
Abstract
Purpose
Approximately, 30% patients after radical prostatectomy (RP) will undergo post-operative biochemical recurrence (BCR). Present stratification method by TNM staging and Gleason score was not adequate to screen high-risk patients. In this study, we intended to identify a novel set of differentially expressed gene (DEG) signature that can predict BCR after RP.
Materials/patients
358 patients after RP with follow-up data were extracted from The Cancer Genome Atlas (TCGA), among which 61 patients had undergone BCR. Key DEGs were confirmed by the intersection of GSE35988 and TCGA_PCa dataset, and their gene expression data were also extracted from TCGA_PCa dataset. Kaplan–Meier plot and Cox proportion hazard regression model were applied to assess the relationship between risk score and survival outcome (BCR).
Results
310 DEGs were confirmed in two prostate cancer dataset. 6 DEGs (SMIM22, NINL, NRG2, TOP2A, REPS2, and TPCN2) were selected to construct a risk score formula. The risk score was a powerful predictive factor independent of TNM stage (HR 3.045, 95% CI 1.655–5.602, p < 0.001).
Conclusion
In this study, a novel 6-gene signature with robust predictive ability on post-operative BCR was constructed and 4 genes (SMIM22, NRG2, NINL and TPCN2) in the 6-gene signature were not reported to be associated with prostate cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Approximately, 23–34% prostate cancer patients will undergo post-operative disease relapse, initially with increased serum prostate specific antigen (PSA) value. Some patients with BCR will progress to local recurrence and distant metastasis. Although androgen deprivation therapy and salvage radiation therapy are effective management for these patients, with a period of disease control, part of the hormone-sensitive prostate cancer patients develops into the stage of castration resistance (castration resistance prostate cancer/CRPC) [1]. In this regard, BCR is an earlier intervention time point than CRPC, and effective assessment of the risk of BCR is a key clinical issue in prostate cancer management. Certain clinical and pathological indicators such as TNM stage, Gleason score and serum PSA have been employed in prediction of BCR. Nevertheless, in virtue of the heterogeneity in prostate cancer, patients with same clinical pathologic parameters always progressed to diverse consequences. Hence, the discovery of addictive prognostic factors to improve patients’ management after RP is desirable.
Numerous factors have been investigated for enhancing the predictive ability of clinical and pathological parameters. High serum alkaline phosphatase [2], lncRNA TMPO-AS1 [3], and NAP1L6 [4] were reported to be significantly associated with prostate cancer survival. Besides single molecule, multiple gene signatures such as Oncotype DX [5], Prolaris [6], Decipher [7], and sigMuc1NW [8] have also been explored the association with prostate cancer prognosis after RP. Although these indicators and others not mentioned here contributed to improve clinical decision and patients’ management, their clinical utilization deserves further validation [9, 10]. So far, there has no assay recommended by EAU or AUA guidelines for clinical prediction.
Nowadays, the application of microarray and RNA-sequencing technology has deepened our recognition of the tumorigenesis and development of prostate cancer. The gene expression omnibus (GEO) provides substantial information about gene expression profile. Besides gene expression profile, The Cancer Genomic Atlas (TCGA) is also a follow-up dataset of prostate cancer (PCa) patients after RP, which facilitates the survival analysis. In this study, we first identified the key DEGs by combining the GEO and TCGA dataset and then constructed a 6-gene signature associated with BCR by survival analysis. Finally, its independent prognostic value was further investigated.
Materials and methods
Data acquisition and pretreatment
The gene expression profile of GSE35988 [11] was obtained from GEO (http://www.ncbi.nlm.nih.gov/geo). Then, we compared the differential expression between the prostate cancer tissue and normal prostate tissue via online tool: GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/). The RNA-sequencing data of TCGA prostate adenocarcinoma (TCGA_PCa) were available on the website of Gene Expression Profiling Interactive Analysis/GEPIA [12] (http://gepia.cancer-pku.cn/index.html). And the differential expression analysis of TCGA_PCa was conducted using GEPIA online tool. The statistical analysis of these two online tools: GEO2R and GEPIA was based on limma R package. The significant level: adjusted p value (adj. p value) was set as 0.05 to reduce the false-positive rate. The criterion of Fold change was set as |logFC| ≥ 1.
The Clinical data and RNA expression data of TCGA prostate adenocarcinoma [13] (up to Aug 11, 2017) were downloaded from TCGA official website (https://portal.gdc.cancer.gov/). The downloaded data type of gene expression was fragments per kilobase of exon per million fragments mapped (FPKM); then, this data type was converted to transcripts per million (TPM) by a bioinformatics engineer [14]. The exclusion criteria of PCa patients were used as follows: (1) pathologic result is not prostate adenocarcinoma, (2) patients with clinical data but not biochemical recurrence data, and (3) patients whose vital clinical information involving American Joint Committee on Cancer (AJCC) TNM stage [15] is missed. At last, 358 patients, both having clinical data and gene expression data, were obtained in our study for survival analysis, as shown in supplementary material 1 (http://dx.doi.org/10.13140/RG.2.2.13131.44324).
The series matrix file of GSE55945 [16] was downloaded from website GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse55945). And the probe IDs were converted into gene symbols via online tool g:profile [17] (http://biit.cs.ut.ee/gprofiler/).
Statistics analysis and data mining
The association between expression level of DEGs and biochemical free survival was analyzed by univariable cox proportional hazard regression model and log-rank test (median as cutoff point) [18]. DEGs were considered with prognostic values if their p values were less than 0.05. The DEGs with statistical significance were assessed in multivariable cox regression model to construct a predictive model. Then, a risk score formula was constructed using the expression level of DEGs and their coefficients calculated in the multivariable Cox regression model. The risk score of each patient was then calculated and patients were ranked into low-risk group and high-risk group using median as cutoff point. The prognostic effect of risk score was assessed in Kaplan–Meier estimate using log-rank test to evaluating its statistical significance. Univariable and multivariable Cox regression analysis was also conducted. The association between the risk score and clinical pathological characteristic used Chi-square test. All analysis conducted above were taken in SPSS 16.0 and the criteria of statistical significance was set as p < 0.05.
Functional enrichment analysis was conducted in Funrich software version 3.1.1 [19].
The normalized gene expression data were extracted from series matrix of GSE55945 and the differential expression analysis was conducted in SPSS using two-tailed student’s t test with p < 0.05 as statistical test criteria.
All the graphs in this study were drawn in GraphPad Prism 7.0 software.
Results
Identification of key DEGs
The GEO2R analysis for GSE35988 was conducted between 49 samples of localized prostate cancer and 12 samples of benign prostate tissue, using data in platform GPL6480. According to the filtering criteria mentioned above, the result showed that there were 767 DEGs, among which 312 genes were upregulated and 455 genes were downregulated.
The differential expression analysis in TCGA_PCa dataset showed that 3017 genes were selected, among which 690 genes were upregulated and 2327 genes were downregulated.
At last, 310 DEGs were confirmed to be appeared in two dataset as shown in Venn diagram (Fig. 1) and supplementary material 2 (http://dx.doi.org/10.13140/RG.2.2.28230.93766). Among them, there were 96 upregulated genes and 214 downregulated genes.

Venn diagram of differentially expressed genes. Venn diagram of differentially expressed genes in the datasets: GSE35988 and TCGA_PCa
Establishment of gene signature with prognostic value
Patients’ characteristics involved in this study are shown in Table 1. The median follow-up time of censored patients was 512 days. To investigate whether the DEGs were related to BCR survival outcome, the 310 genes were taken into statistical analysis using univariable Cox regression and log-rank test. And the result showed that 19 DEGs were significantly related to BCR-free survival (p < 0.05), as shown in Table 2 and Fig. 2.

Kaplan–Meier plots based on the 6 differentially expressed genes. Kaplan–Meier plots of BCR-free survival for 358 PCa patients grouped by single gene constituting the 6-gene signature. Patients were divided into two groups: high expression group and low expression group, based on the gene expression level using the median as cutoff point. The comparison method of two survival curves was Log-rank test. a SMIM22, b NINL, c TPCN2, d NRG2, e TOP2A, f REPS2
All these 19 DEGs were taken into multivariable cox regression model and the method of variables entering into equation was Forward Stepwise (Likelihood Ratio)/Forward:LR in SPSS. When the 6 genes: SMIM22, REPS2, TPCN2, NINL, TOP2A, NRG2 entered into equation, the model was successfully established and all 6 genes had statistical significance (all p < 0.05, Fig. 2; Table 3).
According to the coefficients from multivariable cox regression model and the gene expression levels, risk score formula was created as follows: risk score = (− 0.744 * expression level of SMIM22) + (− 0.809 * expression level of REPS2) + (0.568 * expression level of TPCN2) + 0.681 * expression level of NINL + 0.686 * expression level of TOP2A + (− 0.962 * expression level of NRG2). The gene expression level used in risk score formula was 0 or 1, representing low expression and high expression, respectively. Patients were then ranked into two groups by risk score. Furthermore, the relationship between risk score and clinical pathological parameters was analyzed (Table 4). The result showed that pathologic T stage, N stage, TNM stage and Gleason score had significant difference between the low-risk group and high-risk group, but age was not.
Kaplan–Meier plots showed that the patients with high-risk score inclined to present a worse BCR-free survival probability (Fig. 3a). To evaluate the independent prognostic effect of the 6-gene signature in predicting BCR, the univariable and multivariable cox proportion hazard regression model was applied (Table 5). In light of the TNM stage integrated with the information of pT and pN, pT and pN were not involved in survival analysis. The results showed that in univariable analysis, risk score, Gleason score and TNM stage instead of age had predictive ability on BCR. And in multivariable stepwise cox regression analysis, the risk score still kept its prognostic effect independent of TNM stage.

Kaplan–Meier plots of Risk score in the entire patients group and Gleason score subgroups. a Kaplan–Meier plots of BCR-free survival in the entire patient group (358 patients). Patients were divided into two groups: high-risk score group and low-risk score, using median-risk score as cutoff point. b Comparing means of risk score in different Gleason score subgroups by Students’ t test. **Represents p < 0.01. c Kaplan–Meier plots of BCR-free survival in the subgroup 1: Gleason score ≤ 7 (197 patients). Patients were divided into two groups: high-risk score group and low-risk score, using median risk score as cutoff point. d Kaplan–Meier plots of BCR-free survival in the subgroup 2: Gleason score > 7 (161 patients). Patients were divided into two groups: high-risk score group and low-risk score, using median-risk score as cutoff point
The prognostic effect of risk score in different Gleason score subgroup
Due to that Gleason score was not permitted to enter into multivariable cox regression equation and the association between Gleason score and risk score, a subgroup analysis was conducted to justify whether the prognostic effect of risk score is suitable for all patients regardless of Gleason score. The survival analysis demonstrated that in both subgroups: Gleason score ≤ 7 (n = 197) and Gleason score > 7 (n = 161), the survival difference between high-risk score and low-risk score was significant (all p < 0.01, Fig. 3b–d).
Functional enrichment analysis
The Gene ontology and KEGG pathway analysis was conducted (Fig. 4). The results showed that the 6 genes were enriched in biological process (BP) including cell growth, regulation of nucleic acid metabolism, transport and cell communication. For molecular function (MF), these genes were enriched in growth factor activity, DNA topoisomerase activity, calcium ion binging and ion channel activity. And for cell component (CC) analysis, these genes were located in centriole, nuclear chromosome, kinetochore, and DNA topoisomerase complex. In addition, these genes were involved in ErbB2/ErbB3 signaling, ErbB4 signaling, cell cycle, mitotic and et al.

Functional enrichment analysis on the 6 differentially expressed genes (DEGs). a Biological pathways the 6 DEGs involved in. b Biological process the 6 DEGs involved in. c Cellular component the 6 DEGs involved in. d Molecular function of the 6 DEGs
Validation of 6 DEGs
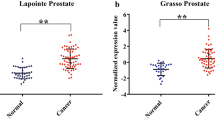
The expression difference between cancer group and normal prostate tissue group of these 6 genes was compared in GSE55945. As shown in Fig. 5, their differential expression was consistent with that of GSE35988 and TCGA PCa.

The expression of six differentially expressed genes in GSE55945. The normalized gene expression values of the 6 DEGs in prostate cancer tissue group and normal prostate tissue group were compared by Student’s t test. *Represents p < 0.05
Discussion
At present, the indication for post-operative adjuvant therapy of prostate cancer is confined to pathological TNM staging as pT3 and pN+, positive surgical margins and Gleason score ≥ 7 [1]. However, some patients without these signs still develop to BCR, the risk stratification needs to be updated.
TCGA-PCa dataset, a large-scale, prospective post-operative follow-up cohort research was exploited in our study. In order to thoroughly explore the high dimensional gene expression data and not just focus on genes at a specific pathway [6] or genes author interested in [17], the strategy for variable selection was set as follows: (a) Using the strict criteria of adj. p < 0.05 and |logFC| ≥ 1 to get DEGs and then identification of common DEGs in the two datasets: GSE35988 and TCGA-PCa. (b) The common DEGs were under univariate survival analysis and, subsequently, the DEGs with statistical significance were further analyzed in multivariate cox regression model to achieve optimal model and risk score formula. (c) The independent predictive value was then investigated for the new variable: risk score, combined with clinical and pathological parameters. This strategy for predictive factors selection could avoid the variable elimination of clinical and pathological parameters when they entered into multivariate cox regression model with the high dimensional transcriptomic data at the same time [20]. After the analysis above, the 6-gene signature was eventually constructed with powerful predictive ability on BCR, independent of TNM stage. In the light of the association between risk score and Gleason score, we divided patients into two subgroups: Gleason ≤ 7, and Gleason score > 7 and conducted subgroup survival analysis. Surprisingly, in the subgroup: Gleason score ≤ 7, risk score was demonstrated to be a predictive factor (Log-rank test p < 0.01), and the result was also statistically significant in subgroup: Gleason score > 7. This result revealed that the predictive effect of risk score may be independent of Gleason score, although this needs further validation in another follow-up cohort.
Downregulation of SMIM22, or named CASIMO1 [21] was reported in breast cancer to decreased cell proliferation and restraint of cell motility, by affecting its downstream phosphorylation of ERK. NRG2 [22, 23] was shown to interact with ErbB family of receptors promoting cell growth and differentiation. The primary function of NINL/Nlp [24] is to promote microtubule nucleation. Its aberrant expression in cancer cells could render cell tumorigenic. TPCN2 [25] belonged to a recently described class of NAADP- and PI(3,5)P2-sensitive Ca2+—permeable cation channels in the endolysosomal system of cells whose downregulation induced abrogating migration of metastatic cancer cells in vitro. In our knowledge, there was no research reported the relationship between the 4 genes (SMIM22, NINL, NRG2, and TPCN2) and prostate cancer, and the exact molecular mechanism of these four genes in prostate cancer development and progression deserved further investigation.
The decreased expression of REPS2/POB1 [26,27,28,29] in androgen-independent prostate cancer cell lines results in loss control of growth factor signaling and, therefore, in loss control of cell proliferation. Our finding that high expression of RESP2 in patients after RP may be associated with decreased risk of BCR (Table 2, HR 0.550, p = 0.025) was consistent with the previous study. TOP2A [30] was demonstrated to enhance androgen signaling by promoting transcription of androgen responsive genes, therefore contributing to hormone-independent cell growth and proliferation. Collectively, the 6-gene signature was involved in various signaling pathway and participated in diverse biological process. These may contribute to its powerful ability in predicting BCR. In addition, we further validated the differential expression of the 6 genes in another public dataset: GSE55945 (all p < 0.05).
Some limitation of this research should take into account. Firstly, the exact molecular mechanism of SMIM22, NRG2, NINL and TPCN2 in prostate cancer had not been explored in our research. Secondly, the variables: serum PSA and surgical margin were not provided by the TCGA_PCa dataset, so the relationship between the novel gene signature and the two variables was not explored. Thirdly, the construction of this gene signature was based on TCGA-PCa follow-up cohort, without validation in a prospective clinical cohort study.
Conclusion
We derived the key DEGs using public dataset: GSE35988 and TCGA-PCa, and then thoroughly analyzed their predictive ability on post-operative biochemical recurrence. Eventually, a novel multi-gene set was constructed with robust prediction of BCR, and its predictive ability was independent of TNM stage. Among the 6-gene signature, 4 genes (SMIM22, NRG2, NINL and TPCN2) have not been reported the relationship with prostate cancer. This novel predictive system will have attractive applications to improve post-operative patients’ management, if validated in other prospective clinical trial.
References
Cornford P, Bellmunt J, Bolla M, et al. EAU-ESTRO-SIOG guidelines on prostate cancer. Part II: treatment of relapsing, metastatic, and castration-resistant prostate cancer. Eur Urol. 2017;71:630–42.
Li D, Lv H, Hao X, et al. Prognostic value of serum alkaline phosphatase in the survival of prostate cancer: evidence from a meta-analysis. Cancer Manag Res. 2018;10:3125–39.
Huang W, Su X, Yan W, et al. Overexpression of AR-regulated lncRNA TMPO-AS1 correlates with tumor progression and poor prognosis in prostate cancer. Prostate. 2018;78:1248–61.
Zheng Y, Gao Y, Li X, et al. Long non-coding RNA NAP1L6 promotes tumor progression and predicts poor prognosis in prostate cancer by targeting Inhibin-beta A. Onco Targets Ther. 2018;11:4965–77.
Albala D, Kemeter MJ, Febbo PG, et al. Health economic impact and prospective clinical utility of oncotype DX(R) Genomic Prostate Score. Rev Urol. 2016;18:123–32.
Cuzick J, Swanson GP, Fisher G, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol. 2011;12:245–55.
Karnes RJ, Choeurng V, Ross AE, et al. Validation of a genomic risk classifier to predict prostate cancer-specific mortality in men with adverse pathologic features. Eur Urol. 2018;73:168–75.
Jiang Y, Mei W, Gu Y, et al. Construction of a set of novel and robust gene expression signatures predicting prostate cancer recurrence. Mol Oncol. 2018;12:1559–78.
Martin NE. New developments in prostate cancer biomarkers. Curr Opin Oncol. 2016;28:248–52.
Ross AE, D’Amico AV, Freedland SJ. Which, when and why? Rational use of tissue-based molecular testing in localized prostate cancer. Prostate Cancer Prostatic Dis. 2016;19:1–6.
Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43.
Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–102.
The Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–25.
Li B, Ruotti V, Stewart RM, et al. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500.
Thompson IM, Andriole GL, Blumenstein B, et al. AJCC cancer staging manual. 6th ed. New York: Springer; 2003.
Arredouani MS, Lu B, Bhasin M, et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin Cancer Res. 2009;15:5794–802.
Reimand J, Arak T, Adler P, et al. g:profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016;44:W83–9.
Huang TB, Dong CP, Zhou GC, et al. A potential panel of four-long noncoding RNA signature in prostate cancer predicts biochemical recurrence-free survival and disease-free survival. Int Urol Nephrol. 2017;49:825–35.
Pathan M, Keerthikumar S, Ang CS, et al. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15:2597–601.
Molinari C, Matteucci F, Caroli P, et al. Biomarkers and molecular imaging as predictors of response to neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer. Clin Colorectal Cancer. 2015;14:227–38.
Polycarpou-Schwarz M, Gross M, Mestdagh P, et al. The cancer-associated microprotein CASIMO1 controls cell proliferation and interacts with squalene epoxidase modulating lipid droplet formation. Oncogene. 2018;37:4750–68.
Ring HZ, Chang H, Guilbot A, et al. The human neuregulin-2 (NRG2) gene: cloning, mapping and evaluation as a candidate for the autosomal recessive form of Charcot-Marie-Tooth disease linked to 5q. Hum Genet. 1999;104:326–32.
Dunn M, Sinha P, Campbell R, et al. Co-expression of neuregulins 1, 2, 3 and 4 in human breast cancer. J Pathol. 2004;203:672–80.
Li J, Zhan Q. The role of centrosomal Nlp in the control of mitotic progression and tumourigenesis. Br J Cancer. 2011;104:1523–8.
Nguyen ON, Grimm C, Schneider LS, et al. Two-pore channel function is crucial for the migration of invasive cancer cells. Cancer Res. 2017;77:1427–38.
Oosterhoff JK, Penninkhof F, Brinkmann AO, et al. REPS2/POB1 is downregulated during human prostate cancer progression and inhibits growth factor signalling in prostate cancer cells. Oncogene. 2003;22:2920–5.
Penninkhof F, Grootegoed JA, Blok LJ. Identification of REPS2 as a putative modulator of NF-kappaB activity in prostate cancer cells. Oncogene. 2004;23:5607–15.
Oosterhoff JK, Kuhne LC, Grootegoed JA, et al. EGF signalling in prostate cancer cell lines is inhibited by a high expression level of the endocytosis protein REPS2. Int J Cancer. 2005;113:561–7.
Badway JA, Baleja JD. Reps2: a cellular signaling and molecular trafficking nexus. Int J Biochem Cell Biol. 2011;43:1660–3.
Schaefer-Klein JL, Murphy SJ, Johnson SH, et al. Topoisomerase 2 alpha cooperates with androgen receptor to contribute to prostate cancer progression. PLoS ONE. 2015;10:e0142327.
Acknowledgements
This research was based on public database: TCGA, GEO and GEPIA, and we are grateful for the extraordinary works of these project groups. We thank Bioinformatics Engineer Rang-Fei Zhu for his excellent pretreatment of TCGA-PCa data.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The patients’ information involved in our research was obtained from The Cancer Genome Atlas (TCGA). All the patients and treatments were complied with the principles laid down in the Declaration of Helsinki in 1964 and its later amendments or comparable ethical standards.
Informed consent
Informed consent was confirmed by all the patients participated in the TCGA-Prostate adenocarcinoma project.
Rights and permissions
About this article

Cite this article
Li, F., Ji, JP., Xu, Y. et al. Identification a novel set of 6 differential expressed genes in prostate cancer that can potentially predict biochemical recurrence after curative surgery. Clin Transl Oncol 21, 1067–1075 (2019). https://doi.org/10.1007/s12094-018-02029-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-018-02029-z