
Abstract
Biomechanical stresses are closely associated with cardiovascular development and diseases. In vivo, vascular smooth muscle cells are constantly stimulated by biomechanical factors caused by increased blood pressure leading to the non-specific activation of cell transmembrane proteins. Thus, various intracellular signal molecules are simultaneously activated via signaling cascades, which are closely related to alterations in the differentiation, phenotype, inflammation, migration, pyroptosis, calcification, proliferation, and apoptosis of vascular smooth muscle cells. Meanwhile, mechanical stress-induced miRNAs and epigenetics modification on vascular smooth muscle cells play critical roles as well. Eventually, the overall pathophysiology of the cells is altered, resulting in the development of many major clinical diseases, including hypertension, atherosclerosis, grafted venous atherosclerosis, and aneurysm, among others. In this paper, important advances in mechanical signal communication in vascular smooth muscle cells are reviewed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Biomechanical stresses produced by blood flow dynamics play pivotal roles in maintaining normal development, structure, and function of the cardiovascular system. Early in the embryo, blood vessels consist of a single layer of endothelial cells (ECs) called endothelial tubes, not arteries or veins. At the beginning of the first heartbeat, cardiac ejection causes blood to flow into the endothelial tubes. The force of outward expansion from blood flow continuously stimulates the mesenchymal cells outside the ECs to differentiate into vascular smooth muscle cells (VSMCs), closely surrounding the ECs, resulting in the development of the endothelial tube into an artery. However, due to the lower pressure in the endothelial tubes connected to the cardiac blood backflow, only a small number of mesenchymal cells outside the ECs differentiate into VSMCs and become veins, acting as conduits for blood reflux. As such, it is the blood pressure that causes the mesenchymal cells to differentiate into VSMCs, resulting in the structural differences between arteries and veins. Similarly, normal blood pressure in adulthood is essential for the maintenance of the structure and function of VSMCs. Shear stress and stretch stress (SS) are two main mechanical forces that occur when blood flows through the blood vessels. Shear stress acts on the ECs, while SS acts on all the cells (ECs, VSMCs, fibroblasts, and undifferentiated mesenchymal stem cells) in the blood vessel wall. Mechanical force can induce ECs to secrete both endothelin-derived vasodilation factor and endothelium-derived vasoconstriction factor. The former includes NO, prostacyclin, and prostaglandin E2. These substances can cause vascular smooth muscle relaxation. The latter mainly includes prostaglandin H2, oxygen free radicals, and endothelin, which cause the VSMCs to contract. Therefore, under normal physiological conditions, mechanical stimulation induces ECs to release and balance the effects of vasodilators and vasoconstrictors, thereby maintaining normal vascular tension. However, under pathophysiological conditions, abnormally increased mechanical stress induced by hypertension will lead to altered vascular structure and function.
Hypertension, hyperlipidemia, and hyperglycemia can cause vascular diseases, such as atherosclerosis, alone or in combination. Theoretically, the blood lipid and blood glucose levels in the arteries and veins of the same individuals are essentially the same, or higher in the veins than in the arteries. However, atherosclerotic lesions only occur in the arteries, and not in the veins. Despite this, once a vein is transplanted into an artery, arterialization (normal blood lipid and blood glucose) or atherosclerosis of the transplanted vein (hyperlipidemia and hyperglycemia) occurs rapidly under arterial pressure. These data all indicate that arterial pressure plays a critical role. The question remains as to how the signal of mechanical stimulation is sensed by the vascular cells and transferred into the cells to cause the pathophysiological changes of the cells. Do vascular cells have specific mechanical receptors? What changes occur to the transmembrane proteins on the cell membrane under mechanical stress stimulation? How are important intercellular signaling molecules in the activation or regulation of VSMCs? What pathophysiological changes and clinically important diseases are caused as a result?
In vivo VSMCs are constantly stimulated by biochemical and biomechanical factors. In recent years, several new mechanisms of mechanical transduction have been discovered. Mechanical stimulation elicits a cell response with many important characteristics. For example, biomechanical stress can induce the nonspecific activation of all cell transmembrane proteins, leading to the communication of multiple signaling pathways and the involvement of various signaling molecules within the cells. Similarly, mechanical stress induces simultaneous changes to differentiation, phenotype, inflammation, migration, proliferation, and/or apoptosis in VSMCs, which are closely related to the development of many major clinical cardiovascular diseases. Most importantly, different phenotypes of VSMCs can lead to different responses to mechanical stimuli. Likewise, mechanical stimulation can alter the cell phenotype and accelerating the changes in their vascular structure and function. This review will focus on these issues and the latest developments.
Mechanoreceptors in VSMCs
In VSMCs, multiple molecules have been identified as direct mechanoreceptors. Some trans-membrane proteins, including GPCRs, RTKs, LOX-1, RAGE, integrins and ion channels (TRPV2, Piezos, etc.) are capable of sensing and transducing mechanical signals. Mechanical stress stimulates mechanoreceptors and subsequently activates various downstream signaling cascades, which cause different pathophysiological effects. In this review, we will introduce some of the main mechanoreceptors and their downstream signaling cascades, and also, the related pathophysiological effects.
GPCRs
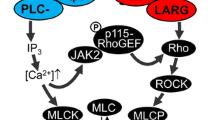
G protein-coupled receptors (GPCRs) play important roles as mechanoreceptors, such as angiotensin II-receptor (AT1R) and adrenergic receptor (AR). They are closely associated with the development of cardiovascular diseases like hypertension and atherosclerosis (Forrester et al. 2018). GPCRs are a group of 7-transmembrane-receptors that are combined with G proteins. G proteins combined with GDP are inactive, and those with GTP are activated, such that they can activate downstream molecules, including adenylate cyclase (AC) and PLC. The activation of AC and PLC produces secondary messengers. AC cleaves ATP into cyclic AMP (cAMP), which continues to activate protein kinase A (PKA). PLC hydrolyzes PIP2 into DAG and IP3. DAG remains on the plasma membrane, and soluble free IP3 promotes the quick release of Ca2+ stored in the cells. The released Ca2+ combine with protein kinase C (PKC) and they cluster near the plasma membrane. DAG, Ca2+ and phosphatidylserine together affect the regulatory domain of PKC, altering the structure of PKC to expose the active domain. Activated PKA and PKC can then phosphorylate some residues of effector proteins. The excess Ca2+ will combine with calmodulin to form Ca2+/ calmodulin complexes, which activate calmodulin -dependent protein kinases (Wootten et al. 2018).
As the first GPCR that was identified as a mechanoreceptor, AT1R is a Gq/11 protein coupled receptor. Besides AT1R, our previous study showed that both mechanical strain stress and norepinephrine could activate AR and the downstream signaling pathway, such as extracellular receptor kinases (ERKs), and increase the proliferation of VSMCs, thus leading to atherosclerosis. When combined, the effect would be even stronger (Liu et al. 2013).
RTKs
Receptor tyrosine kinases (RTKs) are a group of single-subunit transmembrane receptors that contain an extracellular N-terminal domain and an intracellular C-terminal domain. Ligands combine the extracellular N-terminal domain which activates RTKs, and makes them polymerize into dimers or multimers. Mechanical stress can also activate RTKs non-specifically. The intracellular C-terminal kinase domains of activated RTKs would phosphorylate the tyrosine residues of themselves, producing binding sites for Src homology 2 (SH2) and phosphotyrosine binding (PTB) domains. Proteins that contain SH2 or PTB domains bind to RTKs, so that they are activated and transfer signals to downstream molecules.
The platelet derived growth factor receptors (PDGFRs) is one of the first reported mechanoreceptors (Hu et al. 1998; Ma et al. 1999b). PDGFR is composed of alpha and beta subunits, which can be combined into heterodimers and dimers, among other conformations. The monomer is inactive and can be combined with specific ligand of PDGFs to form dimers. Serine, tyrosine, and threonine in the peptide chain can be phosphorylated automatically, giving rise to the initiation of a series of downstream protein phosphorylation cascades.
Mechanical stretching not only increases the expression level of insulin-like growth factor-1 receptor (IGF-1R), but also stimulates and sustains the tyrosine phosphorylation of IGF-1R and insulin receptor substrate-1 (IRS-1). The tyrosine phosphorylation of IGF-1R and IRS-1, and the activation of IRS-1–associated phosphoinositide 3-kinase (PI3K) both result in an increased proliferation of VSMCs (Cheng and Du 2007).
LOX-1
Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) is the main receptor for oxidized low density lipoprotein (ox-LDL) in ECs and VSMCs. The uptake of ox-LDL by ECs and VSMCs is an important process in the development of atherosclerosis. The expression level of LOX-1, as well as autophagy, can be changed by shear stress of different forms and strengths. Low shear stress and inflammatory stimulus lipopolysaccharide could increase autophagy and LOX-1 expression. Furthermore, cells from LOX-1 knockout mice and LOX-1 inhibition exhibited lower levels of autophagy, whereas LOX-1 overexpression enhanced autophagy in ECs. This indicates that LOX-1 may be able to sense mechanical force and induce the autophagy of ECs (Ding et al. 2015; Mao et al. 2015; Zhang et al. 2013b).
Rage
The receptor for advanced glycation end products (RAGE) is a transmembrane receptor of the immunoglobulin super family, which plays an important role in inflammation of diabetes and is upregulated in atherosclerotic plaques. When shear stress is exerted on human aortic ECs, the expression of RAGE visibly changes. High shear stress downregulates RAGE by four-fold, whereas oscillatory SS upregulates RAGE by three-fold. According to our recent research results, RAGE mediates combined signals initiated by diabetes-related advanced glycation end products (AGEs) and hypertension-induced mechanical stress as a common molecular sensor in VSMCs (DeVerse et al. 2012; Li et al. 2012; Ping et al. 2015; Ping et al. 2017).
Integrin
Integrin is a kind of trans-membrane heterodimeric protein which links extracellular matrix and cytoskeleton. Integrin can reduce isolated resistance arterioles dilation consencutively when exposed to step-increase pressure. SS also enhances integrin αvβ3 expression (Balasubramanian et al. 2007; Martinez-Lemus et al. 2005; Sun et al. 2008). Taken together, receptors in the smooth muscular cell (SMC) membrane can be activated by both their ligands specifically and mechanical SS non-specifically, which results in a synergistic increase of the ligand stimulation signal. Thus, it has been demonstrated that receptors play an important role as mechanosensors.
TRPV2
The TRP family includes a wide variety of cation channels, some of them are mechanoreceptors. A transient receptor potential vanilloid (TRPV) homologue, TRPV2 is expressed in VSMCs and is an important stretch sensor in vascular smooth muscles. In hypotonic solutions, isolated cells from the mouse aorta swell because of osmotic pressure. The cell membrane is stretched thus activates TRPV2 inducing a non-selective cation channel current (NSCC) and elevated intracellular Ca2+ ([Ca2+]i). When the external Ca2+ is cleared, cell swelling can’t cause NSCC or elevated [Ca2+]i any more. Treating the mouse aorta with TRPV2 antisense oligonucleotides results in the inhibition of TRPV2 protein expression. At the same time, the NSCC and elevation of [Ca2+]i induced by hypotonic stimulation is suppressed (McGahon et al. 2016; Muraki et al. 2003).
Piezos
Piezos are evolutionary conversed pore-forming ion channels. They were firstly experimentally proven to be mechanosensitive in mouse neuroblastoma N2a cells (Coste et al. 2010; Coste et al. 2012). Piezos also exist in ECs and SMCs, now accumulated evidences indicate their pivotal role in cardiovascular system (Douguet et al. 2019; Rode et al. 2017). During the embryonic period, Piezo1 expressed in ECs can sense shear stress and guarantee the proper alignment and morphology of ECs. Embryos deficient of Piezo1 die midgestation with vascular remodeling defects. Haploinsufficiency of Piezo1 is not lethal, but it results in ECs with a cobblestone-like appearance, compared to normal cells with a linear appearance in the direction of flow (Li et al. 2014; Ranade et al. 2014). Piezo1 of SMCs mediates arterial remodeling under hypertension SMC-specific homozygote knock out of Piezo1 decreases the diameter and thickness of the vascular wall dramatically in chronic hypertension mice. In normotension mice, the deficiency of Piezo1 makes no significant difference in the vascular structure. Increased opening of Piezo1 in SMCs caused by selectively removing smooth muscle filamin A can also increase arterial wall thickness independent of hypertension. Opening of Piezo1 increases intracellular Ca 2+ levels, which may cause calcium-dependent transglutaminases activation within SMCs. This might be the mechanism of how Piezo1 of SMCs mediates vascular remodeling (Douguet et al. 2019; Retailleau et al. 2015; Wu et al. 2017).
The study of ion channels focuses on the observation of the intracellular ion current and requires the observation of living cells. Therefore, it is difficult to simulate the effect of blood pressure on cells in vitro. Until now, the most commonly used method is the patch clamp technique, in which hypoosmosis causes cell swelling and increases intracellular pressure, leading to changes in intracellular ion levels. At the same time, with the help of a specific ion channel inhibitor or agonist, changes in the intracellular ion flow levels can be observed. However, the pressure caused by cell swelling is quite different from the effect of mechanical stretching induced by blood pressure in living cells in the blood vessels. Further experiments are required to confirm whether these results truly reflect the pathophysiological phenomena of vascular cells in hypertension. The development of better instruments that can directly measure the real-time changes of ions in living cells when they are stimulated by mechanical forces is also necessary.
As reviewed above, although abundant work has been done to recognize and identify mechanoreceptors on the VSMCs membrane, there are still many questions waiting to be resolved. Some membrane receptors of VSMCs play pivotal roles in pathophysiological changes induced by hypertension and diabetes, such as thromboxane receptors, endothelin receptors and mineralocorticoid receptors. But whether they are mechanosensitive hasn’t been identified. Further studies are needed to be conducted to explore the separate and joint effect of mechanical stress and their ligands respectively. Moreover, many new techniques as proteomics, epigenetics and big data can also be applied in this area to make more progress.
Pathophysiological effects and related signaling transduction of VSMCs induced by mechanical stimulation
VSMC phenotype transformation
The phenotype transformation in VSMCs is considered to be a key pathophysiological change in various cardiovascular diseases, including aortic dissection, atherosclerosis, and hypertension. A variety of signaling molecules in VSMCs mediate the mechanical stress-induced VSMC phenotype transformation. One type of mechanical stress is SS. SS initiates a signal via MEF2B that enhances Nox1-mediated ROS production through the MEF2B-Nox1-ROS pathway, promoting VSMCs from a contractile to a synthetic phenotype. Thus theNox1 inhibitor could be used as a potential therapeutic treatment for vascular dysfunction in hypertension (Rodriguez et al. 2015). Another type of mechanical stress is laminar shear stress. The exposure of human coronary artery ECs to the shear stress of 5 dyn/cm2 resulted in the dysfunction of Cx40/Cx43 heterotypic myoendothelial gap junctions (Cx43 upregulation and Cx40 downregulation). The junctions were replaced by homotypic Cx43/Cx43 channels, thereby inducing the transition of human coronary artery SMCs into the synthetic phenotype, related to the activation of PDGF receptor signaling (Zhang et al. 2016). The mechanical activation of the effectors of the Hippo pathway, Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ), is involved in the stretch-induced phenotypic switch of human umbilical arterial VSMCs from a contractile to a synthetic state (Wang et al. 2018b). The ERKs pathway may also mediate the shear-stress-stimulated switch from the contractile to synthetic phenotype (Asada et al. 2005; Shi et al. 2010). In sporadic non-syndromic thoracic aortic aneurysms, due to mechanical stress and aortic SMCs loss caused by the increased cystathionine γ-lyase, synthetic aortic SMCs intensify Jagged1/NOTCH1 pathway to counterbalance the weakened aortic wall (Chiarini et al. 2018).
VSMC proliferation
FAK, as a biomechanical sensor and a signaling “switch” stimulated by stretch in vitro (Romer et al. 2006; Schlaepfer et al. 2004; Torsoni et al. 2003), is auto-phosphorylated at Tyr397, activating an attachment site for c-Src that then activates other auto-phosphorylation sites. The FAK/c-Src complex can stimulate MAPKs, such as ERK 1, 2 and p38MAPK (Schlaepfer and Hunter 1997). ERKs promote the proliferation of VSMCs exposed to both pulsatile and sustained SS, while p38MAPK increases apoptosis (Goldman et al. 2003). Moreover, combined SS with or without oxLDL can additively promote the activation of ERKs, leading to the accelerated proliferation of VSMCs via the LOX-1 signaling pathway (Zhang et al. 2013b). In addition, the Rho family members of small GTPases, RhoA, Rac, and Cdc42, contribute to coordinated cell behavior by modulating transcription and the actin cytoskeleton (Hall 2005). Mechanical stretch stimulates RhoA which mediates different downstream effectors, including the MAPKs, thereby initiating signaling pathways that potentiate VSMC proliferation and contractility (Numaguchi et al. 1999). The activation of the ERKs and Akt signaling pathways is involved in inducing VSMC proliferation under mechanical stretch (Fukumoto et al. 2008), where ERK activation is partly mediated by c-Src (Morita et al. 2004). As mentioned above, the Rho/Rho-kinase (ROCK) pathway together with the ERKs and PI3K/Akt pathways are imperative for stretch-induced venous SMC proliferation (Kozai et al. 2005). Rho GDP dissociation inhibitor α (Rho-GDIα) is a dominating sensor of cyclic strain (CS). Pathological CS enhances VSMC proliferation by down-regulating the expression of Rho-GDIα, which is phospho-Rac1-dependent via the p38MAPK pathway, while physiological CS acts the contrary (Qi et al. 2010). Moreover, the tyrosine phosphorylation of signal transducer and activator of transcription-3 (STAT-3) is associated with the positive regulation of cell growth (Horiuchi et al. 2003; Madamanchi et al. 2001; Marrero et al. 1997; Seki et al. 2000; Yu et al. 2003), as well as the inhibition of STAT-3 tyrosine phosphorylation by PP1, which completely blocked the proliferation of SMCs exposed to CS (Kakisis et al. 2005). Mechanical SS stimulated VSMC proliferation in vein graft by increasing the transcription of serum-, glucocorticoid-regulated kinase-1, an injury-responsive kinase and downstream signal of PI3K (Cheng et al. 2010). Besides, mechanical stretch directly regulates Ang II-induced proliferation in VSMCs of spontaneously hypertensive rats via the Ang II type 1/ epidermal growth factor receptor /ERK-dependent signaling pathway (Liu et al. 2010). The strain determinants for SMC proliferation and alignment were the activation of the AC/cAMP/PKA and the PKC pathways in SMC, where the singular inhibition of PKA or PKC failed to inhibit strain-induced SMC proliferation and alignment, indicating either their lack of involvement or the multifactorial feature of these responses (Mills et al. 1997). CS induces the expression of protease-activated receptor-1, leading to an increase in VSMC proliferation in response to thrombin, which may be mediated via the ROS and PKC signaling pathways (Nguyen et al. 2001). In aortic aneurysms and dissections, elevated stress can induce SMC proliferation (e.g., via PDGF), matrix component synthesis (e.g., via TGF-β), and cell signaling increase through AT1Rs (Humphrey et al. 2014). In addition, strain-induced SMC proliferation is regulated by the epidermal growth factor receptor, basic fibroblast growth factor, and PDGF receptor-MAPK-AP-1 signaling pathways (Hu et al. 1999), PI-3 kinase/Akt pathway, NF-κB pathway, mTOR-S6 kinase-eEF2 pathway (Li et al. 2003b), p27Kip1 (Kurpinski et al. 2006), the and repression of emerin and lamin A/C (Qi et al. 2016). At the same time, the phosphorylation of AMP-activated protein kinase (AMPK) exerts an inhibitory effect on Ang II-induced proliferation signaling in VSMCs (Nagata et al. 2004), which is regulated by flow stress-mediated iNOS expression (Kim et al. 2017). As elaborated in our previous review, mechanical stress induces MAPK phosphatase-1 (MKP-1) expression, which is regulated by two signaling pathways, involving growth factor receptor-Ras-ERK and Rac-JNK/SAPK or p38MAPK, where MKP-1 prevents VSMC proliferation via MAPK inactivation (Li et al. 1999). The microRNA-33 (miR-33) - bone morphogenetic protein 3-Smad signaling pathway provides protection against venous SMC proliferation in response to arterial stretching (Huang et al. 2017). Biomechanical strain induces the immediate early response gene iex-1 in VSMCs, putting antiproliferative effects on SMCs (Schulze et al. 2003).
VSMC migration
Mechanical stress in VSMCs may contribute to their migration in the development of vascular diseases, such as atherosclerosis and restenosis. Our earlier research showed that two main signaling pathways link mechanical stress to cell migration: The first is the PDGF receptor–MAPK–matrix metalloproteinase (MMP) pathway, which is responsible for cell detachment from matrix proteins; The second pathway is the PKC δ paxillin–cytoskeleton pathway, which is essential for cell movement PKC δ mediates the actin fiber rearrangement to influence VSMC migration (Li et al. 2003a). Shear stress stimulates ECs to secrete PDGF-BB and interleukin-1α, and both of these mediators stimulate the VSMC ERKs pathway to induce migration (Dardik et al. 2005). Injury-induced ERKs activation is related to the initiation of rat VSMC migration (Moses et al. 2001). Moreover, CS enhances the expression of PDGF-Rβ in human aortic VSMCs (Ma et al. 1999a; Yung et al. 2009). The up-regulation of PDGF signaling- tyrosine kinase pathway (activated by ligand binding to PDGF-R) is likely to be involved in the human aortic VSMC migration under CS (Yung et al. 2009). Apoptosis signal-regulating kinase 1, as a MAPK kinase, activates SMC migration, increases neovascularization, enhances SMC and EC apoptosis and sequentially accelerates mechanical injury-induced vascular remodeling (Tasaki et al. 2013). The up-regulation of MMP-1 plays a critical role in interstitial flow-enhanced VSMC motility, which was further confirmed by the silencing of MMP-1 gene expression (Shi et al. 2009). In addition to the PDGF receptor–MAPK–MMP pathway, pathological CS enhances VSMC migration via down-regulating the expression of Rho-GDIα, the effect of which is dependent on phospho-Rac1 and possibly via the p38MAPK pathway, while physiological CS has the opposite effect (Qi et al. 2010). Low shear stress-induced VSMC migration is mediated by the PI-3 K/Akt pathway, which down-regulates Rho-GDIα, thereby affecting VSMC migration (Qi et al. 2008). Higher cyclic mechanical strain activated-Akt/protein kinase B included pathway is required for VSMC migration and is likely to function via its effects on actin rearrangement (Zhang et al. 2011). The Rho/ROCK pathway regulates mechanical stress-induced VSMC migration (Peyton and Putnam 2005). In addition, CS-induced Nox1 activation decreases actin fiber density and increased matrix metalloproteinase 9 activity, VSMC migration, and vectorial alignment (Rodriguez et al. 2015). DNA microarray, ChiP analysis, and the analysis of isolated mouse femoral arteries exposed to hypertension verified that nuclear factor of activated T-cells 5 translocation to the nucleus induced an increase in tenascin-C abundance in the vessel wall, which in turn stimulates migration of VSMCs (Scherer et al. 2014). As for the inhibition of VSMC migration, the release of vasoactive substances like NO and prostacyclin under laminar shear stress, can decrease the permeability to plasma lipoproteins and the adhesion of leukocytes, and as well as inhibit VSMC proliferation and migration (Pan 2009). Fluid shear stress suppresses SMC migration via the inhibition of the phosphorylation of the ERKs-myosin light chain kinase signaling pathway (Goldman et al. 2007).
VSMC differentiation
VSMC differentiation is considered as a vital event during the progression of VSMC-related diseases, including atherosclerosis, restenosis, and asthma. Two main types of hemodynamic force, shear stress and CS, contribute to the differentiation process in the microenvironment of vascular cells. Under moderate damage, laminar shear stress directly activates growth factor receptors on stem/progenitor cells, thus initiates signaling pathways and leads toward EC differentiation to maintain the vessel integrity. When encountering severe damage or atherosclerotic lesion, the disturbed flow is locally formed; CS stimulates stem cell differentiation to SMC lineages (Potter et al. 2014). Mechanical stress induces MKP-1 expression regulated by growth factor receptor-Ras-ERK and Rac-JNK/SAPK or p38MAPK pathways, which leads to VSMC growth and differentiation (Li et al. 1999). CS activates PDGF-Rβ in a ligand-independent manner, which plays a critical role in the differentiation of VSMCs (Zhang et al. 2013a). Mechanical stimuli play a dominating role in initial ERK pathway, whose activity may play a direct role in SMC differentiation (Dharmarajan et al. 2018). After exposure to CS in both 2D and 3D models, the increased secretion of transforming growth factor-β1 (TGF-β1) into the supernatant of VSMCs, as well as the expression of contractile phenotype markers, including alpha-actin (SMA), calponin, c-fos, phosphorylated Smad2 and Smad5, SIRT6, and smooth muscle protein 22 alpha in VSMCs indicated that CS can guide mesenchymal stem cells (MSCs) to VSMC differentiation via the TGF-β1-Smad-SIRT6 pathway (Dan et al. 2015; Yao et al. 2014). Furthermore, CS of a low magnitude (0.26 Hz, 3%) mediates the differentiation of MSCs into osteogenic cells, whereas greater CS (0.26 Hz, 10%) favors their differentiation into SMCs (Jang et al. 2011). In addition to this, the stretch-induced activation of the AMPK pathway (whose mediators include MO25α, AMPK, AICAR, and ACC) in vascular smooth muscles is in part regulated by reduced levels of miR-144/451, an effect that may play a role in promoting the contractile differentiation of VSMCs (Turczynska et al. 2013). Mechanical CS up-regulates Rac and down-regulate its negative regulator Rho-GDIα in a nonlinear frequency-dependent manner, leading to the activation of the p38 pathway, followed by an increased in the expression of h1-calponin, which is a marker for VSMC differentiation (Qu et al. 2008). Moreover, stretch-induced Rho-ROCK signaling can preserve VSMC differentiation in vascular hypertrophy by influencing the actin dynamics (Albinsson et al. 2004). Mechanical strain can induce a frequency-dependent re-differentiation of synthetic VSMCs in vitro at least partly via the activation of the p38 pathway (Qu et al. 2007). Similarly, laminar shear stress induces synthetic-to-contractile phenotypic switch in VSMCs via the activation of peroxisome proliferator-activated receptor-α/δ mediated by the EC-released prostacyclin (Tsai et al. 2009). However, during hypertension, pathological stretching may induce ER stress in VSMCs, affecting the alternative splicing and activity of large conductance calcium and voltage-activated potassium (BK) channels as a result, subsequently inducing the de-differentiation of VSMCs (Wan et al. 2015). On the other hand, the laminar shear stress suppresses VSMC marker expression by downregulating histone deacetylases 7/8 and the TGF-β signaling pathway, leading to endothelial differentiation (Zhang et al. 2013a).
VSMC inflammation
Proinflammatory stimuli, including cytokines, toll-like receptor (TLR) ligands, oxLDL, and certain matrix proteins etc., all induce inflammatory gene expression through a set of signaling pathways, such as the stress-activated protein kinases p38, JNK, the transcription factors NF-κB, NFAT, and STAT1/3 (O'Neill 2006). Experimental analysis of the smooth muscle response to stretch suggests that too much or little stretch both lead to VSMC proliferation and the induction of inflammatory genes (Birukov et al. 1998; Lehoux et al. 2006). Exposing VSMCs to pulsatile stretch stimulates NF-κB activation through an oxidative stress-dependent pathway (Hishikawa et al. 1997). A key contributor to early atherogenic inflammation, αvβ3, is a primary integrin heterodimer to mediate shear stress-induced proinflammatory signaling (NF-κB, p21-activated kinase), as well as gene expression (ICAM1, VCAM1) (Chen et al. 2015a). SS-induced VSMC-supported thrombin generation is mediated by the integrin αvβ3 signaling pathway (Mao et al. 2012). The stretch-induced production of ROS stimulates monocyte chemoattractant protein-1 (MCP-1) expression in a p38- and ERK- dependent manner (Guest et al. 2006), suggesting that stretch may stimulate inflammatory gene expression via multiple signaling pathways. Biomechanical stress-induced interleukin-6 (IL-6) expression occurs in part via Ras/Rac/p38 MAPK/NF-κB/NF-IL6 signaling pathway, which is downregulated by PKC-δ, contributing to the pathogenesis of atherosclerosis (Zampetaki et al. 2005). Escherichia coli lipopolysaccharide is a potent inducer of NF-κB activity, ERKs phosphorylation, MCP-1 release, and TLR 2 mRNA expression in wild-type mice but not in TLR4-signaling deficient mouse aortic VSMC, which indicated that TLR4 signaling promotes a pro-inflammatory phenotype in VSMCs, such that VSMCs may potentially play an active role in vascular inflammation via the release of chemokines, pro-inflammatory cytokines, and increased TLR 2 expression (Yang et al. 2005). In addition, SS-induced YAP/TAZ activation is followed by a strong pro-inflammatory response in human umbilical arterial SMCs; YAP/TAZ silencing attenuates the expression of cell adhesion molecules and inflammatory cytokines, suggesting that YAP/TAZ activation may exert a pro-inflammatory effect on atherosclerotic lesions (Wang et al. 2018b). Aldosterone promotes early atherosclerosis in areas of turbulent blood flow and an inflammatory plaque phenotype that is associated with rupture in humans, the mechanism of which may involve the VSMC release of soluble factors that recruit activated leukocytes to the vessel wall via proinflammatory placental growth factor signaling (McGraw et al. 2013). Under elevated CS, aortic VSMCs can produce IL-6 and MCP-1, which initiates media macrophage accumulation as well as aortic dilation (Akerman et al. 2018). On the contrary, physiological shear stress is anti-inflammatory, specifically inhibiting MAPK signaling and inhibiting TNFR-associated factor-2 interaction with TNF receptor (TNFR)-1 (Yamawaki et al. 2003).
VSMC apoptosis
Abundant evidence indicates the importance of the apoptosis of VSMCs in the development of vascular lesions, such as atherosclerosis and postangioplasty restenosis. The in vivo SAPK/JNK mediation of apoptosis was involved in the development of atherosclerosis, in which certain cells in the lesions could express higher levels of SAPK/JNK activity, as well as higher levels of p53 (i.e. SAPK/JNK-activated p53) (Shaw and Xu 2003). Moreover, VSMC apoptosis induced by mechanical stress is p53-dependent, where p53 is activated by mechanical stress in a Rac- and p38MAPK-dependent manner (Mayr et al. 2002). Acute cell apoptosis after vascular injury is highly regulated by the activation of the MAPK signaling pathway. Mechanical stress-induced p38MAPK activation is partly responsible for transducing the signals leading to the apoptosis of venous bypass grafts (Xu 2000). Additionally, mechanical stress-induced apoptosis of VSMCs is mediated by the β1-integrin-rac-p38-p53 signaling pathway (Wernig et al. 2003). p53-up-regulated modulator of apoptosis (PUMA) is an important mediator of VSMC apoptosis, wherein CSinduces PUMA expression in cultured human VSMCs via IFN-γ, JNK, and interferon regulatory factor-1 pathways (Cheng et al. 2012). Apoptosis signal-regulating kinase 1 (a MAPK kinase) accelerates mechanical stress-activated VSMC migration via enhanced VSMC and EC apoptosis and/or increased neovascularization, thereby inducing vascular remodeling (Tasaki et al. 2013). Low shear stress-induced VSMC apoptosis is mediated by the suppressed expression of Rho-GDIα (Qi et al. 2008). CS inhibits VSMC growth and enhances VSMC apoptosis, in part, by regulating the Notch receptor and via downstream target gene expression (Morrow et al. 2005). Laminar shear stress stimulates VSMC apoptosis via the Akt pathway (Fitzgerald et al. 2008). The CS-induced decrease in Hedgehog signaling in VSMCs is related to a marked increase in VSMC apoptosis in vitro and in vivo (Morrow et al. 2007). SS, shear stress and AGEs can increase the level of cleaved caspase-3 in VSMCs, inducing the apoptosis of VSMCs (Ekstrand et al. 2010; Fitzgerald et al. 2008; Ping et al. 2017). However, whether other caspases are involved in this process or not has not been fully studied. As such, it may play a fundamental role in arterial remodeling and atherogenesis in vivo. Mechanical stress-induced endoplasmic reticulum stress also promotes VSMC apoptosis and degeneration, providing insight into the formation and progression of thoracic aortic aneurysm/dissection (Jia et al. 2015). CS can significantly induce VSMCs apoptosis and aortic dissection formation, which can be impeded by YAP1 over-expression via Hippo-YAP signal pathway (Liu et al. 2017b).
VSMC pyroptosis
Pyroptosis, a newly discovered form of programmed cell death, is a pro-inflammatory form of regulated cell death, like apoptosis and necrosis. Pyroptosis depends on the enzymatic activity of inflammatory proteases that belong to the family of cysteine-dependent aspartate-specific proteases (caspases), for example, caspase-1. Caspase-1 activation is required for the formation of the protein platform known as the inflammasome (e.g. Absent in melanoma 2 (AIM2)) (Chang et al. 2013). Pyroptosis in VSMCs is mediated via the caspase-1 pathway, indicating that AIM2, a member of the HIN-200 protein family, is an active participant in atherosclerosis (Pan et al. 2018). As mentioned above, pyroptosis may be involved in atherosclerosis, particularly in advanced atherosclerotic lesions, and play an important role in atherosclerotic lesion instability. However, few relevant studies currently exist on pyroptosis, so further research about whether it occurs in atherosclerosis and underlying mechanisms will be needed.
Vascular calcification
Vascular calcification is a pathophysiological cell-regulated process, involving a series of regulatory pathways, factors (e.g. mechanical stimuli) and affecting multiple aspects of the vascular tree, particularly the intima and media of arteries and cardiac valves. Human aortic ECs and SMCs both produce osteoprotegerin under both physiological and pathological conditions, noting a considerably more production of osteoprotegerin by HASMCs under both conditions tested. The treatment of HASMCs with the receptor activator of nuclear factor-κB ligand, derived from nonvascular cells, inhibits basal osteoprotegerin production and completely blocks the strain-mediated upregulation of osteoprotegerin, thereby promoting vascular calcification (Davenport et al. 2018). Mechanical stimulation regulates calcification by calcifying vascular cells. However, the inhibition of the p38 and JNK pathways exerts a direct effect on the osteogenic conversion of the calcifying vascular cells. The inhibition of the ERKs pathway is associated with an increase in alkaline phosphatase expression and a decrease in mineralization, which suggests delayed osteogenic differentiation of the cells, not involved in transduction of the mechanical stimulus (Simmons et al. 2004). High rates of acceleration of calcification is observed in a common model of atherogenesis--apolipoprotein E-deficient [ApoE(−/−)] mice (Wang et al. 2014). Additionally, bone morphogenic protein-2 induced osteogesis in MSCs is controlled by cytoskeletal tension and cell shape via the Rho/ROCK signaling pathway (Wang et al. 2012a). However, this requires further investigation in the context of medial calcification. On the other hand, physiological mechanical strain may protect the arteries from vascular calcification mediated via the calcium-sensing receptor (Molostvov et al. 2015). CS can down-regulate the expression of bone-associated genes (osteopontin, matrix gla protein, alkaline phosphatase, and the transcription factor CBFA-1) in VSMCs, where long-term strain plays a protective role against calcification (Nikolovski et al. 2003). Overall, it has become increasingly clear in recent years that arterial calcification is an active reprogramming of VSMCs by local environmental cues into a dynamic range of phenotypes rather than a passive process.
VSMC cellular component changes
As far as we know, cerebral aneurysm growth is characterized by continuous structural weakness of local VSMCs, the mechanism by which is unclear. After CS, cell proteomics analysis showed that down-expression of 118 proteins and up-expression of 32 proteins in VSMCs, together with decreased expression of collagen type IV/VI and increased expression of MMP-1/MMP-3 (Liu et al. 2018). Decreased expression of fibulin-4 can sensitize SMCs to stimuli and lead to increased expression of early growth response 1 and thrombospondin-1. Thrombospondin-1 is induced by SS and Ang II in SMCs, resulting in elastic fiber organizing disruption and actin cytoskeletal remodeling dysregulation, contributing to the development of ascending aortic aneurysms in vivo (Yamashiro et al. 2018).
In summary, mechanical stress-induced pathophysiological effects of VSMCs in vascular diseases are likely to be dependent on several interacting pathways (Fig. 1), whereby the singular inhibition of one single pathway is ineffective. Currently clarified mechanical stress-induced signaling pathways can provide potential targets for therapeutic intervention in vascular diseases, like atherosclerosis.

Mechanical stretch-induced signaling pathway in VSMCs. Mechanical stretch activates transmembrane proteins including LOX-1, RAGE, integrin, RTKs, GPCRs, TRPV, Piezo and other unknown receptors non-specifically. Then multiple signaling pathways would be activated simultaneously, and subsequently lead to various cellular effects. The kinase system, oxidative stress, ER stress and epidemic changes are involved in these processes. These effects would result in cardiovascular remodeling, such as atherosclerosis, hypertension and grafted venous AS, which are leading cause of the clinical death. FAK: focal adhesion kinase; Grb2: growth factor receptor-bound protein 2; SRC: sarcoma; SOS: Son of Sevenless; G: G protein; PLC: phospholipase C; PI-3 K: phosphoinositide 3-kinases; Ras: a family of small GTPase; MAPKs: mitogen-activated protein kinases; ERK: extracellular regulated protein kinase; JNK: c-Jun N-terminal kinases; P38: P38 MAPK; PDI: protein disulfide isomerase; PIP2: phosphatidylinositol 4,5-bisphosphate; AC: adenylyl cyclase; YAP: Yes-associated protein 1; PIP3: phosphatidylinositol (3,4,5)-trisphosphate; RAF: RAF kinases; IP3: inositol trisphosphate; DAG: diglyceride; cAMP: cyclic adenosine monophosphate; NOX: NADPH oxidase; ROS: reactive oxygen species; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; Akt: protein kinase B; MAPKK: mitogen-activated protein kinase kinase, MMP: matrix metalloproteinase; PKC: protein kinase C; PKA: protein kinase A; ER: endoplasmic reticulum; PERK: protein kinase R (PKR)-like endoplasmic reticulum kinase; IRE1α: inositol-requiring enzyme 1 α; ATF6: activating transcription factor 6; AS: atherosclerosis.
Mechanical stress-induced epigenetic modifications and pathophysiological effects in VSMCs
Epigenetics refers to the study about heritable changes in gene expression with the invariable DNA sequence. Epigenetic modifications are divided into three main categories: DNA methylation, histone modification/chromatin remodeling and RNA-based mechanisms. Hemodynamic forces activate molecular pathways leading to histone modifications, transcription complex activation, and vascular/cardiovascular marker expression in undifferentiated mouse embryonic stem (ES) cells, leading to the onset of a vascular/cardiovascular differentiation program (Illi et al. 2005). Here, we will discuss the functions of methylation, histone modifications and microRNAs (miRNAs) in stretch-induced phenotype transformation, proliferation, migration, differentiation, inflammation, apoptosis and calcification of VSMCs (Table 1).
VSMC phenotype transformation
In atherosclerotic vessels, miR-221/222 is upregulated in initial atherogenic stages and then stimulates VSMCs to switch from the “contractile” to the “synthetic” phenotype, in connection with the induction of proliferation and motility (Chistiakov et al. 2015). Upregulation of miR-31 levels induces downregulation of its target--a cellular repressor of E1A-stimulated genes which is widely expressed in differentiated cells, resulting in the modulation of the phenotype of human VSMCs (Wang et al. 2013). MiR-181a/b is one of the factors involved in the differentiation of VSMCs toward a synthetic phenotype via targeting serum response factor, upregulating synthetic marker genes, and downregulating contractile marker genes (Wei et al. 2017). Mechanical SS increases the expression of miR-29a-3p to reduce the expression of Ten-eleven translocation methylcytosinedioxygenase 1, switching contractile VSMCs to the synthetic phenotype (Jiang et al. 2019). However, several others miRNAs have an inhibitory effect on the modulation of VSMC phenotypic, including miR-15b/16 (Xu et al. 2015), miR-132 (Choe et al. 2013), miR-133 (Torella et al. 2011), miR-195 (Wang et al. 2012b), miR-204 (Courboulin et al. 2011), miR-424/322 (Merlet et al. 2013), and miR-663 (Li et al. 2013).
VSMC proliferation
As for proliferation, miR-31 has been associated with the de-differentiated/proliferative state of VSMCs (Lee et al. 2013). Similarly, miR-221/222 exerts a pro-proliferation effect in VSMCs (Liu et al. 2012). Under conditions of SS, miR-153 and miR-223 are reduced in VSMCs, contributing to resultant IGF-1R activation and VSMC proliferation. miR-21 overexpression can increase the proliferation of VSMCs via decreased phosphatase and tensin and increased Akt activation during the development of aortic aneurysms (Alajbegovic et al. 2017). SS can increase the expression of primary miR-21 and pre-miR-21 in HASMCs, thus regulating VSMC proliferation (Song et al. 2012a). MiR-130a is a novel regulator of proliferation of VSMCs via the inhibition of growth arrest-specific homeobox expression (Wu et al. 2011). In addition, miR-142-5p has been found to promote VSMC proliferation by down-regulating B cell translocation gene 3, which inhibits the expression of cell cycle regulatory genes and cell growth (Kee et al. 2013). MiR-146a targets the Krüppel-like factor 4 3′-untranslated region and forms a feedback loop by which they can regulate each other’s expression, as well as VSMC proliferation (Sun et al. 2011). Likewise, the upregulation of miR-26a can stimulate VSMC proliferation (Leeper et al. 2011). The overexpression of miR-143 significantly inhibits the expression of versican, a component of the extracellular matrix, which is produced by synthetic VSMCs and facilitates VSMC proliferation (Wang et al. 2010). MiR-155 is markedly upregulated in atherosclerotic plaque, thereby accelerating VSMC proliferation by targeting eNOS (Zhang et al. 2015). LncRNA TUG1/miR-145-5p/FGF10 activates the Wnt/β-catenin pathway to promote the proliferation of VSMCs in hypertensive state (Shi et al. 2018). Under pathological conditions (e.g. 15% SS), miR-19b-3p expression is significantly down-regulated, thus VSMC proliferation is promoted causing vascular remodeling via miR-19b-3p/the connective tissue growth factor pathway (Wang et al. 2019b). MiR-155, whose enhancement is related to hypertension, promotes VSMC proliferation by targeting suppressed p27 expression (Xu et al. 2018). Meanwhile, let-7d microRNA, as a vital regulator of cell proliferation, is significantly down-regulated in VSMCs, while let-7d-transfected VSMCs display reduced cell growth, resulting in more cells in the G1 phase than in the G2/M phases of the cell cycle (Yu et al. 2011). It is worth noting that miR-22 overexpression in injured vessels notably reduces the expression of its target genes, decreases VSMC proliferation and inhibits neointima formation in wire-injured arteries. The opposite effect is observed with the local usage of a miR-22 inhibitor to injured arteries (Yang et al. 2018). Endothelial microparticles can deliver functional miR-126 into recipient VSMCs, thus reducing VSMC proliferation and subsequent neointima formation (Jansen et al. 2017). MiR-34a inhibits VSMC proliferation and neointima formation by reducing the expression levels of Notch1 (Chen et al. 2015b). MiR-362-3p represses the VSMC proliferation by directly targeting a disintegrin and metalloproteinase with thrombospondin motifs 1 (ADAMTS1) in atherosclerosis (Li et al. 2017). MiR-133 expression, which is regulated by extracellular signal-regulated kinase 1/2 activation and regulates smooth muscle gene expression through transcription factor Sp-1 repression, can reduce VSMC proliferation and migration in vitro and in vivo (Torella et al. 2011). Furthermore, locally enforced expression of miR-214 in injured vessels was found to significantly reduce the expression levels of NCK-associated protein 1, inhibit VSMC proliferation, and prevent neointima VSMC hyperplasia after injury (Afzal et al. 2016). Extracellular vesicle-derived miR-223 inhibits VSMC proliferation, resulting in a reduction of the atherosclerotic plaque size (Shan et al. 2015).
VSMC migration
With regard to the migration of VSMCs, SS upregulates the levels of acetylated histone H3 and HDAC7, while downregulating the levels of HDAC3/4 in VSMCs, thus inducing the migration of cultured VSMCs (Yan et al. 2009). MiR-221/222 has a pro-migration effect on VSMCs (Liu et al. 2012). The overexpression of miR-26a has also been found to stimulate VSMC migration (Leeper et al. 2011). Likewise, the overexpression of miR-143 facilitates VSMC migration using the same mechanism (Wang et al. 2010). MiR-4463 inhibited the migration of human aortic VSMCs by targeting angiomotin expression, while, in the plasma of patients with atherosclerosis, the expression of miR-4463 was lower than in the control (Wang et al. 2018a). MiR-155 is significantly upregulated in the atherosclerotic plaque, accelerating the migration of VSMCs by targeting eNOS (Zhang et al. 2015). In hypertensive state, LncRNA TUG1/miR-145-5p/FGF10 activates the Wnt/β-catenin pathway to promote VSMC migration (Shi et al. 2018). On the other hand, the overexpression of miR-22 in injured vessels significantly reduces the expression of its target genes, decreases VSMC migration, and inhibits neointima formation as well. In addition, the opposite effect occurs after the local usage of a miR-22 inhibitor to injured arteries (Yang et al. 2018). Endothelial microparticles deliver functional miR-126 into recipient VSMCs to repress VSMC migration and subsequent neointima formation (Jansen et al. 2017). MiR-34a inhibits VSMC migration and neointima formation by reducing Notch1 expression levels (Chen et al. 2015b). MiR-362-3p inhibits VSMC migration by directly targeting ADAMTS1 in atherosclerosis (Li et al. 2017). As mentioned earlier, miR-133 can also reduce VSMC migration in vitro and in vivo, suggesting its potential therapeutic prospect for vascular diseases (Torella et al. 2011). The overexpression of miR-214 in serum-starved VSMCs can significantly decrease VSMC migration by reducing the expression levels of NCK-associated protein 1 (Afzal et al. 2016). Furthermore, extracellular vesicle-derived miR-223 inhibits VSMC migration, resulting in a reduction of atherosclerotic plaque sizes (Shan et al. 2015).
VSMC differentiation
As for VSMC differentiation, miR-143 and miR-145 are associated with promoting the contractile differentiation of VSMCs by negatively regulating the target gene Kruppel-like factor-5 (KLF5) and its downstream myocardin (Alajbegovic et al. 2017; Turczynska et al. 2012). In addition, miR-10a expression has been found to be steadily elevated during the differentiation of mouse embryonic stem cells to VSMCs (Huang et al. 2010). MiR-26a, on the other hand, is known to inhibit VSMC differentiation (Leeper et al. 2011).
VSMC inflammation
As regards the cycle of inflammation, class III HDAC SIRT1 are known to play a protective role in atherosclerosis in VSMCs by suppressing p21, enhancing the replication of senescence-resistant cells and inhibiting inflammatory events, thereby preventing the formation of atherosclerotic plaques (Stein and Matter 2011). As a key regulator of inflammatory gene expression in leukocytes and lymphocytes (Pedersen and David 2008; Urbich et al. 2008), miR-155 is induced by lipopolysaccharides, TNFα, and interferon-γ (IFN-γ) (O'Connell et al. 2007; Tili et al. 2007) in leukocytes, and reduces the expression of the TLR and cytokine signaling pathway components, such as Fas-associated death domain protein, the receptor (TNFR superfamily)-interacting serine-threonine kinase 1, and IκB kinase (Tili et al. 2007), as a negative feedback pathway. Evidence suggests that IL-32α, a cytokine with anti-inflammatory and anti-atherogenic effects, can upregulate the atheroprotective genes Reck and Timp3 by downregulating miR-205 via the Rprd2-Dgcr8/Ddx5-Dicer1 biogenesis pathway (Son et al. 2017). Interleukin-19 (IL-19), a novel anti-inflammatory cytokine, reduces lipid accumulation in VSMCs, as well as low density lipoprotein receptor adaptor protein 1 expression and oxLDL uptake, in a miR133a-dependent manner to attenuate atherosclerosis via anti-inflammatory effects on VSMCs (Gabunia et al. 2017).
VSMC apoptosis
With respect to VSMC apoptosis, stretch increases the expression of primary miR-21 and pre-miR-21 in HASMCs, which is involved in the regulation of stretch-mediated apoptosis (Song et al. 2012a). It is proved that miR-34a expression is increased in the atherosclerotic plaques (Tana et al. 2017; Toba et al. 2014). The overexpression of miR-34 family members represses proliferation and promotes apoptosis of VSMCs and umbilical vein ECs by inhibiting alpha-1 antitrypsin expression (Wang et al. 2019a). However, in VSMCs, miR-221/222 has an anti-apoptosis effect (Liu et al. 2012). MiR-26a can also inhibit VSMC apoptosis (Leeper et al. 2011).
Vascular calcification
In atherosclerotic vessels, miR-221 and miR-222 synergistically contribute to the atherogenic calcification of VSMCs (Chistiakov et al. 2015). As a critical modulator, miR-32 modulates vascular calcification through its targeting of phosphatase and tensin homolog Mrna/Akt/runt-related transcription factor-2 axis in mice. Furthermore, higher miR-32 levels have been found in the plasma of patients with coronary artery calcification compared to non-coronary artery calcification patients (P = 0.016) (Liu et al. 2017a). Another key miRNA, miR-125b, inhibits the process of VSMC calcification (London et al. 2003) by inhibiting osteoblastic proliferation and differentiation (Goettsch et al. 2011; Mizuno et al. 2008; Wen et al. 2014).
Although extensive studies have gone in-depth on the molecular mechanisms of mechanical stress regulating intracellular signals thereby modulating downstream gene expression, research investigating the role of mechanical stress-induced epigenetic pathways has emerged only recently. Numerous studies have uncovered that shear stress-induced epigenetic modifications can affect EC functions, while the effects of mechanical force-induced epigenetic modifications on VSMCs remain unclear. Further research is needed with the goal of shedding light on the mechanisms underlying how the dynamic environment of blood vessels influences vascular cells (especially VSMCs) during the development of vascular diseases.
Simultaneous increases in proliferation and apoptosis of VSMCs induced by mechanical stress
VSMC proliferation and apoptosis play important roles in the pathophysiology of vascular remodeling initiated by mechanical stress. We have been studying the effects of mechanical stress on the proliferation and apoptosis of VSMCs and related mechanisms. So far, most studies have focused only on the unilateral effects of mechanical stress on VSMC proliferation or apoptosis. In our experiments, we found that when mouse venae cavae were transplanted to carotid arteries of non-diabetic mice, the grafted veins underwent the action of arterial pressure. Eight weeks later, the simultaneous increases of cell proliferation and apoptosis were found in the grafted veins wall, which triggered vein graft arterializations. On the other hand, when mouse venae cavae were transplanted to carotid arteries of diabetic mice, the transplanted veins suffered dual roles from arterial pressure and hyperglycemia. After 8 weeks, compared with those of the non-diabetic mice, the wall thickness of the vein grafts and deposition of AGEs were more visibly altered, and more proliferating and apoptotic cells were found, which caused vein graft atherosclerosis (Li et al. 2012; Ping et al. 2015; Ping et al. 2017). In vitro experiments, we found that mechanical SS could non-specially activate transmembrane proteins, including α1-ARs, LOX-1 and RAGE, on cell membranes, leading to the activation of intracellular signaling molecules ERKs and VSMC proliferation. Noradrenaline, ox-LDL, and AGEs could amplify SS-induced signaling respectively, where α1-ARs, LOX1, and RAGE acted as mechanosensors for the transfer of signals (Li et al. 2012; Liu et al. 2013; Zhang et al. 2013b).
Further research found that both SS and AGEs alone were able to induce different levels of MAPKs (including ERK, JNK and P38MAPK) activation and simultaneous increases in the proliferation and apoptosis of VSMCs (Ping et al. 2015; Wang et al. 2017), while the combination of both had a synergistic effect (Ping et al. 2015). The transformation of the VSMC subtypes is closely related to the pathophysiology of vascular disorders. In immunofluorescent staining with SM-α-actin antibody, we found that all cultured VSMCs expressed different levels of SM-α-actin, which suggested the cultured VSMCs existed in different subtypes and with varying degrees of heterogeneity. We found JNK-, and P38MAPK-activated cells with strong SM-α-actin expression were prone to apoptosis, however, ERK-activated cells showed weak SM-α-actin expression which were easy to proliferate. On the contrary, the inhibition of MAPKs signaling pathways resulted in significant decreased proliferation and apoptosis in VSMCs, and treatment with the small interfering RNA of RAGE also significantly inhibited the proliferation and apoptosis of VSMCs. These findings indicate that the same extracellular stimuli could result in different fates in cultured VSMCs, which is mainly related to the selective activation of MAPKs. SM-α-actin may play an important role as a signal target for regulating VSMC proliferation and apoptosis. Consistent with in vitro experiments, more ERK-, JNK-, and P38MAPK-activated cells were observed in the vein grafts of diabetic mice compared to those of non-diabetic mice (Ping et al. 2015).
PDI belongs to the thioredoxin superfamily oxidoreductase, which involves cell survival and death. In a recent study, we found that PDI expression was upregulated in the vein grafts of non-diabetic and diabetic mice, where PDI and SM-α-actin were co-expressed. This strong co-expression of PDI and SM-α-actin may result in VSMC apoptosis, as a weaker co-expression of both contributed to the proliferation of VSMCs. Besides, we found higher co-expression of PDI and SM-α-actin and more proliferative and apoptotic cells in the vein grafts of diabetic mice. In cultured VSMCs, both SS and AGEs rapidly upregulated PDI and NOX1 expression, and ROS production, leading to simultaneous increases of cell proliferation and apoptosis, where combined treatment of both had synergistic effects. Further, the VSMCs with a weak co-expression of PDI and SM-α-actin were proliferating cells, while strong co-expression of both indicated cell apoptosis or death. These results are consistent with those of the in vivo experiments, suggesting that arterial pressure or SS alone can upregulate PDI expression and induce simultaneous increases of VSMC proliferation and apoptosis resulting in vein graft arterializations. Further, diabetic-related AGEs could synergistically amplify arterial pressure/SS- initiated signaling, resulting in higher PDI expression and more proliferative and apoptotic cells, accelerating vein graft arteriosclerosis. Furthermore, different levels of PDI expression indicate cell proliferation or apoptosis (Ping et al. 2017).
Taken together, our study proved that the simultaneous increases of cell proliferation and apoptosis were closely related to VSMC subtypes via PDI-NOX1-ROS signaling in vein graft arterializations or atherosclerosis (Ping et al. 2017) (Fig. 1). However, the mechanism by which PDI and SM-α-actin interact to induce cell proliferation and apoptosis requires further study. In addition, PDI is not only related to cell proliferation and apoptosis, but also closely related to endoplasmic reticulum stress and oxidative stress. The relationship between cell proliferation, apoptosis, endoplasmic reticulum stress and oxidative stress is the subject of our current research. Here, the inhibition of PDI in vitro could simultaneously suppress the proliferation and apoptosis of VSMCs, whether or not the inhibition of PDI in vivo can simultaneously inhibit VSMC proliferation and apoptosis leading to reduced vein graft atherosclerosis need to be further proven. Overall, these results provide a new view on the mechanism of vein graft atherosclerosis, and PDI may role as a new drug target for the prevention and treatment of vascular remodeling diseases.
Conclusions and perspectives
In vitro experiments, animal studies, and human studies have shown that mechanical stresses are powerful determinants in the regulation of arterial function, myocardial remodeling, and the differentiation of progenitor cells in the vascular system. Hypertension-induced mechanical stress non-specifically and receptor ligands specifically co-activate transmembrane proteins, including various receptors, caveolins, ion channels, and pumps on the cell membrane. Activation of transmembrane proteins results in the simultaneous activation of multiple signals of downstream molecules (e.g. intracellular kinases, ions, redox system, and caspases) initiating a profound network of a variety of intracellular multiple signaling pathways, thereby resulting in the multiple functional consequences of pathophysiology, such as cell phenotypic changes, migration, differentiation, inflammation, calcification, proliferation, pyrotosis and apoptosis, etc. The changed pathophysiology is closely associated with cardiovascular remodeling and diseases, leading cause of the clinical death (Fig. 1). Further study should focus on the following aspects: in terms of basic research, studies can be carried out at different levels, such as cell membrane level, cytoplasmic level and nucleus level. There are many kinds of transmembrane proteins on the membrane, and only a few transmembrane proteins such as receptor and mechanical force activation have been reported. Because mechanical force activation is nonspecific, all transmembrane proteins on the membrane may be activated. So, are all of these transmembrane proteins promoting vascular remodeling disease? Are there inhibitory transmembrane proteins activated? Due to the non-specific activation of mechanical forces, many intracellular signaling pathways are activated at the same time, thus forming a complex intracellular signaling network, and the signaling pathways can also regulate each other. So, what molecules in these networks act as network nodes? Can these node-regulating molecules simultaneously block intracellular upstream multi-pathway signals to block mechanical force non-specific multi-pathway activation signals? MiRNA and epigenetic studies have been a hot topic in recent years, and the effects of mechanical stimulation on miRNA and epigenetic regulation can be observed to elucidate the mechanism of mechanical stimulation on vascular remodeling and disease. In terms of drug development, once it is found which membrane proteins mediate mechanical force stimulation signals to promote or inhibit, people can develop corresponding inhibitors or agonists. The regulation of key node molecules can block the damage effect of mechanical forces on blood vessels. VSMCs have very specific functions and physiological characteristics, with different phenotypes. However, the cells with different phenotypes have different intracellular signals and biological behaviors in response to the same mechanical stimulation, just as a group of cells can proliferate and apoptosis under the same in vitro culture. Cell growth and death can occur under the same culture conditions in vitro and in the same internal environment in vivo. However, excessive proliferation or apoptosis is harmful to vascular lesions. Therefore, the clinical prevention and treatment of vascular remodeling and disease (atherosclerosis) need to change from the traditional strategy of inhibiting proliferation and inducing apoptosis of VSMCs to simultaneous inhibition of proliferation and apoptosis. The development of drugs that have multiple functions for the same drug is even more intriguing.
Data Availability
Not applicable.
Abbreviations
- ADAMTS1:
-
A disintegrin and metalloproteinase with thrombospondin motifs 1
- AIM2:
-
Absent in melanoma 2
- AC:
-
Adenylate cyclase
- AT1R:
-
Angiotensin II type 1 receptor
- AR:
-
Adrenergic receptor
- AGEs:
-
Advanced glycation end products
- CS:
-
Cyclic strain
- ECs:
-
Endothelial cells
- eNOS:
-
Endothelial nitric oxide synthase
- ERKs:
-
Extracellular receptor kinases
- FAK:
-
Focal adhesion kinase
- GPCRs:
-
G protein-coupled receptors
- IRS-1:
-
Insulin receptor substrate-1
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- IFN-γ:
-
Interferon-γ
- IL-6:
-
Interleukin-6
- JNKs:
-
C-Jun N-terminal kinases
- LOX-1:
-
Lectin-like oxidized low-density lipoprotein receptor-1
- MCP-1:
-
Monocyte chemoattractant protein-1
- MKP-1:
-
MAPK phosphatase-1
- MMP:
-
Matrix metalloproteinase
- MSCs:
-
Mesenchymal stem cells
- MiRNAs:
-
MicroRNAs
- MAPK :
-
Mitogen-activated protein kinase
- NOX1 :
-
NADPH oxidase subunits 1
- NSCC :
-
Non-selective cation channel current
- OX-LDL :
-
Oxidized low-density lipoprotein
- PUMA :
-
p53-up-regulated modulator of apoptosis
- PI3K :
-
Phosphoinositide 3-kinase
- PLC :
-
Phospholipase C
- PDGFRs :
-
Platelet derived growth factor receptors
- PDI :
-
Protein disulfide isomerase
- PKA :
-
Protein kinase A
- PKC :
-
Protein kinase C
- RhoA :
-
Ras homolog gene family member A
- ROS :
-
Reactive oxygen species
- RAGE :
-
Receptor for advanced glycation end products
- Rho-GDIα :
-
Rho GDP dissociation inhibitor α
- ROCK :
-
Rho/Rho-kinase
- STAT-3 :
-
Signal transducer and activator of transcription-3
- SM-α-actin :
-
Smooth muscle alpha-actin
- SS :
-
Stretch stress
- TAZ :
-
Transcriptional co-activator with PDZ-binding motif
- TGF-β1 :
-
Transforming growth factor-β1
- TRPV :
-
Transient receptor potential vanilloid
- TLR :
-
Toll-like receptor
- VSMC :
-
Vascular smooth muscle cell
- YAP :
-
Yes-associated protein
References
Afzal TA et al. (2016) NCK associated protein 1 modulated by miRNA-214 determines vascular smooth muscle cell migration, proliferation, and Neointima hyperplasia J am heart Assoc 5 10.1161/JAHA.116.004629
Akerman AW et al (2018) Elevated Wall tension initiates Interleukin-6 expression and abdominal aortic dilation. Annals of vascular surgery 46:193–204. https://doi.org/10.1016/j.avsg.2017.10.001
Alajbegovic A, Holmberg J, Albinsson S (2017) Molecular Regulation of Arterial Aneurysms: Role of Actin Dynamics and microRNAs in Vascular Smooth Muscle. Front Physiol 8:569. https://doi.org/10.3389/fphys.2017.00569
Albinsson S, Nordstrom I, Hellstrand P (2004) Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J Biol Chem 279:34849–34855. https://doi.org/10.1074/jbc.M403370200
Asada H et al (2005) Sustained orbital shear stress stimulates smooth muscle cell proliferation via the extracellular signal-regulated protein kinase 1/2 pathway. J Vasc Surg 42:772–780. https://doi.org/10.1016/j.jvs.2005.05.046
Balasubramanian L, Ahmed A, Lo CM, Sham JS, Yip KP (2007) Integrin-mediated mechanotransduction in renal vascular smooth muscle cells: activation of calcium sparks. Am J Physiol Regul Integr Comp Physiol 293:R1586–R1594. https://doi.org/10.1152/ajpregu.00025.2007
Birukov KG, Bardy N, Lehoux S, Merval R, Shirinsky VP, Tedgui A (1998) Intraluminal pressure is essential for the maintenance of smooth muscle caldesmon and filamin content in aortic organ culture. Arteriosclerosis, thrombosis, and vascular biology 18:922–927. https://doi.org/10.1161/01.atv.18.6.922
Chang W, Lin J, Dong J, Li D (2013) Pyroptosis: an inflammatory cell death implicates in atherosclerosis. Med Hypotheses 81:484–486. https://doi.org/10.1016/j.mehy.2013.06.016
Chen J, Green J, Yurdagul A Jr, Albert P, McInnis MC, Orr AW (2015a) alphavbeta3 Integrins mediate flow-induced NF-kappaB activation. Proinflammatory Gene Expression, and Early Atherogenic Inflammation Am J Pathol 185:2575–2589. https://doi.org/10.1016/j.ajpath.2015.05.013
Chen Q, Yang F, Guo M, Wen G, Zhang C, Luong LA, Zhu J, Xiao Q, Zhang L (2015b) miRNA-34a reduces neointima formation through inhibiting smooth muscle cell proliferation and migration. J Mol Cell Cardiol 89:75–86. https://doi.org/10.1016/j.yjmcc.2015.10.017
Cheng JZ, Du J (2007) Mechanical stretch simulates proliferation of venous smooth muscle cells through activation of the insulin-like growth factor-1 receptor. Arteriosclerosis Thrombosis and Vascular Biology 27:1744–1751 https://doi.org/10.1161/Atvbaha.107.147371
Cheng J et al (2010) The mechanical stress-activated serum-, glucocorticoid-regulated kinase 1 contributes to neointima formation in vein grafts. Circ res 107:1265–1274. https://doi.org/10.1161/CIRCRESAHA.110.222588
Cheng WP, Wang BW, Chen SC, Chang H, Shyu KG (2012) Mechanical stretch induces the apoptosis regulator PUMA in vascular smooth muscle cells. Cardiovasc Res 93:181–189. https://doi.org/10.1093/cvr/cvr280
Chiarini A et al (2018) Studies on sporadic non-syndromic thoracic aortic aneurysms: 1. Deregulation of jagged/notch 1 homeostasis and selection of synthetic/secretor phenotype smooth muscle cells. European journal of preventive cardiology 25:42–50. https://doi.org/10.1177/2047487318759119
Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV (2015) Human miR-221/222 in physiological and atherosclerotic vascular remodeling. Biomed res Int 2015:354517. https://doi.org/10.1155/2015/354517
Choe N et al (2013) The microRNA miR-132 targets Lrrfip1 to block vascular smooth muscle cell proliferation and neointimal hyperplasia. Atherosclerosis 229:348–355. https://doi.org/10.1016/j.atherosclerosis.2013.05.009
Coste B et al (2010) Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330:55–60. https://doi.org/10.1126/science.1193270
Coste B et al (2012) Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483:176–181. https://doi.org/10.1038/nature10812
Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Côté J, Simard MJ, Bonnet S (2011) Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 208:535–548. https://doi.org/10.1084/jem.20101812
Dan P, Velot E, Decot V, Menu P (2015) The role of mechanical stimuli in the vascular differentiation of mesenchymal stem cells. J Cell Sci 128:2415–2422. https://doi.org/10.1242/jcs.167783
Dardik A, Yamashita A, Aziz F, Asada H, Sumpio BE (2005) Shear stress-stimulated endothelial cells induce smooth muscle cell chemotaxis via platelet-derived growth factor-BB and interleukin-1alpha. J Vasc Surg 41:321–331. https://doi.org/10.1016/j.jvs.2004.11.016
Davenport C, Harper E, Rochfort KD, Forde H, Smith D, Cummins PM (2018) RANKL Inhibits the Production of Osteoprotegerin from Smooth Muscle Cells under Basal Conditions and following Exposure to Cyclic Strain. J Vasc Res 55:111–123. https://doi.org/10.1159/000486787
DeVerse JS, Bailey KA, Jackson KN, Passerini AG (2012) Shear stress modulates RAGE-mediated inflammation in a model of diabetes-induced metabolic stress. Am J Physiol Heart Circ Physiol 302:H2498–H2508. https://doi.org/10.1152/ajpheart.00869.2011
Dharmarajan A, Floren M, Cox L, Ding Y, Johnson R, Tan W (2018) Mechanochemical Effects on Extracellular Signal-Regulated Kinase Dynamics in Stem Cell Differentiation. Tissue Eng Part A 24:1179–1189. https://doi.org/10.1089/ten.tea.2017.0365
Ding Z, Liu S, Deng X, Fan Y, Wang X, Mehta JL (2015) Hemodynamic shear stress modulates endothelial cell autophagy: role of LOX-1. Int J Cardiol 184:86–95. https://doi.org/10.1016/j.ijcard.2015.01.065
Douguet D, Patel A, Xu A, Vanhoutte PM, Honoré E (2019) Piezo Ion Channels in Cardiovascular Mechanobiology. Trends in pharmacological sciences 40:956–970. https://doi.org/10.1016/j.tips.2019.10.002
Ekstrand J, Razuvaev A, Folkersen L, Roy J, Hedin U (2010) Tissue factor pathway inhibitor-2 is induced by fluid shear stress in vascular smooth muscle cells and affects cell proliferation and survival. J Vasc Surg 52:167–175. https://doi.org/10.1016/j.jvs.2010.02.282
Fitzgerald TN et al (2008) Laminar shear stress stimulates vascular smooth muscle cell apoptosis via the Akt pathway. J cell Physiol 216:389–395. https://doi.org/10.1002/jcp.21404
Forrester SJ et al (2018) Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiological reviews 98:1627–1738. https://doi.org/10.1152/physrev.00038.2017
Fukumoto Y, Hiro T, Fujii T, Hashimoto G, Fujimura T, Yamada J, Okamura T, Matsuzaki M (2008) Localized elevation of shear stress is related to coronary plaque rupture: a 3-dimensional intravascular ultrasound study with in-vivo color mapping of shear stress distribution. J Am Coll Cardiol 51:645–650. https://doi.org/10.1016/j.jacc.2007.10.030
Gabunia K et al (2017) Induction of MiR133a expression by IL-19 targets LDLRAP1 and reduces oxLDL uptake in VSMC. J Mol cell Cardiol 105:38–48. https://doi.org/10.1016/j.yjmcc.2017.02.005
Goettsch C, Rauner M, Pacyna N, Hempel U, Bornstein SR, Hofbauer LC (2011) miR-125b regulates calcification of vascular smooth muscle cells. Am J Pathol 179:1594–1600. https://doi.org/10.1016/j.ajpath.2011.06.016
Goldman J, Zhong L, Liu SQ (2003) Degradation of alpha-actin filaments in venous smooth muscle cells in response to mechanical stretch. Am J Physiol Heart Circ Physiol 284:H1839–H1847. https://doi.org/10.1152/ajpheart.00470.2002
Goldman J, Zhong L, Liu SQ (2007) Negative regulation of vascular smooth muscle cell migration by blood shear stress. Am J Physiol Heart Circ Physiol 292:H928–H938. https://doi.org/10.1152/ajpheart.00821.2006
Guest TM, Vlastos G, Alameddine FM, Taylor WR (2006) Mechanoregulation of monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cells. Antioxid redox signal 8:1461–1471. https://doi.org/10.1089/ars.2006.8.1461
Hall A (2005) Rho GTPases and the control of cell behaviour. Biochem Soc trans 33:891–895. https://doi.org/10.1042/BST20050891
Hishikawa K, Oemar BS, Yang Z, Luscher TF (1997) Pulsatile stretch stimulates superoxide production and activates nuclear factor-kappa B in human coronary smooth muscle. Circ Res 81:797–803. https://doi.org/10.1161/01.res.81.5.797
Horiuchi M, Cui TX, Li Z, Li JM, Nakagami H, Iwai M (2003) Fluvastatin enhances the inhibitory effects of a selective angiotensin II type 1 receptor blocker, valsartan, on vascular neointimal formation. Circulation 107:106–112. https://doi.org/10.1161/01.cir.0000043244.13596.20
Hu Y, Bock G, Wick G, Xu Q (1998) Activation of PDGF receptor alpha in vascular smooth muscle cells by mechanical stress. FASEB J 12:1135–1142. https://doi.org/10.1096/fasebj.12.12.1135
Hu Y, Zou Y, Dietrich H, Wick G, Xu Q (1999) Inhibition of neointima hyperplasia of mouse vein grafts by locally applied suramin. Circulation 100:861–868. https://doi.org/10.1161/01.cir.100.8.861
Huang H, Xie C, Sun X, Ritchie RP, Zhang J, Chen YE (2010) miR-10a contributes to retinoid acid-induced smooth muscle cell differentiation. J Biol Chem 285:9383–9389. https://doi.org/10.1074/jbc.M109.095612
Huang K et al (2017) MicroRNA-33 protects against neointimal hyperplasia induced by arterial mechanical stretch in the grafted vein. Cardiovasc res 113:488–497. https://doi.org/10.1093/cvr/cvw257
Humphrey JD, Milewicz DM, Tellides G, Schwartz MA (2014) Cell biology. Dysfunctional mechanosensing in aneurysms science (New York, NY) 344:477-479 10.1126/science.1253026
Illi B et al (2005) Epigenetic histone modification and cardiovascular lineage programming in mouse embryonic stem cells exposed to laminar shear stress. Circ res 96:501–508. https://doi.org/10.1161/01.RES.0000159181.06379.63
Jang JY, Lee SW, Park SH, Shin JW, Mun CW, Kim SH, Kim DH, Shin JW (2011) Combined effects of surface morphology and mechanical straining magnitudes on the differentiation of mesenchymal stem cells without using biochemical reagents. J Biomed Biotechnol 2011:860652–860659. https://doi.org/10.1155/2011/860652
Jansen F et al (2017) Intercellular transfer of miR-126-3p by endothelial microparticles reduces vascular smooth muscle cell proliferation and limits neointima formation by inhibiting LRP6. J Mol cell Cardiol 104:43–52. https://doi.org/10.1016/j.yjmcc.2016.12.005
Jia LX et al (2015) Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. the journal of pathology 236:373–383. https://doi.org/10.1002/path.4534
Jiang ZL, Liu JT, Liu Z, Chen Y, Qi Y, Yao Q (2019) Microrna-29a involvement in phenotypic transformation of venous smooth muscle cells via Tet1 in response to mechanical cyclic stretch J Biomech Eng 10.1115/1.4044581
Kakisis JD, Pradhan S, Cordova A, Liapis CD, Sumpio BE (2005) The role of STAT-3 in the mediation of smooth muscle cell response to cyclic strain. Int J Biochem cell biol 37:1396–1406. https://doi.org/10.1016/j.biocel.2005.01.009
Kee HJ, Park S, Kwon JS, Choe N, Ahn Y, Kook H, Jeong MH (2013) B cell translocation gene, a direct target of miR-142-5p, inhibits vascular smooth muscle cell proliferation by down-regulating cell cycle progression. FEBS Lett 587:2385–2392. https://doi.org/10.1016/j.febslet.2013.06.005
Kim SA, Sung JY, Woo CH, Choi HC (2017) Laminar shear stress suppresses vascular smooth muscle cell proliferation through nitric oxide-AMPK pathway. Biochem Biophys Res Commun 490:1369–1374. https://doi.org/10.1016/j.bbrc.2017.07.033
Kozai T, Eto M, Yang Z, Shimokawa H, Luscher TF (2005) Statins prevent pulsatile stretch-induced proliferation of human saphenous vein smooth muscle cells via inhibition of Rho/Rho-kinase pathway. Cardiovasc Res 68:475–482. https://doi.org/10.1016/j.cardiores.2005.07.002
Kurpinski K, Park J, Thakar RG, Li S (2006) Regulation of vascular smooth muscle cells and mesenchymal stem cells by mechanical strain. Mol Cell Biomech 3:21–34
Lee J, Wong M, Smith Q, Baker AB (2013) A novel system for studying mechanical strain waveform-dependent responses in vascular smooth muscle cells. Lab Chip 13:4573–4582. https://doi.org/10.1039/c3lc50894c
Leeper NJ, Raiesdana A, Kojima Y, Chun HJ, Azuma J, Maegdefessel L, Kundu RK, Quertermous T, Tsao PS, Spin JM (2011) MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol 226:1035–1043. https://doi.org/10.1002/jcp.22422
Lehoux S, Castier Y, Tedgui A (2006) Molecular mechanisms of the vascular responses to haemodynamic forces. J Intern Med 259:381–392. https://doi.org/10.1111/j.1365-2796.2006.01624.x
Li C, Hu Y, Mayr M, Xu Q (1999) Cyclic strain stress-induced mitogen-activated protein kinase (MAPK) phosphatase 1 expression in vascular smooth muscle cells is regulated by Ras/Rac-MAPK pathways. J biol Chem 274:25273–25280. https://doi.org/10.1074/jbc.274.36.25273
Li C, Wernig F, Leitges M, Hu Y, Xu Q (2003a) Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. FASEB J 17:2106–2108. https://doi.org/10.1096/fj.03-0150fje
Li J et al (2014) Piezo1 integration of vascular architecture with physiological force. Nature 515:279–282. https://doi.org/10.1038/nature13701
Li M, Liu Q, Lei J, Wang X, Chen X, Ding Y (2017) MiR-362-3p inhibits the proliferation and migration of vascular smooth muscle cells in atherosclerosis by targeting ADAMTS1. Biochem Biophys res Commun 493:270–276. https://doi.org/10.1016/j.bbrc.2017.09.031
Li P et al (2013) MicroRNA-663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circ res 113:1117–1127. https://doi.org/10.1161/CIRCRESAHA.113.301306
Li W, Chen Q, Mills I, Sumpio BE (2003b) Involvement of S6 kinase and p38 mitogen activated protein kinase pathways in strain-induced alignment and proliferation of bovine aortic smooth muscle cells. J Cell Physiol 195:202–209. https://doi.org/10.1002/jcp.10230
Li Y, Liu S, Zhang Z, Xu Q, Xie F, Wang J, Ping S, Li C, Wang Z, Zhang M, Huang J, Chen D, Hu L, Li C (2012) RAGE mediates accelerated diabetic vein graft atherosclerosis induced by combined mechanical stress and AGEs via synergistic ERK activation. PLoS One 7:e35016. https://doi.org/10.1371/journal.pone.0035016
Liu G et al (2010) Mechanical stretch potentiates angiotensin II-induced proliferation in spontaneously hypertensive rat vascular smooth muscle cells. Hypertens res 33:1250–1257. https://doi.org/10.1038/hr.2010.187
Liu J et al (2017a) MicroRNA-32 promotes calcification in vascular smooth muscle cells: implications as a novel marker for coronary artery calcification. PLoS one 12:e0174138. https://doi.org/10.1371/journal.pone.0174138
Liu P et al (2018) Cyclic mechanical stretch induced smooth muscle cell changes in cerebral aneurysm Progress by reducing collagen type IV and collagen type VI levels. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 45:1051–1060. https://doi.org/10.1159/000487347
Liu S et al. (2013) Alpha1-Adrenergic receptors mediate combined signals initiated by mechanical stretch stress and norepinephrine leading to accelerated mouse vein graft atherosclerosis. J Vasc Surg 57:1645–1656, 1656 e1641–1643 10.1016/j.jvs.2012.09.061
Liu T et al (2017b) YAP1 up-regulation inhibits apoptosis of aortic dissection vascular smooth muscle cells. Eur Rev Med Pharmacol Sci 21:4632–4639
Liu X, Cheng Y, Yang J, Xu L, Zhang C (2012) Cell-specific effects of miR-221/222 in vessels: molecular mechanism and therapeutic application. J Mol Cell Cardiol 52:245–255. https://doi.org/10.1016/j.yjmcc.2011.11.008
London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H (2003) Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 18:1731–1740. https://doi.org/10.1093/ndt/gfg414
Ma YH, Ling S, Ives HE (1999a) Mechanical strain increases PDGF-B and PDGF beta receptor expression in vascular smooth muscle cells. Biochem Biophys Res Commun 265:606–610. https://doi.org/10.1006/bbrc.1999.1718
Ma YH, Ling SH, Ives HE (1999b) Mechanical strain increases PDGF-B and PDGF beta receptor expression in vascular smooth muscle cells. Biochemical and Biophysical Research Communications 265:606–610. https://doi.org/10.1006/bbrc.1999.1718
Madamanchi NR, Li S, Patterson C, Runge MS (2001) Thrombin regulates vascular smooth muscle cell growth and heat shock proteins via the JAK-STAT pathway. J Biol Chem 276:18915–18924. https://doi.org/10.1074/jbc.M008802200
Mao X et al (2012) Cyclic stretch-induced thrombin generation by rat vascular smooth muscle cells is mediated by the integrin alphavbeta3 pathway. Cardiovasc res 96:513–523. https://doi.org/10.1093/cvr/cvs274
Mao X, Xie L, Greenberg DA (2015) Effects of flow on LOX-1 and oxidized low-density lipoprotein interactions in brain endothelial cell cultures. Free Radic biol med 89:638–641. https://doi.org/10.1016/j.freeradbiomed.2015.10.403
Marrero MB, Schieffer B, Li B, Sun J, Harp JB, Ling BN (1997) Role of Janus kinase/signal transducer and activator of transcription and mitogen-activated protein kinase cascades in angiotensin II- and platelet-derived growth factor-induced vascular smooth muscle cell proliferation. J Biol Chem 272:24684–24690. https://doi.org/10.1074/jbc.272.39.24684
Martinez-Lemus LA, Crow T, Davis MJ, Meininger GA (2005) alphavbeta3- and alpha5beta1-integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am J Physiol heart Circ Physiol 289:H322–H329. https://doi.org/10.1152/ajpheart.00923.2003
Mayr M, Hu Y, Hainaut H, Xu Q (2002) Mechanical stress-induced DNA damage and rac-p38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. FASEB J 16:1423–1425. https://doi.org/10.1096/fj.02-0042fje
McGahon MK et al (2016) TRPV2 channels contribute to stretch-activated Cation currents and myogenic constriction in retinal arterioles invest. Ophthalmol Vis Sci 57:5637–5647. https://doi.org/10.1167/iovs.16-20279
McGraw AP et al (2013) Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J Am Heart Assoc 2:e000018. https://doi.org/10.1161/JAHA.112.000018
Merlet E et al (2013) miR-424/322 regulates vascular smooth muscle cell phenotype and neointimal formation in the rat. Cardiovasc res 98:458–468. https://doi.org/10.1093/cvr/cvt045
Mills I, Cohen CR, Kamal K, Li G, Shin T, Du W, Sumpio BE (1997) Strain activation of bovine aortic smooth muscle cell proliferation and alignment: study of strain dependency and the role of protein kinase A and C signaling pathways. J Cell Physiol 170:228–234. https://doi.org/10.1002/(SICI)1097-4652(199703)170:3<228::AID-JCP2>3.0.CO;2-Q
Mizuno Y et al (2008) miR-125b inhibits osteoblastic differentiation by down-regulation of cell proliferation. Biochem Biophys res Commun 368:267–272. https://doi.org/10.1016/j.bbrc.2008.01.073
Molostvov G, Hiemstra TF, Fletcher S, Bland R, Zehnder D (2015) Arterial Expression of the Calcium-Sensing Receptor Is Maintained by Physiological Pulsation and Protects against Calcification. PLoS One 10:e0138833. https://doi.org/10.1371/journal.pone.0138833
Morita N, Iizuka K, Murakami T, Kawaguchi H (2004) N-terminal kinase, and c-Src are activated in human aortic smooth muscle cells by pressure stress. Mol cell Biochem 262:71–78. https://doi.org/10.1023/b:mcbi.0000038218.09259.1c
Morrow D, Sweeney C, Birney YA, Cummins PM, Walls D, Redmond EM, Cahill PA (2005) Cyclic strain inhibits Notch receptor signaling in vascular smooth muscle cells in vitro. Circ Res 96:567–575. https://doi.org/10.1161/01.RES.0000159182.98874.43
Morrow D et al (2007) Biomechanical regulation of hedgehog signaling in vascular smooth muscle cells in vitro and in vivo. Am J Physiol cell Physiol 292:C488–C496. https://doi.org/10.1152/ajpcell.00337.2005
Moses S, Dreja K, Lindqvist A, Lovdahl C, Hellstrand P, Hultgardh-Nilsson A (2001) Smooth muscle cell response to mechanical injury involves intracellular calcium release and ERK1/ERK2 phosphorylation. Exp cell res 269:88–96. https://doi.org/10.1006/excr.2001.5308
Muraki K, Iwata Y, Katanosaka Y, Ito T, Ohya S, Shigekawa M, Imaizumi Y (2003) TRPV2 is a component of osmotically sensitive cation channels in murine aortic myocytes. Circ res 93:829–838. https://doi.org/10.1161/01.RES.0000097263.10220.0C
Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, Hirata Y (2004) AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation 110:444–451. https://doi.org/10.1161/01.CIR.0000136025.96811.76
Nguyen KT, Frye SR, Eskin SG, Patterson C, Runge MS, McIntire LV (2001) Cyclic strain increases protease-activated receptor-1 expression in vascular smooth muscle cells. Hypertension 38:1038–1043. https://doi.org/10.1161/hy1101.092840
Nikolovski J, Kim BS, Mooney DJ (2003) Cyclic strain inhibits switching of smooth muscle cells to an osteoblast-like phenotype. FASEB J 17:455–457. https://doi.org/10.1096/fj.02-0459fje
Numaguchi K, Eguchi S, Yamakawa T, Motley ED, Inagami T (1999) Mechanotransduction of rat aortic vascular smooth muscle cells requires RhoA and intact actin filaments. Circ Res 85:5–11. https://doi.org/10.1161/01.res.85.1.5
O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D (2007) MicroRNA-155 is induced during the macrophage inflammatory response. Proceedings of the National Academy of Sciences of the United States of America 104:1604–1609. https://doi.org/10.1073/pnas.0610731104
O'Neill LA (2006) Targeting signal transduction as a strategy to treat inflammatory diseases. Nat rev drug Discov 5:549–563. https://doi.org/10.1038/nrd2070
Pan J et al (2018) AIM2 accelerates the atherosclerotic plaque progressions in ApoE−/− mice. Biochem Biophys res Commun 498:487–494. https://doi.org/10.1016/j.bbrc.2018.03.005
Pan S (2009) Molecular mechanisms responsible for the atheroprotective effects of laminar shear stress. Antioxid redox signal 11:1669–1682. https://doi.org/10.1089/ARS.2009.2487
Pedersen I, David M (2008) MicroRNAs in the immune response. Cytokine 43:391–394. https://doi.org/10.1016/j.cyto.2008.07.016
Peyton SR, Putnam AJ (2005) Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol 204:198–209. https://doi.org/10.1002/jcp.20274
Ping S, Li Y, Liu S, Zhang Z, Wang J, Zhou Y, Liu K, Huang J, Chen D, Wang J, Li C (2015) Simultaneous Increases in Proliferation and Apoptosis of Vascular Smooth Muscle Cells Accelerate Diabetic Mouse Venous Atherosclerosis. PLoS One 10:e0141375. https://doi.org/10.1371/journal.pone.0141375
Ping S, Liu S, Zhou Y, Li Z, Li Y, Liu K, Bardeesi ASA, Wang L, Chen J, Deng L, Wang J, Wang H, Chen D, Zhang Z, Sheng P, Li C (2017) Protein disulfide isomerase-mediated apoptosis and proliferation of vascular smooth muscle cells induced by mechanical stress and advanced glycosylation end products result in diabetic mouse vein graft atherosclerosis. Cell Death Dis 8:e2818. https://doi.org/10.1038/cddis.2017.213
Potter CM, Lao KH, Zeng L, Xu Q (2014) Role of biomechanical forces in stem cell vascular lineage differentiation. Arteriosclerosis, thrombosis, and vascular biology 34:2184–2190. https://doi.org/10.1161/ATVBAHA.114.303423
Qi YX, Qu MJ, Long DK, Liu B, Yao QP, Chien S, Jiang ZL (2008) Rho-GDP dissociation inhibitor alpha downregulated by low shear stress promotes vascular smooth muscle cell migration and apoptosis: a proteomic analysis. Cardiovasc Res 80:114–122. https://doi.org/10.1093/cvr/cvn158
Qi YX, Qu MJ, Yan ZQ, Zhao D, Jiang XH, Shen BR, Jiang ZL (2010) Cyclic strain modulates migration and proliferation of vascular smooth muscle cells via rho-GDIalpha, Rac1, and p38 pathway. J cell Biochem 109:906–914. https://doi.org/10.1002/jcb.22465
Qi YX et al (2016) Nuclear envelope proteins modulate proliferation of vascular smooth muscle cells during cyclic stretch application. Proceedings of the National Academy of Sciences of the United States of America 113:5293–5298. https://doi.org/10.1073/pnas.1604569113
Qu MJ, Liu B, Qi YX, Jiang ZL (2008) Role of Rac and rho-GDI alpha in the frequency-dependent expression of h1-calponin in vascular smooth muscle cells under cyclic mechanical strain. Ann biomed Eng 36:1481–1488. https://doi.org/10.1007/s10439-008-9521-0
Qu MJ, Liu B, Wang HQ, Yan ZQ, Shen BR, Jiang ZL (2007) Frequency-dependent phenotype modulation of vascular smooth muscle cells under cyclic mechanical strain. J Vasc Res 44:345–353. https://doi.org/10.1159/000102278
Ranade SS et al (2014) Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proceedings of the National Academy of Sciences of the United States of America 111:10347–10352. https://doi.org/10.1073/pnas.1409233111
Retailleau K et al (2015) Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell reports 13:1161–1171. https://doi.org/10.1016/j.celrep.2015.09.072
Rode B et al (2017) Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nature communications 8:350. https://doi.org/10.1038/s41467-017-00429-3
Rodriguez AI et al (2015) MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology 35:430–438. https://doi.org/10.1161/ATVBAHA.114.304936
Romer LH, Birukov KG, Garcia JG (2006) Focal adhesions: paradigm for a signaling nexus. Circ Res 98:606–616. https://doi.org/10.1161/01.RES.0000207408.31270.db
Scherer C, Pfisterer L, Wagner AH, Hodebeck M, Cattaruzza M, Hecker M, Korff T (2014) Arterial wall stress controls NFAT5 activity in vascular smooth muscle cells. J Am Heart Assoc 3:e000626. https://doi.org/10.1161/JAHA.113.000626
Schlaepfer DD, Hunter T (1997) Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J biol Chem 272:13189–13195. https://doi.org/10.1074/jbc.272.20.13189
Schlaepfer DD, Mitra SK, Ilic D (2004) Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta 1692:77–102. https://doi.org/10.1016/j.bbamcr.2004.04.008
Schulze PC, de Keulenaer GW, Kassik KA, Takahashi T, Chen Z, Simon DI, Lee RT (2003) Biomechanically induced gene iex-1 inhibits vascular smooth muscle cell proliferation and neointima formation. Circ res 93:1210–1217. https://doi.org/10.1161/01.RES.0000103635.38096.2F
Seki Y, Kai H, Shibata R, Nagata T, Yasukawa H, Yoshimura A, Imaizumi T (2000) Role of the JAK/STAT pathway in rat carotid artery remodeling after vascular injury. Circ Res 87:12–18. https://doi.org/10.1161/01.res.87.1.12
Shan Z et al (2015) An endocrine genetic signal between blood cells and vascular smooth muscle cells: role of MicroRNA-223 in smooth muscle function and Atherogenesis. J am Coll Cardiol 65:2526–2537. https://doi.org/10.1016/j.jacc.2015.03.570
Shaw A, Xu Q (2003) Biomechanical stress-induced signaling in smooth muscle cells: an update. Curr Vasc Pharmacol 1:41–58. https://doi.org/10.2174/1570161033386745
Shi L, Tian C, Sun L, Cao F, Meng Z (2018) The lncRNA TUG1/miR-145-5p/FGF10 regulates proliferation and migration in VSMCs of hypertension. Biochem Biophys res Commun 501:688–695. https://doi.org/10.1016/j.bbrc.2018.05.049
Shi ZD, Abraham G, Tarbell JM (2010) Shear stress modulation of smooth muscle cell marker genes in 2-D and 3-D depends on mechanotransduction by heparan sulfate proteoglycans and ERK1/2. PLoS one 5:e12196. https://doi.org/10.1371/journal.pone.0012196
Shi ZD, Ji XY, Qazi H, Tarbell JM (2009) Interstitial flow promotes vascular fibroblast, myofibroblast, and smooth muscle cell motility in 3-D collagen I via upregulation of MMP-1. Am J Physiol heart Circ Physiol 297:H1225–H1234. https://doi.org/10.1152/ajpheart.00369.2009
Simmons CA, Nikolovski J, Thornton AJ, Matlis S, Mooney DJ (2004) Mechanical stimulation and mitogen-activated protein kinase signaling independently regulate osteogenic differentiation and mineralization by calcifying vascular cells. J Biomech 37:1531–1541. https://doi.org/10.1016/j.jbiomech.2004.01.006
Son DJ et al (2017) Interleukin-32alpha inhibits endothelial inflammation, vascular smooth muscle cell activation, and atherosclerosis by Upregulating Timp3 and Reck through suppressing microRNA-205. Biogenesis Theranostics 7:2186–2203. https://doi.org/10.7150/thno.18407
Song J, Hu B, Qu H, Bi C, Huang X, Zhang M (2012a) Mechanical stretch modulates microRNA 21 expression, participating in proliferation and apoptosis in cultured human aortic smooth muscle cells. PLoS one 7:e47657. https://doi.org/10.1371/journal.pone.0047657
Song L, Duan P, Guo P, Li D, Li S, Xu Y, Zhou Q (2012b) Downregulation of miR-223 and miR-153 mediates mechanical stretch-stimulated proliferation of venous smooth muscle cells via activation of the insulin-like growth factor-1 receptor. Arch Biochem Biophys 528:204–211. https://doi.org/10.1016/j.abb.2012.08.015
Stein S, Matter CM (2011) Protective roles of SIRT1 in atherosclerosis. Cell cycle 10:640–647. https://doi.org/10.4161/cc.10.4.14863
Sun SG et al (2011) miR-146a and Kruppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO rep 12:56–62. https://doi.org/10.1038/embor.2010.172
Sun Z, Martinez-Lemus LA, Hill MA, Meininger GA (2008) Extracellular matrix-specific focal adhesions in vascular smooth muscle produce mechanically active adhesion sites. Am J Physiol Cell Physiol 295:C268–C278. https://doi.org/10.1152/ajpcell.00516.2007
Tana C, Giamberardino MA, Cipollone F (2017) microRNA profiling in atherosclerosis, diabetes, and migraine. Ann med 49:93–105. https://doi.org/10.1080/07853890.2016.1226515
Tasaki T et al (2013) Apoptosis signal-regulating kinase 1 deficiency attenuates vascular injury-induced neointimal hyperplasia by suppressing apoptosis in smooth muscle cells. am J Pathol 182:597–609. https://doi.org/10.1016/j.ajpath.2012.10.008
Tili E et al (2007) Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol 179:5082–5089. https://doi.org/10.4049/jimmunol.179.8.5082
Toba H, Cortez D, Lindsey ML, Chilton RJ (2014) Applications of miRNA technology for atherosclerosis. Curr Atheroscler Rep 16:386. https://doi.org/10.1007/s11883-013-0386-9
Torella D et al (2011) MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ res 109:880–893. https://doi.org/10.1161/CIRCRESAHA.111.240150
Torsoni AS, Constancio SS, Nadruz W Jr, Hanks SK, Franchini KG (2003) Focal adhesion kinase is activated and mediates the early hypertrophic response to stretch in cardiac myocytes. Circ Res 93:140–147. https://doi.org/10.1161/01.RES.0000081595.25297.1B
Tsai MC et al (2009) Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circ res 105:471–480. https://doi.org/10.1161/CIRCRESAHA.109.193656
Turczynska KM, Bhattachariya A, Sall J, Goransson O, Sward K, Hellstrand P, Albinsson S (2013) Stretch-sensitive down-regulation of the miR-144/451 cluster in vascular smooth muscle and its role in AMP-activated protein kinase signaling. PLoS one 8:e65135. https://doi.org/10.1371/journal.pone.0065135
Turczynska KM, Sadegh MK, Hellstrand P, Sward K, Albinsson S (2012) MicroRNAs are essential for stretch-induced vascular smooth muscle contractile differentiation via microRNA (miR)-145-dependent expression of L-type calcium channels. J Biol Chem 287:19199–19206. https://doi.org/10.1074/jbc.M112.341073
Urbich C, Kuehbacher A, Dimmeler S (2008) Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc res 79:581–588. https://doi.org/10.1093/cvr/cvn156
Wan XJ et al (2015) Involvement of BK channel in differentiation of vascular smooth muscle cells induced by mechanical stretch. Int J Biochem cell biol 59:21–29. https://doi.org/10.1016/j.biocel.2014.11.011
Wang H et al (2019a) Friend or foe: a Cancer suppressor MicroRNA-34 potentially plays an adverse role in vascular diseases by regulating cell apoptosis and extracellular matrix degradation med. Sci Monit 25:1952–1959. https://doi.org/10.12659/MSM.915270
Wang J et al (2017) Role of nifedipine and hydrochlorothiazide in MAPK activation and vascular smooth muscle cell proliferation and apoptosis. Herz 42:573–584. https://doi.org/10.1007/s00059-016-4489-2
Wang J et al (2013) MicroRNA-31 controls phenotypic modulation of human vascular smooth muscle cells by regulating its target gene cellular repressor of E1A-stimulated genes. Exp cell res 319:1165–1175. https://doi.org/10.1016/j.yexcr.2013.03.010
Wang WB et al (2019b) CTGF regulates cyclic stretch-induced vascular smooth muscle cell proliferation via microRNA-19b-3p. Exp cell res 376:77–85. https://doi.org/10.1016/j.yexcr.2019.01.015
Wang X, Du C, He X, Deng X, He Y, Zhou X (2018a) MiR-4463 inhibits the migration of human aortic smooth muscle cells by AMOT Biosci rep 38 https://doi.org/10.1042/BSR20180150
Wang X, Hu G, Zhou J (2010) Repression of versican expression by microRNA-143. J biol Chem 285:23241–23250. https://doi.org/10.1074/jbc.M109.084673
Wang Y, Cao W, Cui J, Yu Y, Zhao Y, Shi J, Wu J, Xia Z, Yu B, Liu J (2018b) Arterial Wall Stress Induces Phenotypic Switching of Arterial Smooth Muscle Cells in Vascular Remodeling by Activating the YAP/TAZ. Signaling Pathway Cell Physiol Biochem 51:842–853. https://doi.org/10.1159/000495376
Wang Y, Krishna SM, Moxon J, Dinh TN, Jose RJ, Yu H, Golledge J (2014) Influence of apolipoprotein E, age and aortic site on calcium phosphate induced abdominal aortic aneurysm in mice. Atherosclerosis 235:204–212. https://doi.org/10.1016/j.atherosclerosis.2014.04.033
Wang YK, Yu X, Cohen DM, Wozniak MA, Yang MT, Gao L, Eyckmans J, Chen CS (2012a) Bone morphogenetic protein-2-induced signaling and osteogenesis is regulated by cell shape, RhoA/ROCK, and cytoskeletal tension. Stem Cells Dev 21:1176–1186. https://doi.org/10.1089/scd.2011.0293
Wang YS et al (2012b) MicroRNA-195 regulates vascular smooth muscle cell phenotype and prevents neointimal formation. Cardiovasc res 95:517–526. https://doi.org/10.1093/cvr/cvs223
Wei X, Hou X, Li J, Liu Y (2017) miRNA-181a/b regulates phenotypes of vessel smooth muscle cells through serum response factor. DNA cell biol 36:127–135. https://doi.org/10.1089/dna.2016.3525
Wen P et al (2014) miR-125b/Ets1 axis regulates transdifferentiation and calcification of vascular smooth muscle cells in a high-phosphate environment. Exp cell res 322:302–312. https://doi.org/10.1016/j.yexcr.2014.01.025
Wernig F, Mayr M, Xu Q (2003) Mechanical stretch-induced apoptosis in smooth muscle cells is mediated by beta1-integrin signaling pathways. Hypertension 41:903–911. https://doi.org/10.1161/01.HYP.0000062882.42265.88
Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM (2018) Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nature reviews Molecular cell biology 19:638–653. https://doi.org/10.1038/s41580-018-0049-3
Wu J, Lewis AH, Grandl J (2017) Touch, Tension, and Transduction - The Function and Regulation of Piezo Ion Channels. Trends Biochem Sci 42:57–71. https://doi.org/10.1016/j.tibs.2016.09.004
Wu WH, Hu CP, Chen XP, Zhang WF, Li XW, Xiong XM, Li YJ (2011) MicroRNA-130a mediates proliferation of vascular smooth muscle cells in hypertension. Am J Hypertens 24:1087–1093. https://doi.org/10.1038/ajh.2011.116
Xu D, Liao R, Wang XX, Cheng Z (2018) Effects of miR-155 on hypertensive rats via regulating vascular mesangial hyperplasia. Eur rev med Pharmacol Sci 22:7431–7438. https://doi.org/10.26355/eurrev_201811_16283
Xu F, Ahmed AS, Kang X, Hu G, Liu F, Zhang W, Zhou J (2015) MicroRNA-15b/16 attenuates vascular Neointima formation by promoting the contractile phenotype of vascular smooth muscle through targeting. YAP arteriosclerosis, thrombosis, and vascular biology 35:2145–2152. https://doi.org/10.1161/ATVBAHA.115.305748
Xu Q (2000) Biomechanical-stress-induced signaling and gene expression in the development of arteriosclerosis. Trends Cardiovasc med 10:35–41. https://doi.org/10.1016/s1050-1738(00)00042-6
Yamashiro Y et al (2018) Role of Thrombospondin-1 in Mechanotransduction and development of thoracic aortic aneurysm in mouse and humans. Circulation research 123:660–672. https://doi.org/10.1161/circresaha.118.313105
Yamawaki H, Lehoux S, Berk BC (2003) Chronic physiological shear stress inhibits tumor necrosis factor-induced proinflammatory responses in rabbit aorta perfused ex vivo. Circulation 108:1619–1625. https://doi.org/10.1161/01.CIR.0000089373.49941.C4
Yan ZQ, Yao QP, Zhang ML, Qi YX, Guo ZY, Shen BR, Jiang ZL (2009) Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J Biomech 42:945–948. https://doi.org/10.1016/j.jbiomech.2009.01.012
Yang F et al (2018) miR-22 is a novel mediator of vascular smooth muscle cell phenotypic modulation and Neointima formation. Circulation 137:1824–1841. https://doi.org/10.1161/CIRCULATIONAHA.117.027799
Yang X, Coriolan D, Murthy V, Schultz K, Golenbock DT, Beasley D (2005) Proinflammatory phenotype of vascular smooth muscle cells: role of efficient toll-like receptor 4 signaling. Am J Physiol heart Circ Physiol 289:H1069–H1076. https://doi.org/10.1152/ajpheart.00143.2005
Yao QP et al (2014) The role of SIRT6 in the differentiation of vascular smooth muscle cells in response to cyclic strain. Int J Biochem cell biol 49:98–104. https://doi.org/10.1016/j.biocel.2014.01.016
Yu ML, Wang JF, Wang GK, You XH, Zhao XX, Jing Q, Qin YW (2011) Vascular smooth muscle cell proliferation is influenced by let-7d microRNA and its interaction with KRAS. Circ J 75:703–709. https://doi.org/10.1253/circj.cj-10-0393
Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX (2003) PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol cell Physiol 284:C316–C330. https://doi.org/10.1152/ajpcell.00125.2002
Yung YC, Chae J, Buehler MJ, Hunter CP, Mooney DJ (2009) Cyclic tensile strain triggers a sequence of autocrine and paracrine signaling to regulate angiogenic sprouting in human vascular cells. Proceedings of the National Academy of Sciences of the United States of America 106:15279–15284. https://doi.org/10.1073/pnas.0905891106
Zampetaki A, Zhang Z, Hu Y, Xu Q (2005) Biomechanical stress induces IL-6 expression in smooth muscle cells via Ras/Rac1-p38 MAPK-NF-kappaB signaling pathways. Am J Physiol heart Circ Physiol 288:H2946–H2954. https://doi.org/10.1152/ajpheart.00919.2004
Zhang BC, Zhou ZW, Li XK, Xu YW (2011) PI-3K/AKT signal pathway modulates vascular smooth muscle cells migration under cyclic mechanical strain. Vasa 40:109–116. https://doi.org/10.1024/0301-1526/a000080
Zhang C, Zeng L, Emanueli C, Xu Q (2013a) Blood flow and stem cells in vascular disease. Cardiovasc Res 99:251–259. https://doi.org/10.1093/cvr/cvt061
Zhang J, Zhao F, Yu X, Lu X, Zheng G (2015) MicroRNA-155 modulates the proliferation of vascular smooth muscle cells by targeting endothelial nitric oxide synthase. Int J Mol med 35:1708–1714. https://doi.org/10.3892/ijmm.2015.2181
Zhang Z, Chen Y, Zhang T, Guo L, Yang W, Zhang J, Wang C (2016) Role of Myoendothelial Gap Junctions in the Regulation of Human Coronary Artery Smooth Muscle Cell Differentiation by Laminar Shear Stress. Cell Physiol Biochem 39:423–437. https://doi.org/10.1159/000445636
Zhang Z et al (2013b) Simvastatin inhibits the additive activation of ERK1/2 and proliferation of rat vascular smooth muscle cells induced by combined mechanical stress and oxLDL through LOX-1 pathway. Cell signal 25:332–340. https://doi.org/10.1016/j.cellsig.2012.10.006
Funding
This work was supported by the National Natural Science Foundation of China (81070124, 81870219 to CL; 81500337 to SL); the Natural Science Foundation of Guangdong Province (2017A030313574 to CL; 2014A020212109 to SL), and the Fundamental Research Funds for the Central Universities (19ykpy171 to SL).
Author information
Authors and Affiliations
Contributions
J. C. and Y. Z. draft the manuscript, S. L. and C. L. edited the manuscript to its final version. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, J., Zhou, Y., Liu, S. et al. Biomechanical signal communication in vascular smooth muscle cells. J. Cell Commun. Signal. 14, 357–376 (2020). https://doi.org/10.1007/s12079-020-00576-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-020-00576-1