safety and efficacy profile of the drug.
A total of 406 patients were included in this trial, with patients diagnosed with MDS being the largest proportion of this cohort. The median age was 73 and most patients suffered from concomitant diseases such as cardiovascular disorders, diabetes and gastrointestinal diseases, respectively. Taken together, the cohort represented the typical patients that are commonly seen in daily routine practice.
The transfusion burden was high in the majority of the patients with 71% of the patients receiving more than two pRBC per month at study enrollment. However, only few patients had received IC prior to enrollment. Moreover, the mean initial daily dose was rather low with 13.6 mg/kg bw per day which was mainly due to preplanned dose-escalation strategies by the treating physician and reported for 124 of the patients who experienced dose increases. This most likely reflects the awareness for adverse events and application of means to prevent AEs particularly gastrointestinal disturbances associated with DFX treatment.
Most of the patients experienced one or more adverse events. As expected and reported by other groups, AEs affecting the gastrointestinal system and renal system were the most frequent AEs. Both gastrointestinal and renal AEs were considered drug related in the majority of cases. Although dose-escalation strategies were followed in a substantial proportion of the patients, overall incidences of AE do not seem to be affected in our study. In addition, 290 (71%) patients discontinued the treatment prematurely after a median time of 235 days with AEs being the reason for discontinuation in 23% of the patients. Noteworthy, of 290 patients who discontinued treatment 70% of those discontinued within the first 12 months. This indicates that tolerability of DFX still is a substantial obstacle to a thorough and efficient IC. Whether the new DFX formulation (film-coated tablet) will result in a significantly greater adherence to the treatment is not yet clear, although results from a recently published trial suggest better compliance. However, this trial used descriptive statistics only without testing for statistical significance between a dispersible and film-coated formulation of DFX (Taher et al. 2017). There was no substantial difference in the overall frequency of AEs between the different starting dose group nor was the frequency of specific gastrointestinal disturbances or renal events. However, the number of patients in the higher dose groups were small, i.e., the conclusions that can be drawn from these data are limited.
Overall, iron overload was moderate indicated by a mean LIC of 9.5 mg Fe/kg dw measured by liver MRI. Unfortunately, no information was available on the course of LIC in single patients.
The mean serum ferritin level decreased by 254 µg/L (± 1761 µg/L) from baseline to follow-up at 24 months, which seemed to be dose dependent since the reduction in SF increased with increasing initial dose levels. This is in line with previously reported data on the effect of IC on SF.
Transfusion dependency remained stable during treatment, i.e., providing no evidence for erythroid improvement under IC with DFX in our cohort. However, in our trial and other preceding trials hematologic improvement was not the primary endpoint.
In conclusion, our data confirm the tolerability and efficacy profile of DFX. Still, tolerability remains an important issue when considering treatment adherence. The question whether or not elderly patients with IOL do benefit from IC remains elusive and should be a subject of randomized clinical trials.
References
Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC et al (2004) Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 89(10):1187–1193
Delforge M, Selleslag D, Beguin Y, Triffet A, Mineur P, Theunissen K et al (2014) Adequate iron chelation therapy for at least six months improves survival in transfusion-dependent patients with lower risk myelodysplastic syndromes. Leuk Res 38(5):557–563
Gattermann N, Rachmilewitz EA (2011) Iron overload in MDS-pathophysiology, diagnosis, and complications. Ann Hematol 90(1):1–10
Gattermann N, Finelli C, Porta MD, Fenaux P, Ganser A, Guerci-Bresler A et al (2010) Deferasirox in iron-overloaded patients with transfusion-dependent myelodysplastic syndromes: results from the large 1-year EPIC study. Leuk Res 34(9):1143–1150
Kim IH, Moon JH, Lim SN, Sohn SK, Kim HG, Lee GW et al (2015) Efficacy and safety of deferasirox estimated by serum ferritin and labile plasma iron levels in patients with aplastic anemia, myelodysplastic syndrome, or acute myeloid leukemia with transfusional iron overload. Transfusion 55(7):1613–1620
Komrokji RS, Ali NHA, Padron E, Lancet JE, List AF (2011) Impact of iron chelation therapy on overall survival and AML transformation in lower risk MDS patients treated at the moffitt cancer center. Blood 118(21):2776
Latagliata R, Montagna C, Porrini R, Di Veroli A, Leonetti SC, Niscola P et al (2016) Chelation efficacy and erythroid response during deferasirox treatment in patients with myeloproliferative neoplasms in fibrotic phase. Eur J Haematol 96(6):643–649
Lee JW, Yoon SS, Shen ZX, Ganser A, Hsu HC, Habr D et al (2010) Iron chelation therapy with deferasirox in patients with aplastic anemia: a subgroup analysis of 116 patients from the EPIC trial. Blood 116(14):2448–2454
Leitch HALC., Goodman TA et al (2008) Improved survival in patients with myelodysplastic syndrome receiving iron chelation therapy. Clin Leuk 2(3):205–211
Leitch HA, Parmar A, Wells RA, Chodirker L, Zhu N, Nevill TJ et al (2017) Overall survival in lower IPSS risk MDS by receipt of iron chelation therapy, adjusting for patient-related factors and measuring from time of first red blood cell transfusion dependence: an MDS-CAN analysis. Br J Haematol 179(1):83–97
List AF, Baer MR, Steensma DP, Raza A, Esposito J, Martinez-Lopez N et al (2012) Deferasirox reduces serum ferritin and labile plasma iron in RBC transfusion-dependent patients with myelodysplastic syndrome. J Clin Oncol 30(17):2134–2139
Modell B, Khan M, Darlison M (2000) Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet 355(9220):2051–2052
Neukirchen J, Fox F, Kundgen A, Nachtkamp K, Strupp C, Haas R et al (2012) Improved survival in MDS patients receiving iron chelation therapy—a matched pair analysis of 188 patients from the Dusseldorf MDS registry. Leuk Res 36(8):1067–1070
Nolte F, Hochsmann B, Giagounidis A, Lubbert M, Platzbecker U, Haase D et al (2013) Results from a 1-year, open-label, single arm, multi-center trial evaluating the efficacy and safety of oral Deferasirox in patients diagnosed with low and int-1 risk myelodysplastic syndrome (MDS) and transfusion-dependent iron overload. Ann Hematol 92(2):191–198
Rose C, Brechignac S, Vassilief D, Pascal L, Stamatoullas A, Guerci A et al (2010) Does iron chelation therapy improve survival in regularly transfused lower risk MDS patients? A multicenter study by the GFM (Groupe Francophone des Myelodysplasies). Leuk Res 34(7):864–870
Taher AT, Origa R, Perrotta S, Kourakli A, Ruffo GB, Kattamis A et al (2017) New film-coated tablet formulation of deferasirox is well tolerated in patients with thalassemia or lower-risk MDS: Results of the randomized, phase II ECLIPSE study. Am J Hematol 92(5):420–428
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
FN has received research funding and honoraria from Novartis Pharma GmbH; HN, BS, TG, OR, HH, AJ and CS declare no conflict of interest; SA, CJ are employees of Novartis Pharma GmbH; WKH has received research funding from Novartis Pharma GmbH.
Ethical approval
All procedures performed in the trial involving human participants were in accordance with ethical standards of the national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Nolte, F., Nückel, H., Schmidt, B. et al. Tolerability and efficacy of deferasirox in patients with transfusional iron overload: results from a German 2-year non-interventional study. J Cancer Res Clin Oncol 144, 1531–1538 (2018). https://doi.org/10.1007/s00432-018-2665-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-018-2665-x