
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that leads to movement disorders. In motor neurons of ALS patients, intracellular aggregates of superoxide dismutase 1 (SOD1) have often been observed. To elucidate the aggregation mechanism, it is important to analyze the folding equilibrium of SOD1 between folded and aggregation-prone unfolded states. However, in most cases, this folding equilibrium has been studied in dilute solution even though the aggregate formation occurs in a highly crowded intracellular environment. Indeed, a recent study reported that the folding stability of SOD1 decreased in an environment containing protein crowder molecules. To understand such a destabilization effect due to protein crowders, it is necessary to obtain more precise structural information on SOD1 in the presence of protein crowders. Here, we report the 1H, 13C, and 15N backbone resonance assignments of monomeric SOD1 in the absence and presence of the protein crowder lysozyme. The chemical shift differences caused by addition of lysozyme suggest that SOD1 associated with lysozyme via negatively charged surfaces. Based on the assigned chemical shifts, the presence of lysozyme has a limited influence on the secondary structure of SOD1. We anticipate that our assignments will provide an important basis for elucidation of the crowding-induced folding destabilization of SOD1.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Biological context
Cu/Zn-superoxide dismutase 1 (SOD1) is a cytoplasmic radical scavenger that catalyzes the dismutation of superoxide to dioxygen and hydrogen peroxide. In the cytosol, SOD1 forms an enzymatically active homodimer. Each monomeric subunit in a dimer coordinates one catalytic Cu+/2+ ion and one structural Zn+ ion; in addition, one intra-subunit disulfide bond is formed. Loss of both metal binding and disulfide bridge causes dissociation of the dimer into monomeric apoSOD1 that has a high aggregation propensity (Stathopulos et al. 2006). Intracellular aggregates of SOD1 have often been observed in patients of amyotrophic lateral sclerosis (ALS) and the SOD1-mediated degeneration of motor neurons causes progressive weakness of muscle strength, which leads to difficulties in speaking, swallowing, and breathing within 3 to 5 years after disease onset (Armon 1994). Although 90% of ALS cases are sporadic (sALS), the remaining 10% are familial (fALS). Approximately 20% of fALS are associated with genetic mutations in the SOD1 gene (Rosen et al. 1993) and most of these mutations are thought to increase dimer dissociation (Broom et al. 2015). To reveal the mechanism of formation of the pathological aggregates in ALS patients, it is thus important to analyze the folding stability of monomeric apoSOD1.
To simplify the investigation of the folding stability of monomeric apoSOD1, a loop-truncated form, in which all cysteine residues are mutated to serine residues (SOD1ΔIV,ΔVII), has been established previously (Danielsson et al. 2011). Since SOD1ΔIV,ΔVII lacks the ability to bind metals and to form disulfide bonds, it exists as a monomer and its folded state is in equilibrium only with the unfolded state that is a possible precursor of pathological aggregation. The in vitro dynamic exchange between the folded and unfolded states has been quantitatively analyzed by relaxation dispersion NMR spectroscopy (Danielsson et al. 2013). Furthermore, crowded environments affect the folding equilibrium of SOD1ΔIV,ΔVII and the equilibrium is shifted toward the unfolded state in mammalian and bacterial cells (Danielsson et al. 2015). Remarkably, the presence of lysozyme exerts a similar effect on the stability of SOD1ΔIV,ΔVII as the intracellular environment (Danielsson et al. 2015). As a result, it would be intriguing to examine the crowding effect of lysozyme on SOD1ΔIV,ΔVII in more detail.
In this study, we present the 1H, 13C, 15N backbone resonance assignments of SOD1ΔIV,ΔVII in the absence and presence of the protein crowder lysozyme. These assignments will be valuable for identification of the interface between SOD1ΔIV,ΔVII and lysozyme and for further structural analyses.
Methods and experiments
Expression and purification
A pET3a vector encoding the H46W mutant of the loop-truncated human SOD1ΔIV,ΔVII (hereafter: SOD1) was transformed into Escherichia coli strain BL21(DE3). Cells were cultured in M9 minimum media containing 2 g/L U-13C glucose (Cambridge Isotope Laboratory), 0.5 g/L U-15N ammonium chloride (Cambridge Isotope Laboratory) and 50 mg/L ampicillin (Wako). Protein expression was induced with 0.5 mM isopropyl 1-thio-β-d-galactopyranoside (Nacalai Tesque) overnight at 23 °C. All protein purification steps were performed at 4 °C. Harvested cells were resuspended and sonicated in 50 mM Tris–HCl (pH 7.5 at 25 °C). The cell lysate was centrifuged at 38,750×g for 25 min and the supernatant was purified using a two-step ammonium sulfate precipitation. The supernatant was first subjected to a 50% w/v ammonium sulfate precipitation, followed by centrifugation at 38,750×g for 25 min. After a second precipitation at 90% w/v saturation, the pellet was resuspended in 10 mM Tris-HCl (pH 7.5 at 25 °C) and then the solution was dialyzed against 10 mM Tris-HCl (pH 7.5 at 25 °C) to remove residual ammonium sulfate. The dialyzed sample was loaded on a HiTrap Q column (GE Healthcare) and the protein was eluted by a 0 to 300 mM sodium chloride gradient. Fractions containing SOD1 were loaded on a Superdex 75 16/60 gel filtration column (GE Healthcare). The final fractions containing purified SOD1 were concentrated to 1 mM and stored at −80 °C. Protein purity was checked by SDS–PAGE.
NMR measurements and data analysis
All NMR spectra were obtained at 25 °C on a 600 MHz Bruker Avance DRX spectrometer equipped with a 5-mm TXI triple resonance cryoprobe (Bruker BioSpin). The uniformly 13C, 15N-labeled samples in 10 mM Bis-Tris HCl pH 6.3, 5% D2O with and without 50 mg/mL (3.5 mM) hen egg white lysozyme (Nacalai Tesque) were used for the NMR experiments. The samples were prepared at a protein concentration of 1 mM. Protein concentration was determined by absorbance at 280 nm using NanoDrop 2000c (Thermo Fisher Scientific). For the sequential backbone assignments, 1H-15N HSQC, HNCO, HN(CA)CO, HN(CO)CA, HNCA, CBCA(CO)NH, HNCACB, and CC(CO)NH spectra were acquired. 1H chemical shifts were calibrated with sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS: Tokyo Chemical Industry) and both 13C and 15N chemical shifts were calibrated indirectly (Markley et al. 1998). Data were processed by NMRPipe (Delaglio et al. 1995) and sequential assignments were performed manually using MagRO NMRView (Johnson and Blevins 1994; Kobayashi et al. 2007).
Assignments and data deposition
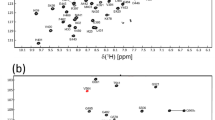
We assigned 98% of the backbone HN, N, Cα, Cβ, and CO resonances for SOD1 in the absence of lysozyme (Fig. 1a). The resonances of the residues Lys92 and Gly96 were missing because of severe line broadening. For SOD1 in the presence of lysozyme, 100% of the backbone HN, N, Cα, Cβ, and CO resonances were assigned (Fig. 1b). These resonance assignments have been deposited in BioMagResBank (BMRB, http://www.bmrb.wisc.edu) under the accession numbers 26893 and 26894 in the absence and presence of lysozyme, respectively. The mean-weighted chemical shift differences of the HN and N resonances indicate that SOD1 interacts with lysozyme via its negatively charged surfaces centered around Asp53 and Asp71 (Fig. 2b). Note that lysozyme is positively charged at neutral pH. Furthermore, we estimated the secondary structure propensities of SOD1 in the absence and presence of lysozyme based on the respective backbone chemical shifts of HN, N, Cα, Cβ, and CO (Marsh et al. 2006). As a result, there was no significant change of the secondary structure propensities of SOD1 due to the addition of lysozyme (Fig. 3). Taken together, these results argue that lysozyme associates with SOD1 through formation of electrostatic interactions; however, it induces little conformational changes in the backbone of SOD1. Our assignments will contribute to elucidating the folding destabilization of SOD1 in macromolecular-crowded environments.

1H-15N HSQC spectra of SOD1 in the presence and absence of a macromolecular crowding agent. The spectra of 1 mM 13C/15N-labeled SOD1 without (a) and with (b) 50 mg/mL lysozyme. The dashed inset displays an enlarged view of the middle region of the full spectrum

Backbone chemical shift differences induced by addition of lysozyme. a Mean-weighted chemical shift differences calculated according to the equation Δδ = {(ΔδH)2 + (0.15ΔδN)2}1/2 where ΔδH and ΔδN are the differences between 1H and 15N backbone chemical shifts in the absence and presence of lysozyme, respectively. Magenta and purple lines represent the average Δδav of the chemical shift differences and one standard deviation above the average (Δδav + 1σ), respectively. The residues exhibiting Δδ > (Δδav + 1σ) are colored purple and those exhibiting Δδav ≤ Δδ ≤ (Δδav + 1σ) are colored magenta. b Structural mapping of the residues exhibiting Δδ ≥ Δδav (upper). Electrostatic potential surface (lower). PDB database accession code: 4BCZ

Secondary structure propensities. Secondary structure propensities of SOD1 without (black) and with (red) lysozyme were estimated using the program SSP (Marsh et al. 2006)
References
Armon C (1994) Motor neuron disease. In: Gorelick PB, Alter M (eds) Handbook of neuroepidemiology. Marcel Dekker, New York, pp 407–454
Broom HR, Rumfeldt JAO, Vassall KA, Meiering EM (2015) Destabilization of the dimer interface is a common consequence of diverse ALS-associated mutations in metal free SOD1. Protein Sci 24:2081–2089. doi:10.1002/pro.2803
Danielsson J, Kurnik M, Lang L, Oliveberg M (2011) Cutting off functional loops from homodimeric enzyme superoxide dismutase 1 (SOD1) leaves monomeric β-barrels. J Biol Chem 286:33070–33083. doi:10.1074/jbc.M111.251223
Danielsson J et al (2013) Global structural motions from the strain of a single hydrogen bond. Proc Natl Acad Sci USA 110:3829–3834. doi:10.1073/pnas.1217306110
Danielsson J et al (2015) Thermodynamics of protein destabilization in live cells. Proc Natl Acad Sci USA 112:12402–12407. doi:10.1073/pnas.1511308112
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Johnson BA, Blevins RA (1994) NMR View: a computer program for the visualization and analysis of NMR data. J Biomol NMR 4:603–614. doi:10.1007/Bf00404272
Kobayashi N et al (2007) KUJIRA, a package of integrated modules for systematic and interactive analysis of NMR data directed to high-throughput NMR structure studies. J Biomol NMR 39:31–52. doi:10.1007/s10858-007-9175-5
Markley JL et al (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids (IUPAC Recommendations 1998). Pure Appl Chem 70:117–142. doi:10.1351/pac199870010117
Marsh JA, Singh VK, Jia ZC, Forman-Kay JD (2006) Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: implications for fibrillation. Protein Sci 15:2795–2804. doi:10.1110/ps.062465306
Rosen DR et al (1993) Mutations in Cu/Zn superoxide-dismutase gene are associated with familial amyotrophic-lateral-sclerosis. Nature 362:59–62. doi:10.1038/362059a0
Stathopulos PB, Rumfeldt JAO, Karbassi F, Siddall CA, Lepock JR, Meiering EM (2006) Calorimetric analysis of thermodynamic stability and aggregation for apo and holo amyotrophic lateral sclerosis-associated Gly-93 mutants of superoxide dismutase. J Biol Chem 281:6184–6193. doi:10.1074/jbc.M509496200
Acknowledgements
We thank Dr. Jens Danielsson and Ms. Sarah Leeb for kindly providing the expression plasmid for SOD1. We are also thankful to Dr. Jens Danielsson for constructive discussions. This work was supported by the scholar project of Toyota Physical and Chemical Research Institute, and by JSPS KAKENHI Grant Number JP26119004.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
Naoto Iwakawa and Daichi Morimoto have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Iwakawa, N., Morimoto, D., Walinda, E. et al. Backbone resonance assignments of monomeric SOD1 in dilute and crowded environments. Biomol NMR Assign 11, 81–84 (2017). https://doi.org/10.1007/s12104-016-9724-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12104-016-9724-5