
Abstract
Matrin-3 is a multifunctional protein that binds to both DNA and RNA. Its DNA-binding activity is linked to the formation of the nuclear matrix and transcriptional regulation, while its RNA-binding activity is linked to mRNA metabolism including splicing, transport, stabilization, and degradation. Correspondingly, Matrin-3 has two zinc finger domains for DNA binding and two consecutive RNA recognition motif (RRM) domains for RNA binding. Matrin-3 has been reported to cause amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) when its disordered region contains pathogenic mutations. Simultaneously, it has been shown that the RNA-binding activity of Matrin-3 mediated by its RRM domains, affects the formation of insoluble cytoplasmic granules, which are related to the pathogenic mechanism of ALS/FTD. Thus, the effect of the RRM domains on the phase separation of condensed protein/RNA mixtures has to be clarified for a comprehensive understanding of ALS/FTD. Here, we report the 1H, 15N, and 13C resonance assignments of the two RNA binding domains and their solution structures. The resonance assignments and the solution structures obtained in this work will contribute to the elucidation of the molecular basis of Matrin-3 in the pathogenic mechanism of ALS and/or FTD.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Biological context
Matrin-3, which was first identified as a protein component of the nuclear matrix (Nakayasu and Berezney 1991; Belgrader et al. 1991), is a multiple functional protein. As a DNA-binding protein, it controls transcriptional regulation (Belgrader et al. 1991; Hibino et al. 1993a, b, 1998, 2000; Niimori-Kita et al. 2018). Furthermore, as an RNA-binding protein, it plays important roles in mRNA metabolism including splicing, transport, stabilization, and degradation (Salton et al. 2011; Kula et al. 2013; Uemura et al. 2017; Coelho et al. 2015; Boehringer et al. 2017; Banerjee et al 2017; Ahmed and Barmada 2021).
Matrin-3 is well-conserved among vertebrates. Human Matrin-3 is composed of 847 amino-acid residues and possesses two CCHH-type zinc finger (ZF) domains (the regions spanning residues H291-Y326 for ZF1 and P797-T847 for ZF2, respectively), which were reportedly responsible for the DNA-binding activity of Matrin-3 (Hibino et al. 2000). In the peptide region between the two ZF domains, there are two tandemly-linked RNA recognition motif domains (RRM domains) spanning residues R398-I477 and R496-V576, respectively (referred to as RRM1 and RRM2, hereafter in the text) (Supplementary Fig. 1a) (Hibino et al. 2006). It has been reported that RRM2 could bind to the RNA sequence (5′-AUCUU-3′) (Ray et al. 2013), but RRM1 does not show any substantial RNA binding activity (Ayala et al. 2005; Kuo et al. 2009; Buratti and Baralle 2001). On the other hand, enhanced cross-linking immunoprecipitation (eCLIP) experiments showed another RNA sequence bound to Matrin-3 other than the pyrimidine-rich sequence described above (Ramesh et al. 2020a, b; Van Nostrand et al. 2016). The remaining amino-acid sequences in Matrin-3, namely the N-terminal region, and that region between RRM2 and ZF2, consist of two intrinsically disordered regions, termed N- and C-IDR, respectively.
Recently, Matrin-3 has attracted remarkable attention, since functional abnormalities of Matrin-3 cause hard-to-treat neuromuscular human diseases, amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD) (Brown and Al-Chalabi 2017; Ito et al. 2017; Xue et al. 2020; Ahmed and Barmada 2021; Malik and Barmada 2021). For familial ALS, a growing number of genetic mutations have been identified on several genes. In particular, pathogenic mutants of TAR DNA-binding protein of 43 kDa (TDP-43) and fused in sarcoma (FUS) form insoluble granules in the cytoplasm, leading to neuronal cell death (Kamelgarn et al. 2016; Picchiarelli and Dupuis 2020). Matrin-3 has also been associated with familial ALS. The S85C, F115C, P154S, and T622A mutants within the N- and C-IDR of Matrin3 have been identified as pathogenic (Lin et al. 2015; Leblond et al. 2016; Xu et al. 2016; Marangi et al. 2017). In this case, the inclusion bodies of wild-type TDP-43 proteins with the mutated Matrin-3 are frequently formed in the cytoplasm. Moreover, even in the sporadic ALS cases, the insoluble granules of wild-type TDP-43 formed in the neural cytoplasm also frequently contain wild-type Matrin-3 (Tada et al. 2018).
In normal neurons, Matrin-3 adopts a granular nuclear localization. However, in spinal cord samples obtained from patients with ALS, Matrin-3 diffuses in the cytoplasm and is involved in the formation of insoluble granules in spinal motor neurons. Recent studies have suggested that the formation of cytoplasmic aggregates of TDP-43 is dependent on a liquid–liquid phase separation (LLPS) and dysfunctional Matrin-3 affects the LLPS of TDP-43. In this case, it has been deduced that the solubility of Matrin-3 is increased upon the interaction with RNA, which suppresses the formation of an abnormal LLPS (Maharana et al. 2018; Gallego-Iradi et al. 2019; Česnik et al. 2020).
On the other hand, it has been also reported that the RNA-binding activity of Matrin-3 could accelerate ALS pathogenesis. The G4C2 repeat expansion in the first intron of the C9orf72 gene is the most common genetic cause of ALS. When its abnormally-transcribed RNA molecules are translocated into the cytoplasm, they are speculated to recruit several RNA binding proteins for the formation of the insoluble granules, and also Matrin-3 through protein-RNA interaction (DeJesus-Hernandez et al. 2011, Renton et al. 2011; Ramesh et al. 2020a).
These evidences suggest that the RNA binding activity mediated by RRMs of Matrin-3 could play an important role in the formation of insoluble granules in the cytoplasm. However, it is not clear yet how their RNA binding activities contribute to ALS pathogenesis, which has been hypothesized to be through protein–protein and/or protein-RNA interactions (Kamelgarn et al. 2016; Iradi et al. 2018; Malik et al. 2018; Ramesh et al. 2020b).
Here we report the 1H, 13C, and 15N chemical shift assignments and the solution structures of the two RRM domains of mouse Matirn-3, since the amino-acid sequences of its RRM domains are entirely identical to the corresponding human domains. These assignments and the structural information obtained in this work will provide insight in the further understanding of the neurodegenerative disease caused by Matrin-3.
Methods and experiments
Sample preparation
A clone of mouse Matrin-3 was utilized, as the primary sequences of the regions corresponding to RRM1 and RRM2 are identical between humans and mice. In our study, a cDNA clone with a natural variation (an S397R mutation), which appeared at a position just preceding the RRM1 region, was used for plasmid construction. Mouse Matrin-3 is composed of 846 amino-acid residues. The protein samples used for the NMR experiments were RRM1 and RRM2 of mouse Matrin-3, corresponding to residues Q390-K478 (RRM1) and K478-V576 (RRM2), respectively (Supplementary Fig. 1b). The folding states of the proteins were checked by 2D 1H–15N HSQC experiments with 15N-labeled samples (Kigawa et al. 2004), and we could produce the two RRM domains in soluble forms.
15N/13C-labeled Matrin-3 RRM1 and RRM2 were synthesized using an Escherichia coli cell-free protein synthesis system (Kigawa et al. 2004; Matsuda et al. 2007) and treated and purified as described previously (Li et al. 2008). The samples were expressed as N-terminal His-tagged fusion proteins. The fusion proteins were purified using a Ni–NTA affinity column. The His-tag was released by TEV protease cleavage and the two RRM domains were further purified using Superdex-75 gel filtration chromatography (GE Healthcare). For structure determination, uniformly 15N/13C-labeled RRM samples were concentrated to nearly 1.0 mM in 20 mM Tris–HCl (Tris-d6) buffer (pH 7.0), containing 100 mM NaCl, 1 mM dithiothreitol, and 0.02% NaN3 with the addition of 2H2O to 10% v/v.
NMR spectroscopy and structure calculations
All NMR data were acquired at 298 K on Bruker 600 MHz and Bruker 800 MHz spectrometers and processed with NMRPipe software (Delaglio et al. 1995). Two-dimensional 1H–13C and 1H–15N HSQC spectra, three-dimensional HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, HBHA(CO)NH, H(CCCO)NH, (H)CC(CO)NH, HCCH-TOCSY, HCCH-COSY, CCH-TOCSY and NOESY spectra (Clore and Gronenborn 1998; Cavanagh et al. 2007) were used to assign all carbon, nitrogen, and hydrogen atoms of the proteins.
NOE peaks from the 15N and 13C-edited 3D NOESY spectra with 80 ms mixing time were converted to distance restraints for the structure calculations of Matrin-3 RRM1 and RRM2. The three-dimensional structures of the proteins were determined by combined automated NOESY cross peak assignment and structure calculation with torsion angle dynamics (Herrmann et al. 2002) implemented in the program CYANA 2.1 (Güntert et al. 1997). The dihedral angle restraints for ϕ and ψ were obtained from the main-chain and the 13Cβ chemical shift values using the program TALOS (Cornilescu et al. 1999) and by analyzing the NOESY spectra. Stereospecific assignments for isopropyl methyl and methylene groups were determined based on the patterns of the inter- and intra-residual NOE intensities (Powers et al. 1993). For each RRM, the structure calculations started from 200 randomized conformers using the standard CYANA simulated annealing schedule, with 40,000 torsion angle dynamics steps per conformer (Güntert and Buchner 2015). Among them, the 20 structures with the lowest CYANA target function values were deposited in the Protein Data Bank (accession codes: 1X4D for RRM1 and 1X4F for RRM2).
Further refinements by restrained molecular dynamics followed by restrained energy minimization were performed for the 40 conformers with the lowest final CYANA target function values, using the Amber12 program with the Amber 2012 force field and a generalized Born model (Case et al. 2005), as described previously (Tsuda et al. 2011). Finally, the 20 conformers with the lowest Amber energy values were selected. They were deposited in the Protein Data Bank (accession codes: 7FBR for RRM1 and 7FBV for RRM2). PROCHECK-NMR (Laskowski et al. 1996) and MOLMOL (Koradi et al. 1996) were used to validate and to visualize the final structures, respectively.
Extent of resonance assignments
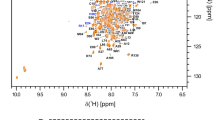
The assigned 1H-15N HSQC spectra of RRM1 and RRM2 are depicted in Fig. 1a and b. In the case of RRM1, the backbone resonance assignments were almost complete, except for the amide protons and nitrogen atoms of Val399, Lys409, Asn410, Lys433, Gln472, Lys473, and Arg476. In total, 98.8%, 100%, and 94.3% of the Cα, Cβ, and C′ chemical shifts were determined, respectively. Furthermore, the chemical shifts of the side-chain resonances except for the Cδ and Cε protons of Tyr454, and the side-chain NH2 resonances of Gln390 were also assigned. The backbone and side-chain resonance assignments for RRM2 are complete except for the amide protons and nitrogen atoms of Gln486, Lys487, Asp489, Glu493, His505, Gly507, Met531, and Lys573, the side-chain resonances of Lys479, Lys 483, Gln486, Arg530, Lys573, and the Cζ protons of Phe536. As described below, the N-terminal segment spanning residues 478–496 adopted a disordered structure, which caused the missing backbone resonances. In total, 98.9%, 98.9%, and 91.9% of the Cα, Cβ, and C′ chemical shifts were determined, respectively. For both RRM domains, all X-Pro peptide bonds were confirmed to be in the trans conformation.

1H-15N HSQC spectra of the two RRM domains of Matrin-3 a RRM1 and b RRM2. Signals are labeled with their assignments. Both data sets were acquired on Bruker 600 MHz spectrometers by the States-TPPI method with the water-flip back pulse sequence. The red-colored peak of V564 is aliased
The quality of the NOESY spectra of RRM1 and RRM2 are appropriate for straight-forward structure calculation. In the 15N- and 13C-edited 3D NOESY spectra, 2063 non-redundant distance restraints including 758 long-range distance restraints for RRM1, and 1905 non-redundant distance restraints including 707 long-range distance restraints for RRM2, were identified. The backbone torsion angle restraints calculated by the TALOS program (Cornilescu et al. 1999) were also used for structure calculations with the program CYANA 2.1 (Herrmann et al. 2002; Güntert et al. 1997; Güntert 2004) and Amber12 (Case et al. 2005). The main chain of the calculated structures of RRM1 and RRM2 were fitted for residues 398–471 and 496–569, respectively (these regions correspond to the canonical secondary structural elements of the RRM domain). A bundle of 20 conformers representing the solution structures of RRM1 and RRM2 are shown in Fig. 2a and b, respectively. Their precisions are characterized by RMSD values to the mean coordinates of 0.25 Å for the backbone atoms and 0.93 Å for all heavy atoms of RRM1, and 0.31 Å for the backbone atoms and 1.18 Å for all heavy atoms of RRM2. For these regions, the structural qualities of both RRMs also reflect that 100.0% of the (ϕ,ψ) backbone torsion angle pairs are in the most favored and additionally allowed regions of the Ramachandran plot, according to the program PROCHECK-NMR (Laskowski et al. 1996). Statistics regarding the quality and precision of the final 20 best conformers that represent the solution structures of RRM1 and RRM2 are given in Supplementary Table I.

Solution structures of the two RRM domain of Matrin-3. Best-fit superposition of the backbone atoms from the 20 structures of Matrin-3 a RRM1 and b RRM2 with the lowest energy, as calculated by CYANA2.1 and Amber12. Ribbon presentation of the lowest energy structure of Matrin-3 c RRM1 and d RRM2. The helices, β-strands, β′–β″ hairpin and loop regions are shown in red, cyan, green and gray, respectively. In addition, the regions corresponding to the C-terminal extensions specific for the PTBP-1 subgroup (Gln472–Ile477 for RRM1 and Glu570-Leu575 for RRM2) were colored brown. Electrostatic surface presentation of Matrin-3 e RRM1 and f RRM2 in the same view as (c) and (d). The back surface of the structures are shown for g RRM1 and h RRM2 (surface models (e) and (f) are rotated 180 degrees around vertical axis). Blue and red represent positive and negative electrostatic surface potentials, respectively
Solution structures of the two RRM domains of Matrin-3
The Matrin-3 RRM1 and RRM2 adopts a β1–α1–β2–β3–α3–β4 topology (β1:V399–M403, α1:N410–V419, β2:I425–L431, β3:E436–E440, α2:T444–T456, and β4:R467–L470 for RRM1, and β1:V497–S501, α1:D510–A517, β2:I523–M529, β3:Q534–E538, α2:R542–K554, and β4:K565–L568 for RRM2) (Supplementary Fig. 1 and Fig. 2a–d). As a canonical RRM fold, the four β-strands form an antiparallel β-sheet with the order of β4–β1–β3–β2. The α1 and α2 helices are packed against the β-sheet structure. The α2 helix and the β′–β″ hairpin structures (L460–P465 for RRM1, and W558–C563 for RRM2) associate with the loop between the β1 strand and the α1 helix (Fig. 2c, d).
For both RRMs, some amino-acid residues in the C-terminal regions just following the β4-strand do not show signals in the 1H-15N HSQC spectra. Thus, the C-terminal regions were determined less well than the core of the RRM-fold in the structural calculations and did not show distinct secondary structure elements. However, the C-terminal regions were located near the β-sheet surface by NOEs between hydrophobic amino-acid residues (Y474 and I477 of RRM1, and Y572 and L575 of RRM2, Supplementary Figs. 1 and 2) and hydrophobic amino-acid residues in the β-sheets (V399, L429, and F438 of RRM1, and V497, I527, and F536 of RRM2) respectively. The C-terminal extension of PTBP1 RRM1 also exhibited the same structural feature, as reported previously (Oberstrass et al. 2005) (Supplementary Fig. 2).
Analysis using the DALI protein structural comparison server (http://ekhidna2.biocenter.helsinki.fi/dali/) showed that the overall structures of Matrin-3 RRM1 and RRM2 are very similar to the first RRM domain of PTBP1 (Z-score: 10.9, RMSD: 1.76 Å for the Cα atoms of matched residues in its best 3D superimposition form PDBID:2N3O) (Supplementary Fig. 2a) and the first RRM domain of hnRNP L (Z-score: 10.3, RMSD: 1.76 Å for the Cα atoms of matched residues in its best 3D superimposition form PDBID:2MQL). Structure superpositions of Matrin-3 RRM1, RRM2, and PTBP1 RRM1 (Oberstrass et al. 2005) revealed the characteristic structural points of these RRMs as mentioned below (for comparison, the structure of Musashi-1 RRM1 is shown in Supplementary Fig. 2b as an example of a “standard” RRM domain). As previously pointed out (Blatter et al. 2015), these RRMs comprise the sub-group (hereafter referred to as the PTBP-1 subgroup).
First, the α2 helices of the PTBP-1 subgroup members are longer than in average RRM-folds. In most RRMs, the α2 helices are composed of ten or eleven residues, while the α2 helices of the Matrin-3 RRMs are composed of fourteen residues. Thus, they are one turn longer at the C-terminus than other canonical RRMs. Second, the members of the PTBP-1 subgroup have a C-terminal extension that covers the β-sheet surface (Supplementary Fig. 2). Aliphatic amino-acid residues are located in the C-terminal fragment just following the β4 strands (I477 of Matrin-3 RRM1 and L575 of Matrin-3 RRM2) and interact with hydrophobic amino-acid residues on the β-sheet surface, as it was discussed by Blatter et al. based on our deposited solution structures of the Matrin-3 RRMs (PDBID: 1X4D and 1X4F) (Blatter et al. 2015). However, the lengths between the end of the β4-strand and the key aliphatic amino-acid residues (I477 of RRM1 and L575 of RRM2) are different from that of PTBP1 RRM1 (L136 of PTBP1 RRM1). Based on a comparison of the tertiary structures, the positions corresponding to the side-chains of Lys473 of RRM1 and Lys571 of RRM2 in Matrin-3 were occupied by His133 in PTBP1 RRM1. Then, the aromatic amino-acid residue corresponding to Y474 (RRM1) and Y572 (RRM2) was not identified in PTBP1 RRM1. In the calculated structures of Matrin-3 RRM1 and RRM2, the side-chains of the preceding Lys residues (K473 of RRM1 and K571 of RRM2) seem to stack with the aromatic rings of the tyrosine residues (Y474 of RRM1 and Y572 of RRM2) and the side-chains of the aromatic amino-acid residues located on the β3-strand (F438 of RRM1 and F536 of RRM2), respectively, through the cation-π interactions (Supplementary Fig. 2c). These interactions were specific for Matrin-3 RRMs among the PTBP-1 subgroup members. In the case of Martin-3 RRMs, the aromatic side chains of Y474 of RRM1 and Y572 of RRM2 seem to occupy the space utilized for recognition of the uracil base in PTBP-1 RRM1. Instead, the spaces could be found on the opposite side of the aromatic ring of Y474 of RRM1 and Y572 of RRM2. In many RRMs, the stacking interactions between the exposed aromatic ring and RNA bases were utilized for RNA recognition (Burd and Dreyfuss 1994; Maris et al. 2005). Thus, it is probable that the spaces are utilized for the accommodation of the RNA bases.
The canonical RRM sequence has two well-conserved consensus sequences, RNP1 [(R/K)-G-(F/Y)-(G/A)-(F/Y)-V-X-(F/Y)] and RNP2 [(L/I)-(F/Y)-(V/I)-X-(N/G)-L], which correspond to the β3 strand and β1 strand, respectively (Bandziulis et al. 1989; Burd and Dreyfuss 1994; Mulder et al. 2007). In the canonical RRMs, aromatic amino-acid residues are located at the third and fifth positions of RNP1 and at the second position of RNP2. They are exposed to solvent and play important roles in the RNA binding activity of RRM domains. However, in the PTBP-1 subfamily including Matrin-3 RRM1 and RRM2, hydrophilic amino-acid residues (Glu436 for Matrin-3 RRM1 and Gln534 for Matrin-3 RRM2) are located at the third position of RNP1. In addition, His residues are located at the second position of RNP2. These features were rare in the canonical RRMs (Muto and Yokoyama 2012). Furthermore, in the case of Matrin-3 RRM1, an acidic amino-acid residue (Asp404) is also located at the position immediately following the β1 strand. Consequently, with Glu436, a negatively-charged patch was formed at the edge of the β-sheet surface of Matrin-3 RRM1 in contrast to Matrin-3 RRM2 (Supplementary Fig. 1b and Fig. 2e, f). This could be the reason that Matrin-3 RRM1 reportedly could not bind to RNA molecules. On the other hand, when the surface models of Fig. 2e, f are viewed from behind, acidic amino-acid residues were clustered and formed a wide negatively-charged patch on the upper surface formed by the C-terminus region of the α1 helix and N-terminus region of the α2 helix in Matrin-3 RRM1 and RRM2 (Fig. 2g, h), which was not obvious in PTBP1 RRM1. Therefore, this negatively-charged surface may mediate the specific protein/protein interaction of Matrin-3. We expect that the assignments and the structural information obtained in this study will provide the insight on the further understanding of the pathogenesis of ALS and/or FTD involving Matrin-3.
References
Ahmed MM, Barmada SJ (2021) Matrin 3 in neuromuscular disease: physiology and pathophysiology. JCI Insight 6:e143948. https://doi.org/10.1172/jci.insight.143948
Ayala YM, Pantano S, D’Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE (2005) Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 348:575–588. https://doi.org/10.1016/j.jmb.2005.02.038
Bandziulis RJ, Swanson MS, Dreyfuss G (1989) RNA-binding proteins as developmental regulators. Genes Dev 3:431–437. https://doi.org/10.1101/gad.3.4.431
Banerjee A, Vest KE, Pavlath GK, Corbett AH (2017) Nuclear poly(A) binding protein 1 (PABPN1) and Matrin3 interact in muscle cells and regulate RNA processing. Nucleic Acids Res 45:10706–10725. https://doi.org/10.1093/nar/gkx786
Belgrader P, Dey R, Berezney R (1991) Molecular cloning of matrin 3. A 125-kilodalton protein of the nuclear matrix contains an extensive acidic domain. J Biol Chem 266:9893–9899
Blatter M, Dunin-Horkawicz S, Grishina I, Maris C, Thore S, Maier Y, Binderreif A, Bujnicki JM, Allain FHT (2015) The signature of the five-Stranded vRRM Fold defined by FUnctional, Structural and Computational Analysis of the hnRNP L protein. J Mol Biol 427:3001–3022. https://doi.org/10.1016/j.jmb.2015.05.020
Boehringer A, Garcia-Mansfield K, Singh G, Bakkar N, Pirrotte P, Bowser R (2017) ALS associated mutations in Matrin 3 alter protein-protein interactions and impede mRNA nuclear export. Sci Rep 7:14529–14542. https://doi.org/10.1038/s41598-017-14924-6
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377:162–172. https://doi.org/10.1056/NEJMra1603471
Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276:36337–36343. https://doi.org/10.1074/jbc.M104236200
Burd CG, Dreyfuss G (1994) Conserved structures and diversities of function of RNA binding proteins. Science 265:615–621. https://doi.org/10.1126/science.8036511
Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr, Onufriev A, Simmerling C, Wang B, Woods RJ (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688. https://doi.org/10.1002/jcc.20290
Cavanagh J, Fairbrother WJ, Palmer AG III, Skelton NJ, Rance M (2007) Protein NMR spectroscopy. Principles and practice, 2nd edn. Academic Press, San Diego
Česnik AB, Motaln H, Rogelj B (2020) The impact of ALS-associated genes hnRNPA1, MATR3, VCP and UBQLN2 on the severity of TDP-43 aggregation. Cells 9:1791–1801. https://doi.org/10.3390/cells9081791
Clore GM, Gronenborn AM (1998) New methods of structure refinement for macromolecular structure determination by NMR. Proc Natl Acad Sci USA 95:5891–5898. https://doi.org/10.1073/pnas.95.11.5891
Coelho MB, Attig J, Bellora N, König J, Hallegger M, Kayikci M, Eyras E, Ule J, Smith CWJ (2015) Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J 34:653–668. https://doi.org/10.15252/embj.201489852
Cornilescu G, Delaglio F, Bax A (1999) Backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13:289–302. https://doi.org/10.1023/a:1008392405740
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung G-YR, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGG GCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based in UNIX pipes. J Biomol NMR 6:277–293. https://doi.org/10.1007/bf00197809
Gallego-Iradi MC, Strunk H, Crown AM, Davila R, Brown H, Rodriguez-Lebron E, Borchelt DR (2019) N-terminal sequences in matrin 3 mediate phase separation into droplet-like structures that recruit TDP43 variants lacking RNA binding elements. Lab Invest 99:1030–1040. https://doi.org/10.1038/s41374-019-0260-7
Güntert P (2004) Automated NMR structure calculation with CYANA. Methods Mol Biol 278:353–378. https://doi.org/10.1385/1-59259-809-9:353
Güntert P, Buchner L (2015) Combined automated NOE assignment and structure calculation with CYANA. J Biomol NMR 62:453–471. https://doi.org/10.1007/s10858-015-9924-9
Güntert P, Mumenthaler C, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol 273:283–298. https://doi.org/10.1006/jmbi.1997.1284
Herrmann T, Güntert P, Wüthrich K (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319:209–227. https://doi.org/10.1016/s0022-2836(02)00241-3
Hibino Y, Nakamura K, Tsukada S, Sugano N (1993a) Purification and characterization of nuclear scaffold proteins which bind to a highly repetitive bent DNA from rat liver. Biochim Biophys Acta 1174:162–170. https://doi.org/10.1016/0167-4781(93)90110-y
Hibino Y, Tsukada S, Sugano N (1993b) Properties of a DNA-binding protein from rat nuclear scaffold fraction. Biochem Biophys Res Commun 197:336–342. https://doi.org/10.1006/bbrc.1993.2480
Hibino Y, Ohzeki H, Hirose N, Sugano N (1998) Involvement of phosphorylation in binding of nuclear scaffold proteins from rat liver to a highly repetitive DNA component. Biochim Biophys Acta 1396:88–96. https://doi.org/10.1016/s0167-4781(97)00176-0
Hibino Y, Ohzeki H, Sugano N, Hiraga K (2000) Transcription modulation by a rat nuclear scaffold protein, P130, and a rat highly repetitive DNA component or various types of animal and plant matrix or scaffold attachment regions. Biochem Biophys Res Commun 279:282–287. https://doi.org/10.1006/bbrc.2000.3938
Hibino Y, Usui T, Morita Y, Hirose N, Okazaki M, Sugano N, Hiraga K (2006) Molecular properties and intracellular localization of rat liver nuclear scaffold protein P130. Biochim Biophys Acta 1759:195–207. https://doi.org/10.1016/j.bbaexp.2006.04.010
Iradi MCG, Triplett JC, Thomas JD, Davila R, Crown AM, Brown H, Lewis J, Swanson MS, Xu G, Rodriguez-Lebron E, Borchelt DR (2018) Characterization of gene regulation and protein interaction networks for Matrin 3 encoding mutations linked to amyotrophic lateral sclerosis and myopathy. Sci Rep 8:4049–4063. https://doi.org/10.1038/s41598-018-21371-4
Ito D, Hatano M, Norihiro Suzuki N (2017) RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci Transl Med 9:eaah5436. https://doi.org/10.1126/scitranslmed.aah5436
Kamelgarn M, Chena J, Kuang L, Arenas A, Zhai J, Zhu H, Gala J (2016) Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim Biophys Acta 1862:2004–2014. https://doi.org/10.1016/j.bbadis.2016.07.015
Kigawa T, Yabuki T, Matsuda N, Matsuda T, Nakajima R, Tanaka A, Yokoyama S (2004) Preparation of Escherichia coli cell extract for highly productive cell-free protein expression. J Struct Funct Genomics 5:63–68. https://doi.org/10.1023/B:JSFG.0000029204.57846.7d
Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graphics 14:51–55. https://doi.org/10.1016/0263-7855(96)00009-4
Kula A, Gharu L, Marcello A (2013) HIV-1 pre-mRNA commitment to Rev mediated export through PSF and Matrin 3. Virology 435:329–340. https://doi.org/10.1016/j.virol.2012.10.032
Kuo P-H, Doudeva LG, Wang Y-T, Shen C-KJ, Yuan HS (2009) Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res 37:1799–1808. https://doi.org/10.1093/nar/gkp013
Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR:programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8:477–486. https://doi.org/10.1007/bf00228148
Leblond CS, Gan-Or Z, Spiegelman D, Laurent SB, Szuto A, Hodgkinson A, Dionne-Laporte A, Provencher P, de Carvalho M, Orrù S, Brunet D, Bouchard J-P, Awadalla P, Dupré N, Dion PA, Rouleau GA (2016) Replication study of MATR3 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 37:209.e17-209.e21. https://doi.org/10.1016/j.neurobiolaging.2015.09.013
Li H, Koshiba S, Hayashi F, Tochio N, Tomizawa T, Kasai T, Yabuki T, Motoda Y, Harada T, Watanabe S, Inoue M, Hayashizaki Y, Tanaka A, Kigawa T, Yokoyama S (2008) Structure of the C-terminal phosphotyrosine interaction domain of Fe65L1 complexed with the cytoplasmic tail of amyloid precursor protein reveals a novel peptide binding mode. J Biol Chem 283:27165–27178. https://doi.org/10.1074/jbc.M803892200
Lin K-P, Tsai P-C, Liao Y-C, Chen W-T, Tsai C-P, Soong B-W, Lee Y-C (2015) Mutational analysis of MATR3 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol Aging 36:2005.e1-2005.e4. https://doi.org/10.1016/j.neurobiolaging.2015.02.008
Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillén-Boixet J, Franzmann TM, Jahnel M, Marrone L, Chang YT, Sterneckert J, Tomancak P, Anthony Hyman A, Simon Alberti S (2018) RNA buffers the phase separation behavior of prion-like RNA-binding proteins. Science 360:918–921. https://doi.org/10.1126/science.aar7366
Malik AM, Barmada SJ (2021) Matrin 3 in neuromuscular disease: physiology and pathophysiology. JCI Insight 11:e143948. https://doi.org/10.1172/jci.insight.143948
Malik AM, Miguez RA, Li X, Ho Y-S, Eva L, Feldman EL, Barmada SJ (2018) Matrin 3-dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. Elife 7:e35977. https://doi.org/10.7554/eLife.35977
Marangi G, Lattante S, Doronzio PN, Conte A, Tasca G, Monforte M, Patanella AK, Bisogni G, Meleo E, Salvatore La Spada SL, Zollino M, Sabatelli M (2017) Matrin 3 variants are frequent in Italian ALS patients. Neurobiol Aging 49:218. https://doi.org/10.1016/j.neurobiolaging.2016.09.023
Maris C, Dominguez C, Allain FH (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J 272:2118–2131. https://doi.org/10.1111/j.1742-4658.2005.04653.x
Matsuda T, Koshiba S, Tochio N, Seki E, Iwasaki N, Yabuki T, Inoue M, Yokoyama S, Kigawa T (2007) Improving cell-free protein synthesis for stable-isotope labeling. J Biomol NMR 37:225–229. https://doi.org/10.1007/s10858-006-9127-5
Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Buillard V, Cerutti L, Copley R, Courcelle E, Das U, Daugherty L, Dibley M, Finn R, Fleischmann W, Gough J, Haft D, Hulo N, Hunter S, Kahn D, Kanapin A, Kejariwal A, Labarga A, Langendijk-Genevaux PS, Lonsdale D, Lopez R, Letunic I, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Nikolskaya AN, Orchard S, Orengo C, Petryszak R, Selengut JD, Sigrist CJ, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C (2007) New developments in the InterPro database. Nucleic Acids Res 35:D224–D228. https://doi.org/10.1093/nar/gkl841
Muto Y, Yokoyama S (2012) Structural insight into RNA recognition motifs: versatile molecular Lego building blocks for biological systems. Wires RNA 3:229–246. https://doi.org/10.1002/wrna.1107
Nakayasu H, Berezney R (1991) Nuclear matrins: identification of the major nuclear matrix proteins. Proc Natl Acad Sci USA 88:10312–10316. https://doi.org/10.1073/pnas.88.22.10312
Niimori-Kita K, Tamamaki N, Koizumi D, Niimori D (2018) Matrin-3 is essential for fibroblast growth factor 2-dependent maintenance of neural stem cells. Sci Rep 8:13412–13421. https://doi.org/10.1038/s41598-018-31597-x
Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir-Ahmady B, Pitsch S, Black DL, Allain FH-T (2005) Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 309:2054–2057. https://doi.org/10.1126/science.1114066
Picchiarelli G, Dupuis L (2020) Role of RNA binding proteins with prion-like domains in muscle and neuromuscular diseases. Cell Stress 4:76–91. https://doi.org/10.15698/cst2020.04.217
Powers R, Garrett DS, March CJ, Frieden EA, Gronenborn AM, Clore GM (1993) The high-resolution, three-dimensional solution structure of human interleukin-4 determined by multidimensional heteronuclear magnetic resonance spectroscopy. Biochemistry 32:6744–6762. https://doi.org/10.1021/bi00077a030
Ramesh N, Daley EL, Gleixner AM, Mann JR, Kour S, Mawrie D, Anderson EN, Kofler J, Donnelly CJ, Kiskinis E, Pandey UB (2020a) RNA dependent suppression of C9orf72 ALS/FTD associated neurodegeneration by Matrin-3. Acta Neuropathol Commun 8:177–197. https://doi.org/10.1186/s40478-020-01060-y
Ramesh N, Kour S, Anderson EN, Rajasundaram D, Pandey UB (2020b) RNA-recognition motif in Matrin-3 mediates neurodegeneration through interaction with hnRNPM. Acta Neuropathol Commun 8:138–159. https://doi.org/10.1186/s40478-020-01021-5
Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, Na H, Irimia M, Matzat LH, Dale RK, Smith SA, Yarosh CA, Kelly SM, Nabet B, Mecenas D, Li W, Laishram RS, Qiao M, Lipshitz HD, Piano F, Corbett AH, Carstens RP, Frey BJ, Anderson RA, Lynch KW, Penalva LOF, Lei EP, Fraser AG, Blencowe BJ, Morris QD, Hughes TR (2013) A compendium of RNA-binding motifs for decoding gene regulation. Nature 499:172–177. https://doi.org/10.1038/nature12311
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita V-M, Kaivorinne A-L, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, ITALSGEN Consortium, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Salton M, Elkon R, Borodina T, Davydov A, Yaspo M-L, Halperin HE, Shiloh Y (2011) Matrin 3 binds and stabilizes mRNA. PLoS ONE 6:e23882. https://doi.org/10.1371/journal.pone.0023882
Tada M, Doi H, Koyano S, Kubota S, Fukai R, Hashiguchi S, Hayashi N, Kawamoto Y, Kunii M, Tanaka K, Takahashi K, Ogawa Y, Iwata R, Yamanaka S, Takeuchi H, Tanaka F (2018) Matrin 3 Is a component of neuronal cytoplasmic inclusions of motor neurons in sporadic amyotrophic lateral sclerosis. Am J Pathol 188:507–514. https://doi.org/10.1016/j.ajpath.2017.10.007
Tsuda K, Someya T, Kuwasako K, Takahashi M, He F, Unzai S, Inoue M, Harada T, Watanabe S, Terada T, Kobayashi N, Shirouzu M, Kigawa T, Tanaka A, Sugano S, Güntert P, Yokoyama S, Muto Y (2011) Structural basis for the dual RNA-recognition modes of human Tra2-β RRM. Nucleic Acids Res 39:1538–1553. https://doi.org/10.1093/nar/gkq854
Uemura Y, Oshima T, Yamamoto M, Reyes CJ, Costa Cruz PH, Shibuya T, Kawahara Y (2017) Matrin3 binds directly to intronic pyrimidine-rich sequences and controls alternative splicing. Genes Cells 22:785–798. https://doi.org/10.1111/gtc.12512
Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, Stanton R, Frank Rigo F, Guttman M, Yeo GW (2016) Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13:508–514. https://doi.org/10.1038/nmeth.3810
Xu L, Li J, Tang L, Zhang N, Fan D (2016) MATR3 mutation analysis in a Chinese cohort with sporadic amyotrophic lateral sclerosis. Neurobiol Aging 38:218. https://doi.org/10.1016/j.neurobiolaging.2015.11.023
Xue YC, Ng CS, Xiang P, Liu H, Kevin Zhang K, Mohamud Y, Luo H (2020) Dysregulation of RNA-binding proteins in amyotrophic lateral sclerosis. Front Mol Neurosci 13:1–11. https://doi.org/10.3389/fnmol.2020.00078
Acknowledgements
We thank Dr. Yoshihide Hayashizaki of the RIKEN Omics Science Center for providing the cDNA clone of mouse Matrin-3 RRM1 and RRM2.
Accession codes
The chemical shift assignments for the two RRM domains of Matrin-3 have been deposited in the BMRB database with the accession numbers 36430 for RRM1 and 36431 for RRM2, respectively. The atomic coordinates for the ensemble of 20 NMR structures of Matrin-3 RRM1 and RRM2 calculated by CYANA 2.1 (accession codes 1X4D and 1X4F, respectively) as well as those with Amber12 refinement (accession cods: 7FBR and 7FBV, respectively).
Funding
This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI) and by the National Project on Protein Structural and Functional Analyses of the Ministry of Education, Culture, Sports, Science and Technology of Japan. This work was also supported by grants from Musashino University Gakuin Tokubetsu Kenkyuhi to YM.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interest to report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
He, F., Kuwasako, K., Takizawa, M. et al. 1H, 13C and 15N resonance assignments and solution structures of the two RRM domains of Matrin-3. Biomol NMR Assign 16, 41–49 (2022). https://doi.org/10.1007/s12104-021-10057-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12104-021-10057-0