
Abstract
The long-term goal of a breeder is to increase genetic variation by bringing desirable genes from natural populations into the breeding population. With the advancement in genomics, molecular marker tools have become the breeder’s choice for genotypic selection, facilitating quick and reliable selection of individuals in the segregating populations. Various marker-assisted breeding (MAB) strategies are needed in different crop systems for the rapid development of cultivars. The advancement of genomic resources has led to the development of multi-parent and multi-trait improvement strategies such as marker-assisted gene pyramiding (MAGP), marker-assisted recurrent selection (MARS), and genomic selection (GS). MARS is an important population improvement method that focuses on cyclically choosing and enriching favorable alleles from biparental or multiparent introgression at several loci. MARS begins with a heterogeneous base population and exploits superior recombinants during each cycle to produce a broad-based improved population, an inbred line or a hybrid. Realizing the MARS potentiality, various public and private sectors have successfully applied it in many commercial crops. Here we present the merits of MARS with other marker-assisted selection schemes, the procedure involved, and key factors to be considered for its successful implementation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The science of plant breeding has progressive cutting-edge innovations, with numerous tactics, concepts, and practices, from domestication to the contemporary method of genomic selection or genome editing. The whole-genome and gene-targeted surveys in combination with the skill of accurate selection through precision breeding are made possible by the development of molecular marker technology. A long-term objective of plant breeders is to create varieties that can adapt to environmental and agricultural difficulties in order to offer sustained and enhanced crop yields. Commercial breeding populations have a narrow genetic base due to domestication bottleneck, and selection pressure operated during evolution (Smýkal et al. 2018). Modern elite lines have been bred by utilizing one or a few parents that resulted in rapid fixation of genes leading to genetic vulnerability. Lack of diversity in modern germplasm may further restrict our ability to breed for increased nutritional levels, and resistance to pests and diseases (Smýkal et al. 2018; Larkan et al. 2013). Identifying the intrinsically superior recombinants or transgressive segregants from the diverse gene pool or segregating progenies with superior alleles is truly challenging (Chaitra et al. 2020). Breeding operations should be rigorously planned to support molecular and genomics approaches that assist the process of introducing favorable alleles from crop wild relatives (CWRs). Multi-parent population (MPPs) or multi-trait enhancement approaches are acquiring greater space, especially in high-resolution mapping studies (Scott et al. 2020) or gene stacking practices such as MARS, MAGP, Marker Assisted Back Crossing (MABC), and GS. The eventual goal of these breeding programs is to enrich the population with favorable and diverse alleles by intensifying the rate of genetic gain for the genes underlying economic traits in the context of faster development of climate-resilient varieties (Prasanna et al. 2013; Kole 2013; Varshney et al. 2018).
Indispensable evaluation of germplasm source material or later segregating generations is mandatory in breeding programs for various traits governed by major or polygenes. Even with the huge availability of plant resources, and several genetic models, success through traditional phenotype-based breeding methods is inadequate due to time and G x E interactions (Hallauer et al. 2010). In recent years, an increase in selection accuracy based on genotype knowledge has added weightage to the development of varieties or hybrids with complex traits. On the other hand, even with the availability of high-throughput sequencing platforms and genomic resources, genomics-assisted breeding may fail in the absence of high-quality phenotypic data. Uncovering the genetic basis of complex traits, cloning, and sequencing the candidate genes will not benefit economically deprived farmers unless researchers transform them into superior varieties. It is essential to adapt the breeding strategy that reflects on the power of phenotype as well as the precision of genotype to resolve the complexity encountered in multi-trait and multi-stage selection for economically important traits (Sonnino et al. 2007). Henceforth, marker-assisted recurrent selection (MARS) received greater importance for multi-trait and multi-parent population improvement, which provides greater advantage of a cyclic selection of recombinants for complex traits (Sandhu et al. 2018a, b). Recurrent selection (RS) (Sprague and Eberhart 1977) is a population improvement approach wherein, the target genes from multiple genotypes or heterogeneous populations are combined by repeated selection of desirable recombinants in a cyclic manner (Chahal and Gosal 2006). Lande and Thompson (1990) proposed MARS scheme, for the selection of the desirable individuals for complex traits based on the identified QTL in the same population, and the relative preciseness depends on the genetic variation explained by associated markers (Ceballos et al. 2015; Beyene et al. 2016; Bankole et al. 2017). MARS comprises of selection, evaluation and recombination at every cycle among selected plants with the expectation of increased frequency of beneficial marker alleles for target traits and in turn the genetic gain in the progeny population (Mayor and Bernardo 2009a, b; Stam 1995; Peleman and van Der Voort 2003; Abdulmalik et al. 2017). It is ascertained that a genetic gain achieved through MARS is higher than that of phenotypic-recurrent selection (Moreau et al. 2004; Openshaw and Frascaroli 1997). The genetic gain of different traits under MARS scheme was almost twice than that of phenotypic-recurrent selection (Eathington 2005; Crosbie et al. 2006; Marcón et al. 2020). Many cross-pollinated crops have benefited from genomic and marker-assisted recurrent selection. However, due to time-consuming crossing procedures, such selection is not possible in self-pollinated crops (Sekine et al. 2021). MARS is mainly, regarded as a genotype-driven approach that favor faster development of varieties or hybrids to achieve “ideal genotypes” (Peleman and van der Voort 2003). Presently, in many private sectors, MARS research programs have been initiated for genetic modeling of breeding populations for different complex traits (Ragot et al 2000; Eathington 2005; Crosbie et al. 2006). Breeders are utilizing germplasm knowledge and marker-trait associations to improve breeding populations with multiple traits. Both phenotypic selection in various cycles (Dhliwayo et al. 2014) and the efficiency of MARS over phenotypic selection (Beyene et al. 2016; Bankole et al. 2017) have highlighted the potential of recurrent selection on yield, abiotic stress, and quality parameters in plants. With 2–3 cycles of recurrent selection, the genetic gain for the target traits had been enhanced to a desirable level saving time and valuable resources during the development of varieties for various quantitative traits in maize, soybean, and sunflower (Johnson 2004; Eathington et al. 2007). MARS was utilized to pyramid leaf rust and coffee berry disease resistance alleles in coffea arabica L. (Saavedra et al. 2023). MARS can be an efficient strategy for designing future crops by integrating multiple desirable traits from several plants (Varshney et al. 2021). This review paper aims to provide the details about the potentiality of MARS on population improvement and on current status of MARS in different crops, and its merits over other MAS schemes.
MARS in Comparison to MAS Schemes
MAS v/s MARS
MAS is an indirect selection process where a trait of interest is selected based on a marker linked to that trait of interest (Song et al. 2023). MAS is realistic if the trait of interest is governed by one or two major genes, and it is ineffective and impractical for complex traits governed by polygenes (Bernardo 2008; Budhlakoti et al. 2022b). MARS is a type of MAS used in recurrent selection (Bankole et al. 2017). Recurrent selection is a type of selection that basically involves increasing the frequency of superior genes for various characters in a population (Saavedra et al. 2023). MARS is regarded as an effective strategy for improving polygenic traits (Suvarna et al. 2023).
The markers that are tightly linked to the QTL region on a chromosome are consistently used to predict the performance of elite breeding lines in both animals and crop plants (Goddard and Hayes 2009; Bernardo 2008). Although many successful MAS programs were conducted worldwide and many varieties for various traits have been developed in numerous crop systems, few major impediments of MAS curtail its application in a breeding program (Hallauer 1999; Heffner et al. 2009). MAS relies on a limited number of molecular markers (Sinha et al 2023). Most of the markers used for the selection of target traits were based on major-effect QTLs that explained the highest phenotypic variation by ignoring many other QTLs with minor effects (Bernardo 2002; Eathington et al. 2007; Xu 2012; Kushanov et al. 2021). Another constraint of MAS is that the marker tightly linked with target traits identified in one population may not show polymorphism for other populations (Lande and Thompson 1990; Schuster 2011; Sakiyama et al. 2014; Platten et al. 2019). Besides that, the selected individuals are simply advanced by inbreeding or backcrossing. Consequently, the efficiency of markers linked to target genes decreases with an increase in recombination frequency due to single crossover or double crossover events between marker and QTL regions (Jiang 2013). Whereas, MARS can exploit the advantage of multi-trait improvement (Peleman and van Der Voort 2003) in a single population by successfully intermating selected individuals in every cycle. So, the chances of getting superior recombinants would also be increased in the population as the selection of plants is critically based on significantly linked markers to QTL regions. Hence, the genetic gain expected from MARS would also be higher than MAS (Ribaut and Ragot 2007; Bankole et al. 2017).
MABC v/s MARS
MABC employs two parents (recurrent and donor) contrasting for a single trait to improve an already superior cultivar for one or a few traits. Here, the resulting variety would be just a recurrent parent improved for the single trait, and the markers used for target trait selection would generally be marker linked with mono or oligogenic traits or major effect QTL. MABC overlooks polygenic traits. Consequently, the success of MABC crucially depends on the number of factors viz., the population size in each generation of a back cross, marker-gene association (phenotypic variation explained, and the genetic distance). The number of markers used for recombinant selection is proportionate to minimize linkage drag in the vicinity of the target segment of the chromosome (Jiang 2013) and the recurrent parent genome recovery during background selection for non-target segments of chromosome play critical roles in MABC. Hospital (2003) suggested that the MABC population should contain at least one genotype (recurrent parent) with all desirable alleles except for QTL under question. Nonetheless, the number of QTLs cannot be increased gradually beyond six because of difficulty in handling, and a greater chance of linkage drag is expected as unwanted alleles from donor parent could be present on target chromosome even after BC6 generation (Newbury 2003). Two QTL regions for high seed protein content in soyabean were introduced through MABC (Sebolt et al. 2000), on the contrary, only one QTL was confirmed in BC3F4:5 and it was found to be unstable in different genetic background and multiple environments. Pi9 gene responsible for resistance to blast in rice (Scheuermann and Jia 2016), was obstructing the incorporation either by MAS or MABC resulted in contrary grain hull color due to the probable linkage drag from wild progenitor (Amante-Bordeos et al. 1992).
The critical difference between MAS/MABC and MARS is that the former methods analyze the markers linked to QTL at only one generation and strive to firmly unite with genomic variations, while, the MARS evaluates the significantly linked flanking markers of QTL region at each cycle of recurrent selection (Johnson 2004; Eathington 2005; Crosbie et al. 2006) and identifies the linked QTL with higher precision. MARS is useful for capturing a large number of major as well as minor QTLs; hence the genetic gain achieved by MARS is higher than that by MABC (Bernardo and Charcosset 2006; Kushwah et al. 2020).
MAS in Pedigree Breeding vs MARS
MAS has been deliberately used in a pedigree breeding program wherein plants' selection is merely based on few loci at early generations, like F2 or F3 (Eathington 2005; Dudley and Lambert 2010) which need indispensable use of large population (Ribaut and Betrán, 1999). Accordingly, MAS is inadequate for the accuracy of QTL parameters such as QTL position on genomic region and contemporary relevance across environments or gene pools. However, in various breeding programs for disease resistance, namely, cyst nematode (Heterodera glycines) in soybean (Cregan et al. 1999) and for nutritional quality underlining β-glucan content in oat, MAS was extensively employed (Asoro et al. (2013). However, in some cases like the aroma in rice is governed by a recessive gene coding for betaine aldehyde dehydrogenase2 (Bradbury et al. 2005), and high lysine content in maize governed by double recessive opaque-2 gene (Babu and Prasanna 2014; Yang et al.2005) the success rate diminished when these QTLs were proposed for introgression. MARS program facilitated multifaceted improvements by bringing together elite alleles from two or more parental lines with low heritable traits (Bernardo 2002). Positive interaction between QTLs over multiple selection cycles helped to isolate high yielding superior plants under various biotic and abiotic stresses (Sandhu et al. 2018a, b). MARS would allow for the rapid expansion of the gene pool of existing cultivars (Moreau et al. 1998; Xu and Crouch 2008).
Marker-assisted Gene Pyramiding (MAGB) vs MARS
MAS success paved the way for emergence of gene pyramiding schemes with the ambitious achievement of a variety stacked with multiple genes from multiple donor parents (Joshi and Nayak 2010). However, gene pyramiding success relies on key factors viz., no. of genes to be transferred, no. of founder parents involved, compatibility of the recurrent parent with the donor parents, the distance between the genes, flanking markers and linkage drags. Besides, population size maintained, the scheme involved (stepwise, simultaneous or combined method), and the number of markers used for background genome recovery are the other important factors (Yang et al. 2005). In contrast, MARS allows the continuous addition of more genes onto existing pyramids (Pretorius et al. 2007) during cyclic intermating of progenies to develop an ideal genotype, which under normal selfing and segregation circumstances may not be expected to achieve even after Fn generation (Stam 1995; Chevalet and Mulsant 1992; Knapp 1998; Moreau et al. 1998; Xie and Xu 1998). Therefore, MARS is reflected as an approach of “Genotype construction” (Stam 1995; Peleman and van der Voort 2003) when beneficial alleles are pooled from more than two parents.
GS v/s MARS
Genomic selection (Meuwissen et al. 2001) or Genome-wide selection (Bernardo and Yu 2007) is a black box of genomic prediction through high-density genome-wide markers covering the entire genome. The marker indices across the entire genome are used to assess Genomic Estimated Breeding Values (GEBV) (Nakaya and Isobe 2012) for the selection of appropriate individuals in breeding cycles as an alternative to the genotype of markers in MAS (Jiang 2013). Breeding values (BV) are not considered ideal in plant breeding (Shamshad and Sharma 2018) as BV is estimated as a conditional expected value based on marker genotype than QTL genotype (Goddard and Hayes 2007). It could be an excellent method once high sequence data, and high SNP data are obtained (Goddard and Hayes 2007). The high-throughput genotypic data necessitates high-performance computer simulation models and appropriate statistical analysis methods to calculate BV. However, GS is challenging for crops with an unpredictably complex genome, distinctively higher ploidy levels, high heterozygosity, or transposable elements. Few polymorphic markers obtained across the genome are virtually estimated to have more substantial genetic effects, and additional markers are valued to have weaker genetic effects since these markers are not validated for biological efficacies specific for the target agricultural traits (Bernardo 2014; Arruda et al. 2016; Boeven et al. 2016; Spindel et al. 2016; Bian and Holland 2017). The hypothetical simulation and practical GS studies are routinely practiced in animal breeding (Goddard and Hayes 2007; Jannink et al. 2010). However, GS is not an exciting opportunity in crop plants; besides, large-scale investigations are not available in practical plant breeding (Desta and Ortiz 2014; Jannink et al. 2010; Jiang 2013). The extensive application of GS in plant breeding demands a thorough understanding of complex traits (Nakaya and Isobe 2012; Jiang 2013). GS analyzes the proportion of genetic resemblance between the training population and breeding population through the linkage disequilibrium between marker and trait loci (Desta and Ortiz 2014). However, the breeding populations on which breeders implement research works are individually different from the training population considered.
Consequently, population structure can impact consistent errors in estimates of GEBVs for complex traits (Lyra et al. 2018). The population structure of self-pollinating plants or inbreeding is an essential drawback for GS application in plant breeding (Desta and Ortiz 2014). The additive genetic variance that ignores dominant or related epistatic interactions is crucial in GEBV assessments. Hence, an estimated fraction of breeding values in the GS model achieves higher selection gain in self-pollinating crops due to the use of homozygous lines as founder parents (Heffner et al. 2009). However, GS is not impressive for crops having an advantage of cross-pollination due to dominance and epistatic interactions; it is also ineffective for traits with narrow-sense heritability (Heffner et al. 2009; Nakaya and Isobe 2012). GS is also unrealistic for breeding populations consisting of hundreds and thousands of crosses/populations at the same time (Jiang 2013). However, recently a number of integrated models have been developed to comprehend both additive and non-additive effects and improve the accuracy of GS (Majumdar et al. 2020; Sehgal et al. 2020; Tanaka 2020; Mishra et al. 2021; Budhlakoti et al. 2022a; Sinha et al. 2023). Nevertheless, the marker effects and GEBV estimates may change due to changes in gene frequencies and epistatic interactions over a period of time (Misztal et al. 2021; Budhlakoti et al. 2022b). This would necessitate the updating of the GS model with every breeding cycle (Jighly et al. 2019). The cost of implementation of a new model in GS is more than traditional breeding &/or MAS (Hickey et al. 2017a, b). Moreover, limited knowledge of the genetic architecture of quantitative traits limits our ability to develop appropriate models for GS to achieve maximum prediction accuracy (Bartholomé et al. 2022). Size and genetic relationship of training & breeding population, genetic diversity and heritability of the trait under concern, the influence of genotype-environment (G × E) interaction, and the density of markers affect the prediction accuracy (Hickey et al. 2017a, b; Xu et al. 2020a, b; Budhlakoti et al. 2022b).
Besides, simulation studies, statistical models, and types of breeding populations used for progeny performance assessments (BC1s/F2/RILs/DHs/Inbreds/OPVs) may produce the random effects during the identification of genomic regions/QTLs responsible for complex traits during linkage map construction using training population. All these factors affect accuracies of selections in GS (Liu et al. 2019, 2018; Crossa et al. 2017; Hickey et al. 2017a, b; Schopp et al. 2017; Zhang et al. 2017, 2019; Wang et al. 2018). MARS schemes can be accelerated to have more accurate GEBV estimates since GS coupled with MARS can assess numerous loci, haplotypes or marker impacts in different cycles (Sinha et al. 2023).
MARS is good enough for accumulating desirable alleles of up to 9–10 QTLs in the homozygous lines advanced from selected individuals (Wang et al. 2007). In MARS, the selection comprises of several loci, ultimately evaluated for many successive cycles/generations. The off-season nurseries reallocate breeding materials evaluation in the target environment, and the number of generations can be increased to 3–4 each year in MARS than one per year in phenotypic-recurrent selection. The change in resources is essential to the MARS program meant for complex traits (Edwards and Johnson 1994; Johnson 2004; Crosbie et al. 2006). The enhancement of the population using fast-tracked cycles at prolonged nurseries helps breeders to achieve a higher proportion of plants with favorable alleles and increase the opportunity for selection of haplotype-specific for the target environment/trait (Cobb et al. 2019).
Genomics-assisted Breeding and MARS
Advances in next-generation sequencing (NGS) technologies have made it possible to generate a large number of functional markers (FMs). Marker-assisted breeding programs employ FMs, often referred to as precision markers, which are connected to variance in phenotypic traits (Yang et al. 2015; Salgotra and Stewart 2020). FMs can accumulate beneficial alleles/QTL regions in any genetic background population more efficiently through MARS, without additional calibrations (Abdulmalik et al. 2017; Nawaz et al. 2017; Rodenburg 2018; Kulkarni et al. 2023). Favorable alleles at many (10–40) of the key loci involved in the expression of the target characteristics can be accumulated by MARS (Varshney et al. 2012). MARS optimizes the efficiency of converting genetic diversity into genetic gain through several recurrent selections per cycle (Gorjanc et al. 2018a, b). Using FMs can greatly reduce the number of cycles to select ideal genotype and enable genetic gain for the complex traits (Salgotra and Stewart 2020). NGS technologies are strengthening multiparent marker-assisted recurrent selection programs to find relationships between different traits and genomes (Bohra 2013; Sinha et al. 2023). Using of FMs in MARS further help in designing future crops (Varshney et al. 2021).
MARS was a successful breeding technique for more effectively pyramiding several QTLs with minor effects on wheat crown rot resistance by the use of FMs (Rahman et al. 2020). MARS effectively improved provitamin A content in tropical maize, both α-carotene and β-cryptoxanthin showed increased genetic gain after two rounds of recurrent cycles with a favorable frequency of functional SNP marker alleles (Kebede et al. 2021). Recurrent genomic selection increased the long-term genetic gain by optimal cross-selection quickly. This is accomplished by increasing the efficiency of converting genetic diversity into genetic gain. Meanwhile, minimized the genetic diversity loss and reduced the decline in genomic prediction accuracy with fast cycling (Gorjanc et al. 2018a, b). Multi-trait ensemble genomic prediction and simulations of recurrent selection demonstrate the long-term genetic gains in wheat for complex trait genetic architecture & increased prediction accuracy for almost 90% of traits, improving grain yield prediction accuracy by 3–52% (Fradgley et al. 2023). Since the GS evaluates many loci, haplotypes, or marker effects throughout the entire genome to calculate the GEBV, in GS programmes, recurrent selection techniques may be hastened, enabling farmers to fully utilize genetic influences in the production field (Heffner et al. 2010; Sinha et al. 2023). Using single-step GBLUP over generations in a reciprocal recurrent selection (RRS) program, researchers were able to achieve high prediction accuracies for growth characteristics in the hybrid Eucalyptus grandis x E. urophylla. These results suggested a significantly accelerated RRS program by GS (Grattapaglia 2022). QuMARS tool was developed to combine phenotypic, MARS, and GS for both short and long-term breeding programs (Ali et al. 2020).
MARS was successfully employed in various crop systems; the details are presented in Table 1.
Key Factors to be Considered for MARS
-
1.
Selection of founder parents
The co-ordination between germplasm curator and breeder is necessary to utilize genetic diversity originating from crop wild relatives and other un-explored germplasm that ensures the success in obtaining a novel combination of favorable alleles accumulation at multi-loci during the intermating phase of recurrent selection (Hufford et al. 2013; Sawler et al. 2013; Dempewolf et al. 2017).
Founder parents are usually selected from existing genotypes, cultivars, varieties, landraces, germplasm lines, and evaluated in regular seasons for various traits. Generally, two types of parental selections followed. First, based on plant performance under high × high and high × low crossing panels that can produce the best lines. Second, the genetic diversity among parents and progeny performance is evaluated for several cycles for parental selection (Wang et al. 2005).
Hence, the parents should be selected carefully with the following attributes.
-
All the parents must have synchronization for flowering, which could ease in effective intermating. One of the parent's selections with male sterility was found useful in a few experiments (Dhliwayo et al. 2014).
-
At least two parents must be polymorphic for every marker selected for MARS.
-
Parents be cross-compatible; in other words, progenies should not be sterile.
Each parent selected would be advantageous if it is agronomically superior; otherwise, it takes more cycles of recurrent selection to improve the population for agronomic characters.
-
-
2.
Population development
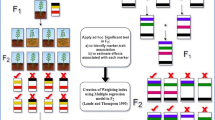
RS plays a prominent role in population improvement, commonly employed as intra-population and inter-population improvement approaches designed to intensify populations per se performance or enhance combining ability. Different recurrent selection schemes were developed like S0, S1, S2, full-sib, half-sib, ear-to-row, reciprocal recurrent, and reciprocal full-sib recurrent selection depending on inter or intrapopulation improvement approaches (Vasal et al. 2004). Figure 1 explains the general MARS schemes with various population improvement approaches. It can be done in one population (intra-population improvement) or two populations (inter-population improvement).
-
(a)
Intra-population RS
Intra-population improvement approaches are conceived to improve populations per se for quantitative traits of commercial importance (Dhillon 1991; Vasal et al. 2004; Malav et al. 2016; Dormatey et al. 2020). The cyclic betterment of plants in advanced generations acquires favorable alleles through a slow but successive process by improving the mean performance of the population. Intra-population improvement approaches may involve individuals, families (Half sibs/Full sibs/selfed progenies), or test crosses. Half-sib and full-sib families have been used and proved effective in improving maize populations (Hallauer and Filho 1988). The evaluation trials are generally replicated experiments in single or multiple locations, and the selected progenies will be intermated in all possible combinations (Eathington 2005).
Intra-population improvement may involve evaluation of individuals (mass selection) or its progenies (full-sibs, half-sibs or selfed progeny selection) that have been advanced within a population or test-cross progenies developed by using a tester (related or unrelated, narrow or broad-genetic base). Different approaches of intra-population improvement aim at enhancement of the performance of the population per se for all random mated or selfed generation, but in case of test-cross progenies evaluation, the importance is on improving combining ability (Vasal et al. 2004).
Unlike RS, MARS eliminates the extra round of testcross progeny evaluation, as the markers have the advantage of genotypic selection. Codominant markers would be more informative than dominant markers in MARS (Nadeem et al. 2018; Cholin et al. 2019; Perveen et al. 2023).
-
(b)
Inter-population RS
Inter-population improvement approaches are strategically utilized to facilitate both population improvement and hybrid development procedures (Vasal et al. 2004; Malav et al. 2016; Dormatey et al. 2020). Besides, two heterotic populations are simultaneously improved, and attention is given to the combining ability. It was recommended that only agronomically superior populations are subjected to inter-population improvement schemes (Vasal et al. 2004). For vigorous and productive improvement of plants, tolerance to inbreeding is critical in inter-population improvements, highlighting both combining ability and crossbred performance. Extensively used inter-population improvement schemes are reciprocal recurrent selection-half sibs (RRS-HS) (Comstock et al. 1949) and reciprocal recurrent selection-full sibs (RRS-FS) (Hallauer and Eberhart 1970; Hallauer 1973). RRS has proved successful in outcrossing species such as maize (Eyherabide and Hallauer 1991; Tardin et al. 2007; Souza et al. 2010; Kolawole et al. 2018) and sugar beetroot (Doney and Theurer 1978; Hecker 1985). Simulations in oil palm have revealed that genomic selection has the potential to reduce the generation time of an RRS breeding cycle from 20 to 6 years (Cros et al. 2015). The incorporation of genomic selection into RRS would also allow to combine RRS and speed breeding approaches (Watson et al. 2018). However, empirical evidence of the superiority of Reciprocal recurrent genomic selection (RRGS) breeding programmes is still lacking (Rembe et al. 2021).
A multi-parent-based MARS scheme would be more rewarding for integrating favorable alleles at multi-loci from 10–15 parents (Ragimekula et al. 2013), to construct the ideal genotype and obtain the greatest possible genetic gain (Stam 1995; Peleman and van Der Voort 2003).
In bi-parental populations, MARS specifies an F2 population's advancement by one cycle of phenotypic and marker genotyping preceding with two or three cycles of only genotyping (Edwards and Johnson 1994; Johnson 2004; Eathington et al. 2007). MARS will be proceeded to pyramid beneficial alleles from various genetic sources (Singh et al. 2016).
If the objective is to obtain open-pollinated varieties or adaptation of exotic germplasm, intra-population methods are recommended. However, inter-population methods are the most appropriate when the purpose is to extract the inbred lines of good combiners or production of intra-population or inter-population synthetics or production of potential hybrids.
-
(a)
-
3.
QTL introgression by MARS
MARS can be initiated without any QTL information, while the selection is based on a significant marker-trait association established during the MARS process (Xu 2012). But the effectiveness of MARS will be higher when the QTL were known (Bernardo and Charcosset 2006; Moreau et al. 2004). MARS is useful for complex traits; however, as the number of QTL increases, fewer known QTL produce the maximum efficiency. The usefulness of having prior knowledge of QTL under genetic models has been examined, including different numbers of QTL, different levels of heritability, unequal gene effects, linkage, and epistasis. It was found that MARS is most beneficial for traits controlled by a moderately large number of QTL (e.g., 40) (Bernardo and Charcosset 2006).
Adequate genotyping and phenotyping are extremely important in MARS for appropriate evaluation and meticulous selection of QTL combinations and ideal genotypes. To realize genetic background of germplasm in a population improvement approaches, SSR or SNP markers were consistently used (Baskaran et al. 2009; Bankole et al. 2017). SSR markers present randomly throughout the genome, provide several advantages. First, it helps to understand genetic differentiation in every recombination cycle and confirm the widespread nature of selection effects (Gallagher et al. 2015). Second, it examines the co-selection of traits. Third, it scans genomic regions under high and low selection pressure during early or advanced generations and determine their role in modifying target traits (Gallagher et al. 2015).
For complex traits under multi-locus control, a significant number of SSR markers present neighboring to QTL regions would also show selection effects (Gallagher et al. 2015). SNPs can instruct change in allele frequencies response to selection (Baskaran et al. 2009). SNP may be present within coding sequences of genes, non-coding regions of genes or in the intergenic regions between genes at different frequencies in different chromosomes, with the availability of genetic information of these SNPs, selection of more number of introgressed QTLs can be done at the same time (Kumpatla et al. 2012; Jiang 2013). MARS scheme using genome-wide SSR/SNP markers covering one marker per 10 cM distance of every chromosome are adequate for the effective selection of favorable alleles analogous to multiple trait combinations (Nayak et al. 2017).
High throughput and precision phenotyping platforms are favorably utilized in MARS to complement cost-efficient genotyping platforms and enhance screening under year-round, off-season nurseries to speed up the development of climate-resilient germplasms with increased productivity and nutritional quality (Gedil and Menkir 2019). MARS, in combination with precise phenotyping, has produced resilient food crops in maize (Xu 2012; Prasanna et al. 2013), facilitating improved genetic gain and rapid cultivar development (Gedil and Menkir 2019).
Strategies involved in precision phenotyping may include biotic stress harmonization using remote sensing, digital/multispectral technologies to evaluate biomass, senescence, anthesis, lodging, plant stand, inflorescence traits, spatial variation in the field, use of ground-penetrating radar to estimate water uptake and root depth and type of root (Xu et al. 2017). Biotic or abiotic stress phenotyping was performed in multiple environments using mobile robotic phenotyping hubs (Xu et al. 2017). PHENObot is an automatic robotic device used for rapid data acquisition and GPS tracking, spectroscopy, and 3D analysis of individual plants used for robust phenotyping in vineyards (Kicherer et al. 2015).
-
4.
Number of Recombination cycles
MARS intends to increase favorable allele frequency while avoiding identity by descent; in a way that genetic recombination remains useful as a source of novel genetic variation (Morais 1997; Bernardo 2010; Müller et al. 2017). Although continuous inbreeding and selection lead to the rapid depletion of genetic variation in a population (Falconer and Mackay 1996), recombination of progenies after each selection cycle leads to breaking gene blocks into smaller pieces, generating many more genetic combinations than expected with large chromosomal segments (Dudley and Lambert 2010).
Genetic variance underlying essential traits of interest should be evaluated at every recombination cycle to confirm the genetic diversity available in the population (Tourrette et al. 2019). After identifying prominent QTLs in early generations like F3 or F4, plants carrying specific flanking markers to the QTL region will be confirmed by marker values. Later, elite lines carrying favorable QTL regions are subjected to controlled pollination under greenhouse conditions at single or multiple environments to develop specific envirotype or best general combiner or potential hybrid (Tourrette et al. 2019).
Prolonged recombination cycles strengthen the response to selection by breaking the linkage between QTLs (Hill and Robertson 1966; Felsenstein 1974). Suppose two QTLs are linked together; having a contrasting effect on each other tends to inherit together, impeding the efficiency of selection. The presence of such negative linkage disequilibrium between various QTL regions are familiar, that appear in population due to continuous selection (Bulmer effect, Bulmer 1971) or genetic drift (Barton 2009). Increased recombination at multiple cycles in MARS gathers preferential mutations in evolving populations (Tourrette et al. 2019). In the absence of recombination cycles, it is challenging to eliminate deleterious mutations even under careful selections during population improvement schemes. Higher recombination (Felsenstein 1974) with smaller loss in genetic variability counterbalance loss of prediction accuracies over generations and substantiating higher genetic gain benefited by continuous intermating and recombination. Long-term selection programs profoundly increased genetic gain of 30% after 20 generations ((Tourrette et al. 2019). As the number of recombinant cycles and selection rate increases after every cycle the genetic gain improves at faster rate (Kushwah et al. 2020; Singh et al. 2023). MARS over a number of generations can result in faster gains particularly for low heritability traits by selection based on estimated breeding values (EBVs) calculated using more complete pedigree information in best linear unbiased prediction (BLUP) analysis (Slater et al. 2014).
-
5.
Genetic gain
Genetic gain can be defined as the total increase in the performance of the population over its parental population that is achieved by careful artificial selection annually (Xu et al. 2017, 2020a, b). In this era of molecular breeding, the rate of genetic gain per unit of time can be increased by speeding up the selection cycles and intensifying the selection pressure and improving the evaluation precision, thus increasing the heritability (Bernardo 2010; Müller et al. 2017).
The genetic gain is usually estimated using the below-mentioned equation (Lush 1937; Stephen and Rita 2008). Commonly known as “the breeder’s equation”.
$$\mathrm{Genetic\;Gain}=\Delta G={h}^{2}\sigma p\; i/L$$where in, ΔG = Expected genetic gain; ‘i’ is the intensity of selection; h2 = narrow sense heritability; L = time of breeding cycle; σp = phenotypic standard deviation or standard deviation of breeding value. The environment plays a major role in creating the difference of improved genetic gain between the breeder's experimental trials and the actual farmer's field. This gap could be minimized by precise genotyping with the controlled environment at the field level (Xu et al. 2017).
These primary factors of genetic gain are highly influenced by many other subfactors as detailed below.
-
(a)
Genetic variation:
-
(i)
The types of germplasm used (ecotypes, landraces, wild relative, introgression lines, or exotic libraries), their origins, number, and relationship with the target trait under selection plays a significant role in improving genetic gain (Xu et al. 2017). A thumb rule for the proportion of plants selected in each generation of recurrent selection can be estimated by ‘Nsel’ (Bernardo et al. 2006). The estimated Nsel be proportionate to the number of cycles for which selection is practiced. On the other side, the type of population used in the MARS program, either permanent segregating populations like DH/RIL or temporary segregating populations like F2/F2:3, is essential components of genetic variation foundation for selection response in subsequent cycles (Xu et al. 2017). A comparative study revealed that the response to selection was higher in the DH population instead of F2, which was further found to be greater in MARS than even in genomic selection (Mayor and Bernardo 2009a, b). Genetic gain achieved in MARS in maize was twice than recurrent phenotypic selection (Earthington 2005; Crosbie et al. 2006).
-
(ii)
The type of markers, number of markers, genome coverage is essential to study different parameters like allele effect (additive, dominance-dominance) and interactions, including GxG and G × E, to reveal genetic variation (Xu et al. 2017). MARS within segregating populations is affected by the genetic makeup of the genes and targeted genetic background of concerned alleles present at each locus that has epistatic interaction with the target locus, while studying quantitative traits (Xu et al. 2017). Marker effects of SNPs calculated with best linear unbiased prediction models (BLUP) that permitted the prediction of genomic estimated breeding values (GEBV) and further selection of 10% of the lines with highest GEBV in F2:3 could improve the genetic gain in the Maize MARS program for drought tolerance along with increased grain yield and agronomic performance (Bankole et al. 2017).
-
(i)
-
(b)
Heritability: Remodeling field experimental areas and alterations used to improve heritability are essential to enhance genetic gain. Heritability is estimated by the ratio of genetic variance (Vg) to phenotypic variance (Vp), the latter being partitioned into Vg and Ve (environmental variance). Vp depends on the type of population (mortal/immortal), population size, high-throughput, and precision of phenotyping and the number of multi-environmental trials. Ve can be studied by environmental assay or envirotyping, which represent all environmental factors that affect plant growth/development and yield (Xu 2011, 2012; Cooper et al. 2014). Envirotyping facilitates increasing selection accuracy, multi-environmental trials, and enhanced varietal evaluation, which in turn increases heritability.
-
(c)
Selection intensity (i): ‘i’ is a constant factor, estimated by the rate of selection, the proportion of plants selected from the total population. Evaluated by the formula i = Nsel/N to measure the selection intensity, where, Nsel is the number of selected individuals, N is total population size, ‘i’ corresponds to strong selection pressure. Increased population size is becoming increasingly important in the MARS scheme for multi-trait improvement approaches. To achieve greater genetic gain by utilizing existing genetic variation present in the population, an excessive number of trials is necessary with large population size (Xu et al. 2017). The population size required for MARS depends on the number of traits considered (Xu et al. 2017). Moreover, many plants are subjected to both genotyping and phenotyping to ensure the presence of genes influencing target traits and discover a novel combination of alleles governing multiple traits (Li et al. 2012). Contrastingly, due to the higher selection pressure in the population, the population size may get randomly reduced. Thereupon, allowing genetic drift to eventuate at non-target loci, abolishing the possibility of selecting a unique combination of traits. So, meticulous selection skill is necessary during every cycle of MARS. Higher selection intensity in turn increases the rate of genetic gain (Xu et al. 2017; Allier et al. 2019). It is also possible to boost selection intensity by choosing fewer parents. However, it is typically more important to make careful decision of number of parents depending on whether the breeding goal is for long- or short-term genetic gain (Bernardo and Charcosset 2006). As a result, in order to improve selection intensity through larger populations, budgets must be increased or the expense of evaluating each candidate for selection must be decreased (Cobb et al. 2019). By phenotyping all candidates for selection, even at low levels of replication, Lorenz (2013) and Riedelsheimer et al. (2013) discovered that the application of genomic prediction often boosted response to selection.
-
(d)
Selection index: Considering the magnitude of marker effects on the target traits, the selection index is widely utilized in MARS programs (Lande and Thompson 1990; Edwards and Johnson 1994). The selection index can be estimated by Mj = ¼ ΣbiXij, where Mj is the marker score assigned to jth individual, bi is prominent to the ith marker locus, and Xij is designated to score 1 if the jth individual has homozygous marker allele with favorable effect. Otherwise, -1 if the individual is homozygous for the unfavorable marker allele. Whereas, the value of bi is determined compared to multiple regressions relevant to trait values (Lande and Thompson 1990; Hospital and Charcosset 1997). Selection index critically evaluates the number of plants to be selected for further generations and indirectly implying the genetic gain to be improved in further cycles (Singh et al. 2023).
-
(e)
Cycle time: The long-term selection schemes are advantageous for improving genetic gain. Genetic gain increases in recurrent cycles with visible effects after 4–5 cycles (Tourrette et al. 2019; Nayak et al. 2017). Screening under off-season nurseries and multiple environments under the greenhouse, phytotrons, and winter nurseries directly influence the cycle time, subsequently the genetic gain (Xu et al. 2017).
Rapid generation advance (RGA) was proposed by Goulden (1939) and undergone many modifications by Grafius (1965). In recent years it has been included in the strategies of “speed breeding” (Watson et al. 2018) wherein, depending on the crop, number of generations can be accelerated and can achieve desired cycle time in MARS program (Cobb et al. 2019).
The significance of the above factors, if understood clearly, the improved genetic gain can be achieved in a given time interval by accelerating the breeding procedure by marker-assisted recurrent breeding strategies. MARS has been suggested for "forward breeding" of inherent genes and pyramiding of several genes/QTLs for complex traits, especially, yield components and various biotic and abiotic stresses. (Singh et al. 2023).
-
(a)

MARS selection procedure with different population improvement approaches
Conclusion
Even after three decades of introducing the concept of MAS by Smith and Simpson (1986), the success of MAS programs is limited to a few crops. Genomic selection is the most advanced prediction model-dependent genome-wide marker strategy employed mostly using SNP markers. The feasibility of its utilization in every crop may be delimited owing to financial constraints and necessary training population especially in cross pollinating crops. In this context, MARS would be more rewarding for crops where the genomic information and budget are limited. MARS apparently makes best use of genetic diversity present in the population. MARS seems to be more robust and cost-efficient, worthwhile for incorporating multiple desirable alleles for multiple QTL regions simultaneously with profound increase in genetic gain. MARS is more promising scheme for obtaining novel gene combinations at every cycle. Collaborative MARS research in public and private sectors at both national and international level could help in precise breeding.
References
Abdulmalik RO, Menkir A, Meseka SK, Unachukwu N, Ado SG, Olarewaju JD, Aba DA, Hearne S, Crossa J, Gedil M (2017) Genetic gains in grain yield of a maize population improved through marker assisted recurrent selection under stress and non-stress conditions in West Africa. Front Plant Sci 8:841
Ali M, Zhang L, DeLacy I, Arief V, Dieters M, Pfeiffer WH, Wang J, Li H (2020) Modeling and simulation of recurrent phenotypic and genomic selections in plant breeding under the presence of epistasis. The Crop Journal 8(5):866–877
Allier A, Lehermeier C, Charcosset A, Moreau L, Teyssèdre S (2019) Improving short-and long-term genetic gain by accounting for within-family variance in optimal cross-selection. Front Genet 10:1006
Amante-Bordeos A, Sitch LA, Nelson R, Dalmacio RD, Oliva NP, Aswidinnoor H, Leung H (1992) Transfer of bacterial blight and blast resistance from the tetraploid wild rice Oryza minuta to cultivated rice, Oryza Sativa. Theor Appl Genet 84(3–4):345–354
Arruda MP, Lipka AE, Brown PJ, Krill AM, Thurber C, Brown-Guedira G, Dong Y, Foresman BJ, Kolb FL (2016) Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat (Triticum aestivum L.). Mol Breed 36(7):84
Asoro FG, Newell MA, Beavis WD, Scott MP, Tinker NA, Jannink JL (2013) Genomic, marker-assisted, and pedigree-BLUP selection methods for β-glucan concentration in elite oat. Crop Sci 53(5):1894–1906
Babu R, Prasanna BM (2014) Molecular breeding for quality protein maize (QPM). In: Genomics of Plant Genetic Resources. Springer, Dordrecht. pp 489–505
Bankole F, Menkir A, Olaoye G, Crossa J, Hearne S, Unachukwu N, Gedil M (2017) Genetic gains in yield and yield related traits under drought stress and favorable environments in a maize population improved using marker assisted recurrent selection. Front Plant Sci 8:808
Bartholomé J, Prakash PT, Cobb JN (2022) Genomic prediction: Progress and perspectives for rice rice improvement. In: Genomic Prediction of Complex Traits: Methods and Protocols, pp 569–617
Barton NH (2009) Why sex and recombination? 187–195 in coldspring harbor symposia on quantitative biology. Cold Spring Harbor Laboratory Press, New York
Baskaran K, Hash CT, Senthilvel S, Raj AB, Muthiah RA (2009) SSR allele frequency changes in response to recurrent selection for pearl millet grain yield and other agronomic traits. J SAT Agric Res 7:8
Bernardo R (2010) Breeding for quantitative traits in plants, 2nd edn. Stemma Press, Woodbury, MN
Bernardo R (2002) Breeding for quantitative traits in plants (Vol. 1, p 369). Woodbury, MN: Stemma Press
Bernardo R (2008) Molecular markers and selection for complex traits in plants: Learning from the last 20 years. Crop Sci 48(5):1649–1664
Bernardo R (2014) Genome wide selection when major genes are known. Crop Sci 54(1):68–75
Bernardo R, Charcosset A (2006) Usefulness of gene information in marker-assisted recurrent selection: a simulation appraisal. Crop Sci 46(2):614–621
Bernardo R, Yu J (2007) Prospects for genome-wide selection for quantitative traits in maize. Crop Sci 47:1082–1090. https://doi.org/10.2135/cropsci2006.11.0690
Bernardo R, Moreau L, Charcosset A (2006) Number and fitness of selected individuals in marker-assisted and phenotypic recurrent selection. Crop Sci 46:1972–1980
Beyene Y, Semagn K, Crossa J, Mugo S, Atlin GN, Tarekegne A, Meisel B, Sehabiague P, Vivek BS, Oikeh S, Alvarado G (2016) Improving maize grain yield under drought stress and non-stress environments in sub-Saharan Africa using marker-assisted recurrent selection. Crop Sci 56(1):344–353
Bian Y, Holland JB (2017) Enhancing genomic prediction with genome-wide association studies in multiparental maize populations. Heredity 118(6):585–593
Boeven PH, Longin CFH, Würschum T (2016) A unified framework for hybrid breeding and the establishment of heterotic groups in wheat. Theor Appl Genet 129(6):1231–1245
Bohra A (2013) Emerging paradigms in genomics-based crop improvement. Sci World J 2013
Bradbury LM, Fitzgerald TL, Henry RJ, Jin Q, Waters DL (2005) The gene for fragrance in rice. Plant Biotechnol J 3(3):363–370
Budhlakoti N, Kushwaha AK, Rai A, Chaturvedi KK, Kumar A, Pradhan AK, Kumar U, Kumar RR, Juliana P, Mishra DC, Kumar S (2022a) Genomic selection: a tool for accelerating the efficiency of molecular breeding for development of climate-resilient crops. Front Genet 13:832153
Budhlakoti N, Mishra DC, Majumdar SG, Kumar A, Srivastava S, Rai SN, Rai A (2022b) Integrated model for genomic prediction under additive and non-additive genetic architecture. Front Plant Sci 13:1027558
Bulmer M (1971) The effect of selection on genetic variability. Am Nat 105(943):201–211
Ceballos H, Kawuki RS, Gracen VE, Yencho GC, Hershey CH (2015) Conventional breeding, marker-assisted selection, genomic selection and inbreeding in clonally propagated crops: a case study for cassava. Theor Appl Genet 128:1647–1667
Cerrudo D, Cao S, Yuan Y, Martinez C, Suarez EA, Babu R, Zhang X, Trachsel S (2018) Genomic selection outperforms marker assisted selection for grain yield and physiological traits in a maize doubled haploid population across water treatments. Front Plant Sci 9:366
Chahal GS, Gosal SS (2006) Principles and procedures of plant breeding. Biotechnological, and conventional approaches. Alpha Sci. International Ltd. Harrow. UK
Chaitra KC, Sarvamangala C, Manikanta DS, Chaitra PA, Fakrudin B (2020) Insights into genetic diversity and population structure of Indian carrot (Daucus carota L.) accessions. J Appl Genet 61:303–312
Charmet G, Robert N, Perretant MR, Gay G, Sourdille P, Groos C, Bernard S, Bernard M (2001) Marker assisted recurrent selection for cumulating QTLs for bread-making related traits. Euphytica 119(1–2):89–93
Chevalet C, Mulsant P (1992) Using markers in gene introgression breeding programs. Genetics 132(4):1199–1210
Cholin SS, Poleshi CA, Manikanta DS, Christopher C (2019) Exploring the genomic resources of carrot for cross-genera transferability and phylogenetic assessment among orphan spices and vegetables of Apiaceae family. Hortic Environ Biotechnol 60:81–93
Cobb JN, Juma RU, Biswas PS, Arbelaez JD, Rutkoski J, Atlin G, Hagen T, Quinn M, Ng EH (2019) Enhancing the rate of genetic gain in public-sector plant breeding programs: Lessons from the breeder’s equation. Theor Appl Genet 132:627–645
Comstock RE, Robinson HF, Harvey PH (1949) A breeding procedure designed to make maximum use of both general and specific combining ability. J Agron 41(8):360–367
Cooper OR, Parrish DD, Ziemke J, Balashov NV, Cupeiro M, Galbally IE, Gilge S, Horowitz L, Jensen NR, Lamarque JF, Naik V (2014) Global distribution and trends of tropospheric ozone: an observation-based review. Elem Sci Anth 2
Cregan PB, Mudge J, Fickus EW, Danesh D, Denny R, Young ND (1999) Two simple sequence repeat markers to select for soybean cyst nematode resistance conditioned by the rhg1 locus. Theor Appl Genet 99(5):811–818
Cros D, Denis M, Bouvet JM, Sánchez L (2015) Long-term genomic selection for heterosis without dominance in multiplicative traits: Case study of bunch production in oil palm. BMC Genomics 16:1–17
Crosbie TM, Eathington SR, Johnson GR, Edwards M, Reiter R, Stark S, Mohanty RG, Oyervides M, Buehler RE, Walker AK, Dobert R (2006) Plant breeding: Past, present, and future. In: Lamkey KR, Lee M (eds) Plant breeding: The Arnel R Hallauer international symposium. Blackwell Publishing, Oxford, UK, pp 3–50
Crossa J, Pérez-Rodríguez P, Cuevas J, Montesinos-López O, Jarquín D, de los Campos G, Burgueño J, González-Camacho JM, Pérez-Elizalde S, Beyene Y, Dreisigacker S (2017) Genomic selection in plant breeding: methods, models, and perspectives. Trends Plant Sci 22(11):961–975
de C. Lara LA, Santos MF, Jank L, Chiari L, Vilela MDM, Amadeu RR, Dos Santos JP, Pereira GDS, Zeng ZB, Garcia AAF (2019) Genomic selection with allele dosage in Panicum maximum jacq. G3: Genes Genomes Genetics 9(8):2463–2475
Dempewolf H, Baute G, Anderson J, Kilian B, Smith C, Guarino L (2017) Past and future use of wild relatives in crop breeding. Crop Sci 57(3):1070–1082
Desta ZA, Ortiz R (2014) Genomic selection: Genome-wide prediction in plant improvement. Trends Plant Sci 19(9):592–601
Dhillon BS (1991) Alternate recurrent selection of S1 and half-sib families for intrapopulation improvement (No. 93-012892. CIMMYT.)
Dhliwayo T, Palacios-Rojas N, Crossa J, Pixley KV (2014) Effects of S1 recurrent selection for provitamin A carotenoid content for three open-pollinated maize cultivars. Crop Sci 54(6):2449–2460
Doney DL, Theurer JC (1978) Reciprocal recurrent selection in sugarbeet. Field Crop Res 1:173–181
Dormatey R, Sun C, Ali K, Coulter JA, Bi Z, Bai J (2020) Gene pyramiding for sustainable crop improvement against biotic and abiotic stresses. Agronomy 10(9):1255
Dudley JW, Lambert RJ (2010) 100 generations of selection for oil and protein in corn. Plant Breed Rev 24:79–110
Eathington SR, Crosbie TM, Edwards MD, Reiter RS, Bull JK (2007) Molecular markers in a commercial breeding program. Crop Sci 47:154–163
Eathington SR (2005) Practical applications of molecular technology in the development of commercial maize hybrids. Proc 60th Ann Corn Sorghum Seed Res Conf Washington, D.C., USA. American Seed Trade Association
Edwards M, Johnson L (1994) RFLPs for rapid recurrent selection: Analysis of molecular marker data. American Society for Horticultural Science, CSSA, Madison, WI, pp 33–40
Eyherabide GH, Hallauer AR (1991) Reciprocal full‐sib recurrent selection in maize: II. Contributions of additive, dominance, and genetic drift effects. Crop Sci 31(6)1442–1448
Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics. 4th ed. Pearson Education, Harlow, UK
Felsenstein J (1974) The evolutionary advantage of recombination. Genetics 78(2):737–756
Fradgley N, Gardner KA, Bentley AR, Howell P, Mackay IJ, Scott MF, Mott R, Cockram J (2023) Multi-trait ensemble genomic prediction and simulations of recurrent selection highlight importance of complex trait genetic architecture for long-term genetic gains in wheat. in silico Plants 5(1):diad002
Gahlaut V, Jaiswal V, Tyagi BS, Singh G, Sareen S, Balyan HS, Gupta PK (2017) QTL mapping for nine drought-responsive agronomic traits in bread wheat under irrigated and rain-fed environments. PLoS ONE 12(8):e0182857
Gallagher JA, Turner LB, Cairns AJ, Farrell M, Lovatt JA, Skøt K, Armstead IP, Humphreys MO, Roldan-Ruiz I (2015) Genetic differentiation in response to selection for water-soluble carbohydrate content in perennial ryegrass (Lolium perenne L.). BioEnergy Res 8(1):77–90
Gedil M, Menkir A (2019) An integrated molecular and conventional breeding scheme for enhancing genetic gain in maize in Africa. Front Plant Sci 10
Goddard ME, Hayes BJ (2007) Genomic selection. J Anim Breed Genet 124(6):323–330
Goddard ME, Hayes BJ (2009) Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet 10(6):381–391
Gorjanc G, Gaynor RC, Hickey JM (2018a) Optimal cross selection for long-term genetic gain in two-part programs with rapid recurrent genomic selection. Theor Appl Genet 131:1953–1966
Gorjanc G, Gaynor RC, Hickey JM (2018b) Optimal cross-selection for long-term genetic gain in two-part programs with rapid recurrent genomic selection. Theor Appl Genet 131:1953–1966
Goulden CH (1939) Problems in plant selection. In: Burnett RC (ed) Proceeding of the seventh genetics congress. Cambridge University Press, Edinburgh, pp 132–133
Grafius JE (1965) Shortcuts in plant breeding. Crop Sci 5:377
Grattapaglia D (2022) Twelve years into genomic selection in forest trees: Climbing the slope of enlightenment of marker assisted tree breeding. Forests 13(10):1554
Grenier C, Cao TV, Ospina Y, Quintero C, Châtel MH, Tohme J, Courtois B, Ahmadi N (2015) Accuracy of genomic selection in a rice synthetic population developed for recurrent selection breeding. PLoS ONE 10(8):e0136594
Hallauer AR (1999) Temperate maize and heterosis. In Coors JG, Pandey S (eds), The genetics and exploitation of heterosis in crops (pp 353–361). Proc Int Symp Heterosis Crops. Mexico City, 18–22 August 1997. ASA, CSSA and SSSA, Madison, WI, USA
Hallauer AR, Eberhart SA (1970) Reciprocal full-sib selection 1. Crop Sci 10(3):315–316
Hallauer AR (1973) Hybrid development and population improvement in maize by reciprocal full-sib selection. Egypt J Genet Cytol
Hallauer AR, Carena MJ, Miranda Filho JD (2010) Quantitative genetics in maize breeding (vol. 6). Springer Science & Business Media
Hallauer R, Filho JBM (1988) Quantitative genetics in maize breeding, 2nd edn. Iowa State University Press, Ames, Iowa, USA
Hecker RJ (1985) Reciprocal recurrent selection for the development of improved sugarbeet hybrids. J ASSBT 23:47–57
Heffner EL, Lorenz AJ, Jannink JL, Sorrells ME (2010) Plant breeding with genomic selection: Gain per unit time and cost. Crop Sci 50(5):1681–1690
Heffner EL, Sorrells ME, Jannink JL (2009) Genomic selection for crop improvement. Crop Sci 49(1):1–12
Hickey JM, Chiurugwi T, Mackay I, Powell W (2017a) Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery. Nat Genet 49(9):1297–1303
Hickey JM, Chiurugwi T, Mackay I, Powell W, Eggen A, Kilian A, Jones C, Canales C, Grattapaglia D, Bassi F, Atlin G (2017b) Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery. Nat Genet 49(9):1297
Hill WG, Robertson A (1966) The effect of linkage on limits to artificial selection. Genet Res 8(3):269–294
Hospital F (2003) Marker-assisted breeding. In: Newbury HJ (ed) Plant molecular breeding. Blackwell Publishing and CRC Press, Oxford/Boca Raton, pp 30–59
Hospital F, Charcosset M (1997) Marker-assisted introgression of quantitative loci. Genetics 147:1469–1485
Hufford MB, Lubinksy P, Pyhäjärvi T, Devengenzo MT, Ellstrand NC, Ross-Ibarra J (2013) The genomic signature of crop-wild introgression in maize. PLoS Genet 9(5):e1003477
Jain N, Singh GP, Singh PK, Ramya P, Krishna H, Ramya KT, Todkar L, Amasiddha B, Kumar KP, Vijay P, Jadon V (2014) Molecular approaches for wheat improvement under drought and heat stress
Jannink JL, Lorenz AJ, Iwata H (2010) Genomic selection in plant breeding: From theory to practice. Brief Funct Genomics 9(2):166–177
Jiang GL (2013) Molecular markers and marker-assisted breeding in plants. Plant breeding from laboratories to fields, pp 45–83
Jighly A, Lin Z, Pembleton LW, Cogan NO, Spangenberg GC, Hayes BJ, Daetwyler HD (2019) Boosting genetic gain in allogamous crops via speed breeding and genomic selection. Front Plant Sci 10:1364
Johnson R (2004) Marker assisted selection. In: Jannick J (ed), Plant Breed Rev 24(1):293–310
Joshi RK, Nayak S (2010) Gene pyramiding-A broad spectrum technique for developing durable stress resistance in crops. Biotechnol Mol Biol Rev 5(3):51–60
Kebede D, Mengesha W, Menkir A, Abe A, Garcia-Oliveira AL, Gedil M (2021) Marker based enrichment of provitamin A content in two tropical maize synthetics. Sci Rep 11(1):14998
Kicherer A, Herzog K, Töpfer R (2015) High-throughput phenotyping for trait detection in vineyards. In: BIO Web of Conferences (vol. 5, p 01018). EDP Sciences
Knapp SJ (1998) Marker-assisted selection as a strategy for increasing the probability of selecting superior genotypes. Crop Sci 38(5):1164–1174
Kolawole AO, Menkir A, Blay E, Ofori K, Kling JG (2018) Genetic advance in grain yield and other traits in two tropical maize composites developed via reciprocal recurrent selection. Crop Sci 58(6):2360–2369
Kole C (ed) (2013) Genomics and breeding for climate-resilient crops. New York: Springer
Kulkarni CC, Cholin SS, Bajpai AK, Ondrasek G, Mesta RK, Rathod S, Patil HB (2023) Comparative root transcriptome profiling and gene regulatory network analysis between Eastern and Western carrot (Daucus carota L.) cultivars reveals candidate genes for vascular tissue patterning. Plants 12:3449
Kumpatla SP, Buyyarapu R, Abdurakhmonov IY, Mammadov JA (2012) Genomics-assisted plant breeding in the 21st century: Technological advances and progress. In: Plant breeding. Intechopen
Kushanov FN, Turaev OS, Ernazarova DK, Gapparov BM, Oripova BB, Kudratova MK, Rafieva FU, Khalikov KK, Erjigitov DS, Khidirov MT et al (2021) Genetic diversity, QTL mapping, and marker-assisted selection technology in cotton (Gossypium Spp.). Front Plant Sci 12:779386
Kushwah A, Gupta S, Bindra S, Johal N, Singh I, Bharadwaj C, Dixit GP, Gaur PM, Nayyar H, Singh S (2020) Gene pyramiding and multiple character breeding. In Chickpea: Crop wild relatives for enhancing genetic gains (pp 131–165). Academic Press
Lande R, Thompson R (1990) Efficiency of marker-assisted selection in the improvement of quantitative traits. Genet 124(3):743–775
Larkan NJ, Lydiate DJ, Parkin IAP, Nelson MN, Epp DJ, Cowling WA, Rimmer SR, Borhan MH (2013) The B rassica napus blackleg resistance gene LepR3 encodes a receptor-like protein triggered by the L eptosphaeria maculans effector AVRLM 1. New Phytol 197(2):595–605
Li X, Zhu C, Wang J, Yu J (2012) Computer simulation in plant breeding. In: Advances in agronomy (Vol. 116, pp 219–264). Academic Press
Liu X, Wang H, Hu X, Li K, Liu Z, Wu Y, Huang C (2019) Enhancing genomic selection with quantitative trait loci and nonadditive effects revealed by empirical evidence in maize. Front Plant Sci 10:1129
Liu X, Wang H, Wang H, Guo Z, Xu X, Liu J, Wang S, Li WX, Zou C, Prasanna BM, Olsen MS (2018) Factors affecting genomic selection revealed by empirical evidence in maize. Crop J 6(4):341–352
Lorenz AJ (2013) Resource allocation for maximizing prediction accuracy and genetic gain of genomic selection in plant breeding: a simulation experiment. G3: Genes Genomes Genetics 3(3):481–491
Lush J (1937) Animal breeding. Iowa State College Press, Ames, Plans
Lyra DH, Granato ÍSC, Morais PPP, Alves FC, dos Santos ARM, Yu X, Guo T, Yu J, Fritsche-Neto R (2018) Controlling population structure in the genomic prediction of tropical maize hybrids. Mol Breeding 38(10):126
Majumdar GS, Rai A, Mishra DC (2020) Integrated framework for selection of additive and nonadditive genetic markers for genomic selection. J Comput Biol 27(6):845–855
Malav AK, Chandrawat I, Chandrawat KS (2016) Gene pyramiding: an overview. Int J Curr Res Biosci Plant Biol 3(7):22–28
Marcón F, Martínez EJ, Zilli AL, Rodríguez GR, Brugnoli EA, Acuña CA (2020) Recurrent phenotypic selection and recurrent selection based on combining ability in tetraploid bahiagrass. Crop Sci 60(3):1386–1397
Mayor PJ, Bernardo R (2009a) Doubled haploids in commercial maize breeding: One-stage and two-stage phenotypic selection versus marker-assisted recurrent selection. Maydica 54(4):439–448
Mayor PJ, Bernardo R (2009b) Genome wide selection and marker-assisted recurrent selection in doubled haploid versus F2 populations. Crop Sci 49(5):1719–1725
Merrick LF, Herr AW, Sandhu KS, Lozada DN, Carter AH (2022) Utilizing genomic selection for wheat population development and improvement. Agronomy 12(2):522
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genet 157:1819–1829
Mishra DC, Budhlakoti N, Majumdar SG, Rai A (2021) Innovations in genomic selection: Statistical perspective 101–111
Misztal I, Aguilar I, Lourenco D, Ma L, Steibel JP, Toro M (2021) Emerging issues in genomic selection. J Animal Sci 99(6):skab092
Morais OP (1997) Effective population size. In: Guimarães EP (ed), Recurrent selection in rice. (In Spanish.) Centro Int. Agric. Trop., Cali, Colombia
Moreau L, Charcosset A, Gallais A (1998) Marker-assisted selection efficiency in populations of finite size. Genetics 148(3):1353–1365
Moreau L, Charcosset A, Gallais A (2004) Experimental evaluation of several cycles of marker-assisted selection in maize. Euphytica 137(1):111–118
Muleta KT, Pressoir G, Morris GP (2019) Optimizing genomic selection for a sorghum breeding program in Haiti: a simulation study. G3: Genes Genomes Genetics 9(2):391–401
Müller D, Schopp P, Melchinger AE (2017) Persistency of prediction accuracy and genetic gain in synthetic populations under recurrent genomic selection. G3: Genes Genomes Genetics 7(3):801
Nadeem MA, Nawaz MA, Shahid MQ, Doğan Y, Comertpay G, Yıldız M, Hatipoğlu R, Ahmad F, Alsaleh A, Labhane N, Özkan H (2018) DNA molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing. Biotechnol Biotechnol Equip 32(2):261–285
Nakaya A, Isobe SN (2012) Will genomic selection be a practical method for plant breeding? Ann Bot 110(6):1303–1316
Nawaz MA, Yang SH, Rehman HM, Baloch FS, Lee JD, Park JH, Chung G (2017) Genetic diversity and population structure of Korean wild soybean (Glycine soja Sieb. and Zucc.) inferred from microsatellite markers. Biochem Syst Ecol 71:87–96
Nayak SN, Singh VK, Varshney RK (2017) Marker-assisted selection
Newbury HJ (2003) Plant molecular breeding. Blackwell Publishing/CRC Press, Birmingham
Ngcamphalala W (2018) Initiation of a wheat pre-breeding effort aimed at yield improvement using male-sterility marker assisted recurrent selection (Doctoral dissertation, Stellenbosch: Stellenbosch University)
Openshaw SJ, Frascaroli E (1997) QTL detection and marker-assisted selection for complex traits in maize. In: Proceedings of the 52nd Annual Corn and Sorghum Research Conference. pp 44–53. (American Seed Trade Association, Washington D.C., USA)
Peleman JD, van der Voort JR (2003) The challenges in marker assisted breeding. Eucarpia leafy vegetables. Center for Genetic Resources, The Netherlands
Perveen N, Cholin SS, Hipparagi K, Prabhuling G, Murthy BNS, Peerjade D (2023) Molecular diversity assessment among the pomegranate genotypes belonging to diverse genetic background using microsatellite markers. Acta Physiol Plant 45:92
Platten JD, Cobb JN, Zantua RE (2019) Criteria for evaluating molecular markers: Comprehensive quality metrics to improve marker-assisted selection. PLoS ONE 14(1):e0210529. https://doi.org/10.1371/journal.pone.0210529
Prasanna BM, Cairns J, Xu Y (2013) Genomic tools and strategies for breeding climate resilient cereals. In: Genomics and Breeding for Climate-resilient Crops (pp 213–239). Springer, Berlin, Heidelberg
Pretorius ZA, Pakendorf KW, Marais GF, Prins R, Komen JS (2007) Challenges for sustainable cereal rust control in South Africa. Aust J Agric Res 58(6):593–601
Ragimekula N, Varadarajula NN, Mallapuram SP, Gangimeni G, Reddy RK, Kondreddy HR (2013) Marker assisted selection in disease resistance breeding. J Plant Breed Genet 1(2):90–109
Ragot M, Gay G, Muller JP, Durovray J (2000) Efficient selection for the adaptation to the environment through QTL mapping and manipulation in maize. Molecular approaches for the genetic improvement of cereals for stable production in water-limited environments, pp 128–130
Rahman M, Davies P, Bansal U, Pasam R, Hayden M, Trethowan R (2020) Marker-assisted recurrent selection improves the crown rot resistance of bread wheat. Mol Breeding 40:1–14
Rembe M, Reif JC, Ebmeyer E, Thorwarth P, Korzun V, Schacht J, Boeven PH, Varenne P, Kazman E, Philipp N, Kollers S (2021) Reciprocal recurrent genomic selection is impacted by genotype-by-environment interactions. Front Plant Sci 12:703419
Rembe M, Zhao Y, Jiang Y, Reif JC (2019) Reciprocal recurrent genomic selection: an attractive tool to leverage hybrid wheat breeding. Theor Appl Genet 132:687–698
Ribaut J, Betrán J (1999) Single large-scale marker-assisted selection (SLS-MAS). Mol Breed 5:531–541
Ribaut JM, Ragot M (2007) Marker-assisted selection to improve drought adaptation in maize: the backcross approach, perspectives, limitations, and alternatives. J Exp Bot 58(2):351–360
Riedelsheimer C, Endelman JB, Stange M, Sorrells ME, Jannink JL, Melchinger AE (2013) Genomic predictability of interconnected biparental maize populations. Genetics 194(2):493–503
Rodenburg RJ (2018) The functional genomics laboratory: F unctional validation of genetic variants. J Inherit Metab Dis 41(3):297–307
Saavedra LM, Caixeta ET, Barka GD, Borém A, Zambolim L, Nascimento M, Cruz CD, Oliveira ACBD, Pereira AA (2023) Marker-assisted recurrent selection for pyramiding leaf rust and coffee berry disease resistance alleles in Coffea arabica L. Genes 14(1):189
Sakiyama NS, Ramos HCC, Caixeta ET, Pereira MG (2014) Plant breeding with marker-assisted selection in Brazil. Crop Breed Appl Biotechnol 14(1):54–60
Salgotra RK, Stewart CN Jr (2020) Functional markers for precision plant breeding. Int J Mol Sci 21(13):4792
Sandhu N, Dixit S, Swamy BM, Vikram P, Venkateshwarlu C, Catolos M, Kumar A (2018a) Positive interactions of major-effect QTLs with genetic background that enhances rice yield under drought. Sci Rep 8(1):1–13
Sandhu N, Dixit S, Swamy BM, Vikram P, Venkateshwarlu C, Catolos M, Kumar A (2018b) Positive interactions of major-effect QTLs with genetic background that enhances rice yield under drought. Sci Rep 8(1):1626
Santa Catarina R, Pereira MG, Vettorazzi JCF, Cortes DFM, de Sousa Poltronieri TP, Azevedo AON, Moreira NF, Miranda DP, de Moraes R, Pirovani AAV, Ramos HCC (2020) Papaya (Carica papaya L.) S1 family recurrent selection: Opportunities and selection alternatives from the base population. Scientia Horticulturae 260:108848
Sawler J, Reisch B, Aradhya MK, Prins B, Zhong GY, Schwaninger H, Simon C, Buckler E, Myles S (2013) Genomics assisted ancestry deconvolution in grape. PLoS ONE 8(11):e80791
Scheuermann KK, Jia Y (2016) Identification of a Pi9-containing rice germplasm with a newly developed robust marker. Phytopathology 106(8):871–876
Schopp P, Müller D, Technow F, Melchinger AE (2017) Accuracy of genomic prediction in synthetic populations depending on the number of parents, relatedness, and ancestral linkage disequilibrium. Genetics 205(1):441–454
Schuster I (2011) Marker-assisted selection for quantitative traits. Crop Breed Appl Biotechnol S1:50–55
Scott MF, Ladejobi O, Amer S, Bentley AR, Biernaskie J, Boden SA, Clark M, Dell’Acqua M, Dixon LE, Filippi CV, Fradgley N (2020) Multi-parent populations in crops: a toolbox integrating genomics and genetic mapping with breeding. Heredity 1–21
Sebolt AM, Shoemaker RC, Diers BW (2000) Analysis of a quantitative trait locus allele from wild soybean that increases seed protein concentration in soybean. Crop Sci 40(5):1438–1444
Sehgal D, Rosyara U, Mondal S, Singh R, Poland J, Dreisigacker S (2020) Incorporating genome-wide association mapping results into genomic prediction models for grain yield and yield stability in CIMMYT spring bread wheat. Front Plant Sci 11:197
Sekine D, Tsuda M, Yabe S, Shimizu T, Machita K, Saruta M, Yamada T, Ishimoto M, Iwata H, Kaga A (2021) Improving quantitative traits in self-pollinated crops using simulation-based selection with minimal crossing. Front Plant Sci 1859
Semagn K, Beyene Y, Babu R, Nair S, Gowda M, Das B, Tarekegne A, Mugo S, Mahuku G, Worku M, Warburton ML (2015) Quantitative trait loci mapping and molecular breeding for developing stress resilient maize for Sub-Saharan Africa. Crop Sci 55(4):1449–1459
Shamshad M, Sharma A (2018) The usage of genomic selection strategy in plant breeding. Next generation plant breeding 93
Singh J, Kaur S, Majithia H (2016) Emerging genetic technologies for improving the security of food crops. In: Emerging Technologies for Promoting Food Security (pp 23–41). Woodhead Publishing
Singh NK, Joshi A, Sahoo S, Tufchi M, Rakshit S (2023) Molecular breeding for improving yield in maize: Recent advances and future perspectives. In: QTL Mapping in Crop Improvement, pp 75–99
Sinha D, Maurya AK, Abdi G, Majeed M, Agarwal R, Mukherjee R, Ganguly S, Aziz R, Bhatia M, Majgaonkar A, Seal S (2023) Integrated genomic selection for accelerating breeding programs of climate-smart cereals. Genes 14(7):1484
Slater AT, Wilson GM, Cogan NO, Forster JW, Hayes BJ (2014) Improving the analysis of low heritability complex traits for enhanced genetic gain in potato. Theor Appl Genet 127:809–820
Smith C, Simpson SP (1986) The use of genetic polymorphisms in livestock improvement. J Anim Breed Genet 103:205–217
Smýkal P, Nelson MN, Berger JD, Von Wettberg EJ (2018) The impact of genetic changes during crop domestication on healthy food development. Agronomy 8(3):26
Song L, Wang R, Yang X, Zhang A, Liu D (2023) Molecular markers and their applications in marker-assisted selection (MAS) in bread wheat (Triticum aestivum L.). Agriculture 13(3):642
Sonnino A, Carena MJ, Guimarães EP, Baumung R, Pilling D, Rischkowsky B (2007) An assessment of the use of molecular markers in developing countries. Marker-assisted selection: Current status and future perspectives in crops, livestock, forestry and fis. Rome, Food and Agriculture Organization of the United Nations, pp 15–26
Souza Jr CLD, Barrios SCL, Moro GV (2010) Performance of maize single-crosses developed from populations improved by a modified reciprocal recurrent selection. Scientia Agricola 67:198–205
Spasibionek S, Mikołajczyk K, Ćwiek–Kupczyńska H, Piętka T, Krótka K, Matuszczak M, Nowakowska J, Michalski K, Bartkowiak-Broda I (2020) Marker assisted selection of new high oleic and low linolenic winter oilseed rape (Brassica napus L.) inbred lines revealing good agricultural value. PLoS One 15(6):e0233959
Spindel JE, Begum H, Akdemir D, Collard B, Redoña E, Jannink JL, McCouch S (2016) Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity 116(4):395–408
Sprague GF, Eberhart SA (1977) Corn breeding in corn and corn improvement (Sprague, GF, ed.: 305–362). Wisconsin, USA: American Society of Agronomy
Stam P (1995) Marker-assisted breeding. In J.W. Van Ooijen & J. Jansen, eds. Biometrics in plant breeding: applications of molecular markers. Proc. 9th Mtg. EUCARPIA Section on Biometrics in Plant Breeding, 32–44. Wageningen, Netherlands, CPRO-DLO
Stephen PM, Rita HM (2008) Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol 147(3):969–977
Suvarna Ashwini K, Yashaswini R (2023) Marker assisted recurrent selection for crop improvement. In: Molecular Marker Techniques: A Potential Approach of Crop Improvement (pp. 55–67). Singapore: Springer Nature Singapore
Tanaka E (2020) Simple outlier detection for a multi-environmental field trial. Biometrics 76(4):1374–1382
Tardin FD, Pereira MG, Gabriel APC, do Amaral Júnior AT, de Souza Filho GA (2007) Selection index and molecular markers in reciprocal recurrent selection in maize
Thudi M, Gaur PM, Krishnamurthy L, Mir RR, Kudapa H, Fikre A, Kimurto P, Tripathi S, Soren KR, Mulwa R, Bharadwaj C (2014) Genomics-assisted breeding for drought tolerance in chickpea. Funct Plant Biol 41(11):1178–1190
Tourrette E, Bernardo R, Falque M, Martin OC (2019) Assessing by modeling the consequences of increased recombination in recurrent selection of Oryza sativa and Brassica rapa. G3: Genes Genomes Genetics 9(12):4169–4181
van Berloo R, Stam P (2001) Simultaneous marker-assisted selection for multiple traits in autogamous crops. Theor Appl Genet 102(6–7):1107–1112
Varshney RK, Bohra A, Yu J, Graner A, Zhang Q, Sorrells ME (2021) Designing future crops: Genomics-assisted breeding comes of age. Trends Plant Sci 26(6):631–649
Varshney RK, Ribaut JM, Buckler ES, Tuberosa R, Rafalski JA, Langridge P (2012) Can genomics boost productivity of orphan crops? Nat Biotechnol 30(12):1172–1176
Varshney RK, Singh VK, Kumar A, Powell W, Sorrells ME (2018) Can genomics deliver climate-change ready crops? Curr Opin Plant Biol 45:205–211
Vasal SK, Singh NN, Dhillon BS, Patil SJ (2004) Population improvement strategies for crop improvement. In: Plant Breeding (pp 391–406). Springer, Dordrecht
Wang B, Ding Q, Fu X, Kang IS, Jin K, Shukla J, Doblas‐Reyes F (2005) Fundamental challenge in simulation and prediction of summer monsoon rainfall. Geophys Res Let 32(15)
Wang J, Wan X, Li H, Pfeiffer WH, Crouch J, Wan J (2007) Application of identified QTL-marker associations in rice quality improvement through a design-breeding approach. Theor Appl Genet 115(1):87–100
Wang X, Xu Y, Hu Z, Xu C (2018) Genomic selection methods for crop improvement: Current status and prospects. The Crop Journal 6(4):330–340
Watson A, Ghosh S, Williams MJ, Cuddy WS, Simmonds J, Rey MD, Asyraf Md Hatta M, Hinchliffe A, Steed A, Reynolds D, Adamski NM (2018) Speed breeding is a powerful tool to accelerate crop research and breeding. Nat Plants 4(1):23–29
Worthington ML, Miles JW (2015) Reciprocal full-sib recurrent selection and tools for accelerating genetic gain in apomictic Brachiaria. In Molecular Breeding of Forage and Turf: The Proceedings of the 8th International Symposium on the Molecular Breeding of Forage and Turf (pp 19–30). Springer International Publishing
Xie C, Xu S (1998) Strategies of marker aided recurrent selection. Crop Sci 38:1526–1535
Xu Y, Crouch JH (2008) Marker-assisted selection in plant breeding: From publications to practice. Crop Sci 48(2):391–407
Xu Y (2011) From line to space: a 3-D profile of molecular plant breeding. In The first congress of cereal biotechnology and breeding
Xu Y (2012) Environmental assaying or e-typing as a key component for integrated plant breeding platform. In Marker-assisted selection workshop, 6th international crop science congress
Xu Y, Li P, Zou C, Lu Y, Xie C, Zhang X, Prasanna BM, Olsen MS (2017) Enhancing genetic gain in the era of molecular breeding. J Exp Bot 68(11):2641–2666
Xu Y, Liu X, Fu J, Wang H, Wang J, Huang C, Prasanna BM, Olsen MS, Wang G, Zhang A (2020a) Enhancing genetic gain through genomic selection: From livestock to plants. Plant Commun 1(1):100005
Xu Y, Liu X, Fu J, Wang H, Wang J, Huang C, Prasanna BM, Olsen MS, Wang G, Zhang A (2020) Enhancing genetic gain through genomic selection: from livestock to plants. Plant Commun 1(1)
Yang H, Li C, Lam HM, Clements J, Yan G, Zhao S (2015) Sequencing consolidates molecular markers with plant breeding practice. Theor Appl Genet 128:779–795
Yang W, Zheng Y, Zheng W, Feng R (2005) Molecular genetic mapping of a high-lysine mutant gene (opaque-16) and the double recessive effect with opaque-2 in maize. Mol Breeding 15(3):257–269
Yi C, Guo W, Zhu X, Min L, Zhang T (2004) Pyramiding breeding by marker assisted recurrent selection in upland cotton II. Selection effects on resistance to Helicoverpa armigera. Scientia Agricultura Sinica 37:801–807
Zhang A, Wang H, Beyene Y, Semagn K, Liu Y, Cao S, Cui Z, Ruan Y, Burgueño J, San Vicente F, Olsen M (2017) Effect of trait heritability, training population size and marker density on genomic prediction accuracy estimation in 22 bi-parental tropical maize populations. Front Plant Sci 8:1916
Zhang H, Yin L, Wang M, Yuan X, Liu X (2019) Factors affecting the accuracy of genomic selection for agricultural economic traits in maize, cattle, and pig populations. Front Genet 10:189
Zhao X, Li B, Zhang K, Hu K, Yi B, Wen J, Ma C, Shen J, Fu T, Tu J (2016) Breeding signature of combining ability improvement revealed by a genomic variation map from recurrent selection population in Brassica napus. Sci Rep 6(1):29553
Zhu Z, Chen L, Zhang W, Yang L, Zhu W, Li J, Liu Y, Tong H, Fu L, Liu J, Rasheed A (2020) Genome-wide association analysis of Fusarium head blight resistance in Chinese elite wheat lines. Front Plant Sci 11:206
Funding
The idea of implementing the MARS research is the extension of the project supported by the Department of Biotechnology (DBT), Government of India under DBT-BIO-CARe (File No: 102/IFD/SAN/3308/2014-15). CCK greatly acknowledges KSTePS, Ph.D. Fellowship of Dept of Science and Technology (AGR08:2019-20)-Govt of Karnataka.
Author information
Authors and Affiliations
Contributions
SSC conceived the idea, prepared the draft, and corrected the final version of the manuscript; KCC collected literature and assisted in the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Communicated by: Ray Ming
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cholin, S.S., Kulkarni, C.C. Prospects of Marker-Assisted Recurrent Selection: Current Insights and Future Implications. Tropical Plant Biol. 16, 259–275 (2023). https://doi.org/10.1007/s12042-023-09348-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-023-09348-8