
Abstract
A permanent mapping population of rice consisting of 65 non-idealized chromosome segment substitution lines (denoted as CSSL1 to CSSL65) and 82 donor parent chromosome segments (denoted as M1 to M82) was used to identify QTL with additive effects for two rice quality traits, area of chalky endosperm (ACE) and amylose content (AC), by a likelihood ratio test based on stepwise regression. Subsequently, the genetics and breeding simulation tool QuLine was employed to demonstrate the application of the identified QTL in rice quality improvement. When a LOD threshold of 2.0 was used, a total of 16 chromosome segments were associated with QTL for ACE, and a total of 15 segments with QTL for AC in at least one environment. Four target genotypes denoted as DG1 to DG4 were designed based on the identified QTL, and according to low ACE and high AC breeding objectives. Target genotypes DG1 and DG2 can be achieved via a topcross (TC) among the three lines CSSL4, CSSL28, and CSSL49. Results revealed that TC2: (CSSL4 × CSSL49) × CSSL28 and TC3: (CSSL28 × CSSL49) × CSSL4 resulted in higher DG1 frequency in their doubled haploid populations, whereas TC1: (CSSL4 × CSSL28) × CSSL49 resulted in the highest DG2 frequency. Target genotypes DG3 and DG4 can be developed by a double cross among the four lines CSSL4, CSSL28, CSSL49, and CSSL52. In a double cross, the order of parents affects the frequency of target genotype to be selected. Results suggested that the double cross between the two single crosses (CSSL4 × CSSL28) and (CSSL49 × CSSL52) resulted in the highest frequency for DG3 and DG4 genotypes in its derived doubled haploid derivatives. Using an enhancement selection methodology, alternative ways were investigated to increase the target genotype frequency without significantly increasing the total cost of breeding operations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rapid progress in the development of polymorphic molecular markers has led to the intensive use of QTL mapping in genetic study for quantitative traits (Lander and Botstein 1989; Zeng 1994; Piepho 2000; Dekkers and Hospital 2002; van Eeuwijk et al. 2002; Wan et al. 2004, 2005; Wang et al. 2006). The mapping populations required for QTL detection, such as F2, backcross, recombination inbred lines (RIL), and doubled haploids (DH), can be classified into two categories, temporary populations and permanent populations. In a permanent population such as RIL and DH, each individual in the population is genetically homozygous at all loci, and the genetic composition will not change during self-pollination. Thus, permanent populations allow precisely determining the phenotype of complex quantitative traits through replicated experiments, and the same genotype can be repeatedly tested under different environments to the study of genotype or QTL × environment interaction (Piepho 2000; van Eeuwijk et al. 2002).
More recently, permanent populations consisting of series of chromosome segment substitution (CSS) lines have been used for gene introgression and fine mapping (Eshed and Zamir 1995; Tanksley and Nelson 1996; Nadeau et al. 2000; Kubo et al. 2002; Belknap 2003; Cowley et al. 2003). The advantage of such mapping population is that segregation in each individual is restricted to a small segment of the genome, whereas the individuals in RIL and DH populations may differ at loci across the entire genome. Consequently, confounding interaction effects are much smaller in CSS lines as compared to RIL and DH populations.
QTL discovery and variety development are in general separate processes, and how QTL mapping results can be used to pyramid desired alleles at various loci has only been rarely addressed in the literature (Tanksley and Nelson 1996; Young 1999). Further, most QTL-mapping results are rarely used by breeders for various reasons. These include the lack of information on QTL by environment interactions, which is related to yield stability and hence one of the major generic-breeding objectives (Cooper et al. 2005). In addition, information on gene action of QTL in the new genetic background which is different from the background in which the original marker discovery research was carried out is often lacking (Mackill and Ni 2001). Hence, the linkage phase is unknown and generally extensive validation of the marker-trait linkage phase is required across all parental stocks commonly used by breeders. Third, breeders prefer diagnostic markers or markers closely linked to target QTL, as both false positives and false negatives can seriously affect the impact of marker-assisted selection in a breeding program. Finally, an additional reason for the reluctance in applying QTL results to crop improvement is the lack of appropriate tools that can combine the different types of biological information and turn complex and voluminous data into knowledge that can be applied in breeding.
QuLine (previously called QuCim) is a genetics and breeding simulation tool that allows integrating various genes with multiple alleles which operate within epistatic networks and differentially interact with the environment interaction, and predict the outcomes from a specific cross following the application of a real selection scheme (Wang et al. 2003, 2004). It therefore has the potential to bridge the vast amount of biological data and breeder’s queries optimizing selection gain and efficiency. QuLine has been used to compare two different selection strategies (Wang et al. 2003), study the effects on selection of dominance and epistasis (Wang et al. 2004), predict cross performance using known gene information (Wang et al. 2005), and optimize marker- assisted selection to efficient pyramid multiple genes (Wang et al. 2007).
The objectives of this study were (1) to re-analyse the rice QTL mapping dataset used in Wan et al. (2004, 2005) by a novel mapping method with non-idealized CSS lines to identify QTL with additive effects for two quality traits, i.e., the area of chalky endosperm (ACE) and amylose content (AC), and (2) to utilize the identified marker-QTL associations in designing a breeding methodology to develop low ACE and high AC rice inbred lines.
Material and methods
A CSS population consisting of 65 non-idealized lines
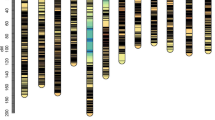
A permanent population consisting of 65 rice CSS lines (denoted as CSSL1 to CSSL65) and 82 chromosome segments (denoted as M1 to M82) was used as the mapping population in this study (Fig. 1). The two parents are the Oryza sbsp. japonica rice variety Asominori (the background or recurrent parent) and the indica rice variety IR24 (the donor parent) (for details about this population, see Tsunematsu et al. 1996; Kubo et al. 2002; Wan et al. 2004). Each chromosome segment was represented by a marker, and the linkage distance between the 82 markers is given in Table 1. Each CSS line in the population contains one or a few segments from the donor parent. On average, each substitution segment exists in 3.7 CSS lines, and each CSS line carries 4.6 segments from the donor parent (calculated from Fig. 1).

A CSS population consisting of 65 CSS lines and 82 chromosome segments derived from the two rice parents, japonica Asominori and indica IR24, where Asominori is the background parent (represented by 0) and IR24 is the donor parent (represented by 2)
Phenotypic data of ACE and AC
Rice quality is a complex trait consisting of many components such as milling, appearance, nutrition, cooking and eating qualities. Among these qualitative properties, consumers pay more attention to the fine appearance and eating quality (Huang et al. 1998; Wan et al. 2004). For the improvement of appearance, milling and edibility, the endosperm of high-quality rice varieties should be free of chalkiness (low or zero ACE), since chalky grains have a lower starch granule density compared to vitreous grains, and are therefore more prone to breakage during milling. In addition, chalkiness reduces the palatability of cooked rice since longitudinal and transverse cracks occur easily in chalky kernels when the grain is steamed or boiled. It has been established that AC is the most important factor affecting edibility of rice, and low ACE and high AC are generally favored in rice quality improvement. It has to be noted that rice end-use quality and the breeding objectives are varied due to different consumer preferences and traditions in different countries.
The 65 CSS lines along with their two parents were grown in two replications in eight different environments, denoted as E1 to E8 (Wan et al. 2004, 2005). ACE and AC were assessed using accepted methods.
A likelihood ratio test based on stepwise regression for QTL mapping with non-idealized chromosome segment substitution lines
The standard t-test used in the idealized case that each CSS line contains a single chromosome segment from the donor parent (Belknap 2003) is not suitable for CSS line carrying more than one segment from the donor parent as shown in Fig. 1. Due to high intensity selection in the process of generating CSS lines, the gene and marker frequencies do not follow the same path as in a standard unselected mapping population. Similarly, standard mapping methods such as interval mapping (Lander and Botstein 1989), composite interval mapping (Zeng 1994), and inclusive composite interval mapping (Li et al. 2007) are not suitable either. We propose a likelihood ratio test based on stepwise regression (abbreviated as RSTEP-LRT hereafter, available from http://www.isbreeding.net/software.html), where stepwise regression is used initially to select the most important chromosome segments for the trait of interest, and followed by the likelihood ratio test to calculate the LOD score of each segment. Three major steps are included in the RSTEP-LRT method (Wang et al. 2006).
Step1: Reducing multicollinearity among marker variables
When using the linear regression model of phenotype on marker variable in a non-idealized CSS population, the multicollinearity among markers is an obvious problem. The level of multicollinearity can be assessed by the variable inflation factor and the condition number (Myers 1990). In this study we consider the condition number, which is defined as k = λmax /λmin, where λmax and λmin are the maximum and minimum eigenvalues of the correlation matrix between markers, respectively. As an informal rule (Myers 1990), 1,000 was used as the threshold value for the condition number in QTL mapping with CSS lines. The sequential process for decreasing multicollinearity was: if marker pairs have a perfect correlation, (i.e., correlation coefficient equal to 1) one of them can be randomly deleted; if the correlation is high, but not perfect, the marker present in more CSS lines than the other marker is deleted, so that the mapping population will gradually approach an idealized one. The procedure was repeated until the threshold condition number of 1,000 was reached (for more details see Wang et al. 2006).
Step 2: Identifying the most important marker variables using stepwise regression
Suppose two parents, P 1 (the background parent), and P 2 (the donor parent) differ in t markers. A total of n CSS lines were derived from the two parents through advanced backcrossing and marker-assisted background selection. As a result, most of the chromosome segments in these CSS lines are expected to derive from the background parent P 1. The phenotypic value for a quantitative trait of interest of the jth CSS line, y j , can be represented by a linear model, i.e., \(y_j = b_0 + \sum_{i = 1}^t {b_i x_{ji}} + e_j ,\) where j = 0 (for the background parent), 1, 2, ..., n, b 0 is the intercept, b i (i = 1, ..., t) is the partial regression coefficient of the phenotype on the ith marker, x ji is the indicator variable for the ith marker in the jth CSS line, which is equal to −1 if the marker type is the same as in P 1, and 1 if the marker type is the same as in P 2, and e j is the random experimental error. Stepwise regression was used to select the most important marker variables and determine the estimates for the parameters in the above-shown linear model. The largest p-value for entering variables was set at 0.05, and the smallest p-value for removing variables was set at 0.10.
Step 3: Calculating LOD score of each chromosome segment
Assuming there is one QTL on the ith chromosome segment with two alleles q and Q, where q is present in P 1, and Q in P 2. The observation values can be adjusted by \(\Delta y_j = y_j - \sum_{k \ne i} {b_k x_{jk}},\) which maintains the QTL information on the current segment, but at the same time eliminates the effects from QTL on other segments. Rearrange Δy j and let j = 0, 1, 2, ..., n 1 represent the CSS lines having P 1 marker type, and j = n 1 + 1, n 1 + 2, ..., n refer to the CSS lines having P 2 marker type. Thus Δy j follows the distribution N(μ1, σ 2 A ) for j = 0, 1, 2, ..., n 1 and the distribution N(μ2, σ 2 A ) for j = n 1 + 1, n 1 + 2, ..., n, where N(μ1, σ 2 A ) and N(μ2, σ 2 A ) represent the normal distributions of the two QTL genotypes qq and QQ, respectively. The existence of QTL in the current chromosome segment can be tested by the following hypotheses H 0 :μ1 = μ2 versus H A :μ1 ≠ μ2. Under the null hypothesis H 0, Δy j follows the same normal distribution denoted by N(μ0, σ 20 ). The mean and variance of this distribution can be estimated as,
The log-likelihood function under H 0 can be calculated as,
where f(Δy j ;μ0, σ 20 ) is the density function for the normal distribution N(μ0, σ 20 ). The log-likelihood function under the alternative hypothesis H A is,
where
Therefore, the likelihood ratio test can be derived from the two likelihoods under the two hypotheses, from which the LOD score of the current chromosome segment can be calculated.
The genetics and breeding simulation tool QuLine
QuLine is a QU-GENE application module that was specifically developed to simulate the wheat-breeding programs at the International Maize and Wheat Improvement Center (CIMMYT) (Podlich and Cooper 1998; Wang et al. 2003, 2004), but has the potential to simulate most, if not all, breeding methodologies for developing inbred lines. The breeding methods that can be simulated in QuLine include mass selection, pedigree breeding (including single seed descent), bulk population breeding, backcross breeding, topcross (or three-way cross) breeding, doubled haploid breeding, marker-assisted selection, and combinations and modifications of these methods. The chromosomal locations of genes and markers, and their occurrence in specific parents can be explicitly and precisely defined. Simulation experiments can therefore be designed to compare the breeding efficiency of different selection strategies under a series of pre-determined genetic models.
QuLine was used in this study to simulate topcrosses among three selected CSS lines, doublecrosses among four selected CSS lines, and to generate the segregating breeding populations. Then various selection schemes were applied in different generations. The target genotype was selected by markers. As an example, the simulation process for a topcross and three selection schemes are shown in Fig. 2. The three selection schemes require the same amount of DNA samples. As marker analysis is becoming increasingly cheaper, the costs of plant/line operations such as handling, tagging, leaf sampling and DNA extraction constitute a major part of the costs in marker-assisted breeding. Therefore the three selection schemes may require similar financial resources.

Diagram of the crossing and selection process of a top cross. For Scheme 1, selection was conducted with 1,000 DH lines derived from 200 TCF1 individuals. For Scheme 2, an enhancement selection was first conducted with 200 TCF1 individuals. Then 800 DH lines were derived from those retained individuals. Finally, the target genotypes were selected with the 800 DH lines. For Scheme 3, an enhancement selection was first conducted with the 200 TCF1 individuals. Then 200 TCF2 individuals were derived from those retained TCF1 individuals. Enhancement selection was applied again with the 200 TCF2 individuals. Finally, the 600 DH lines were derived from those retained TCF2 individuals, from which the target genotypes were selected
Results
Threshold LOD score of QTL mapping with non-idealized CSS lines
A problem common to QTL mapping methods is the difficulty determining the threshold LOD score, or likelihood ratio (LR) test (Lander and Botstein 1989; Zeng 1994; Churchill and Doerge 1994). A LOD threshold between 2 and 3 was proposed in Lander and Botstein (1989) to ensure an overall false positive rate of 5%. A permutation test was recommended by Churchill and Doerge (1994) to determine an appropriate threshold for the experimental data at hand. When permutation tests were applied in RSTEP-LRT, the probability of LOD > 2.0 was found to be lower than 0.05 in any environment for ACE (Table 2). The probability was also lower than 0.05 for AC in all environments except E4 (Table 2).
For a specific mapping population, higher threshold values result in a lower power, and therefore QTL with small effects cannot be readily identified. Lower thresholds result in higher powers, but also endure higher false positive rates. The appropriate significance level to be used depends on the purpose of the mapping experiment. If QTL mapping is performed with the eventual goal of cloning QTL or introgressing a few QTL with large effects, a stringent threshold of LOD such as 3.00 should be used for QTL mapping with a CSS population. However, if the goal is to exploit QTL information in marker-assisted selection for a complex trait, a less stringent LOD threshold such as 2.0 may be appropriate, as the false positive will have limited influence on the results from marker-assisted selection. Therefore, the threshold LOD of 2.0 was applied when we conducted the QTL mapping for ACE and AC, according to our goal to derive rice inbred lines combining multiple favorable alleles through marker-assisted selection.
QTL for ACE and AC estimated from 65 non-idealized CSS lines
The donor parent IR24 has larger ACE and higher AC compared with the background parent Asominori. Transgressive segregation was observed for both traits in the CSS population in all environments (results not shown). ANOVA showed that there were significant differences on ACE and AC among the 65 CSS lines and Asominori. Environmental effects and genotype by environment interactions were also significant. For ACE, the estimated genotypic variance (i.e., 15.21) was almost three times the interaction variance (i.e., 5.39), while the environmental variance was relatively small (i.e., 1.73). For AC, the environmental variance was the largest variance component (i.e., 1.29), and the genotypic variance (i.e., 0.65) was about twice the magnitude of the interaction variance (i.e. 0.31). Due to significant genotype by environment interactions, QTL mapping was separately conducted for each environment.
When the LOD threshold of 2.0 was applied, a total of 16 chromosome segments demonstrated the existence of QTL for ACE in at least one environment. These QTL are distributed on 9 of the 12 rice chromosomes, one each on chromosomes 1, 2, 9, and 10, two each on chromosomes 3, 5, and 12, and three each on chromosomes 7 and 8. QTL on M57 and M59 were detected in seven environments (Table 3), and the alleles from IR24 at the two loci constantly increased ACE in all environments, indicating they both have a high stability for ACE. The QTL on M57 explained from a minimum of 6.60% of phenotypic variance in E6 (the least) to a maximum of 35.08% in E8 (the highest), and the QTL on M59 explained a minimum of 1.00% of phenotypic variance in E1 (the least) to a maximum of 16.93% in E6 (the highest). The remaining QTL were only detected in one to three environments (Table 3). These minor QTL have smaller effects compared with QTL on M57 and M59 and the effects depend on environment (Table 3), which may be called minor QTL. It should be noted that the minor QTL on M35 was detected only in E8, but had high stability, constantly decreasing ACE in all environments. Such minor QTL should also be considered in gene pyramiding to maximize the genetic gain once major QTL have been fixed.
A total of 15 chromosome segments demonstrated the existence of QTL for AC in at least one environment. These QTL are also distributed on nine chromosomes, one each on chromosomes 2, 6, 8, 11 and 12, two each on chromosomes 3, 9 and 10, and four on chromosome 1 (Table 4). The QTL on M57 were detected in eight environments, and the QTL on M59 was detected in seven environments. The alleles from IR24 at the two loci constantly increased AC in all environments, indicating their high stability on AC. The QTL on M57 explained from a minimum of 9.39% of phenotypic variance in E5 (the least) to a maximum of 31.54% in E2 (the highest), and the QTL on M59 explained from a minimum of 0.24% of phenotypic variance in E6 (the least) to a maximum of 16.02% in E7 (the highest). Other QTL were only detected in one to four environments (Table 4). In the case of AC, a minor QTL on M60 was detected in E1, E3, E7, and E8, and the allele from IR24 constantly increased AC in all environments, and can be considered as another stable minor QTL.
QTL on M4, M23, M57, M59, and M79 showed effects on both traits, and QTL on M57 and M59 are the two major genes increasing ACE and AC simultaneously. This is in agreement with the high-positive correlation (r = 0.69**) between ACE and AC in the CSS population.
Design of the target genotype based on the identified marker-QTL associations
Not all QTL in Tables 3 and 4 are useful in designing the target genotype if the breeding objective is to develop inbred lines with improved performance across all environments. Implicitly, the two major QTL on chromosome segments M57 and M59 are most important due to their high stability. To maximize genetic gain, we also considered QTL with significant effects in three or more environments. They are QTL on M23 and M56 affecting ACE, and QTL on M4, M14, and M60 affecting AC (Tables 3, 4). The QTL on M23 decreases ACE in E1 and E3, but increases ACE in other environments. However the effects in E1 and E3, are relatively small compared with those in other environments (Table 3). Similarly, the QTL on M56 has negative effects on ACE in E4 and E8, but these effects are minor compared to its positive effect in other environments (Table 3). The negative effect of the QTL on M14 on AC is relatively small compared to the positive effects in other environment (Table 4), and effects of the two QTL on M14 and M60 have the same direction on AC (Table 4), hence they are relevant in improving the genetic gain on AC. As stated before, the QTL on M35 constantly decreases ACE in all environments and was considered in designing the target genotype.
QTL described above and their average effects on ACE and AC are given in Table 5, which allowed acquiring the target genotype with relatively low ACE and high AC. With these gene effects, the correlation coefficient between observed values and predicted values is 0.812 for ACE, and 0.809 for AC (Fig. 3). The observed ACE and AC of Asominori are 4.76 and 15.12%, while the predicted ACE and AC are 6.05 and 15.58%, respectively (Table 5).

Correlation relationship between observation and prediction from identified QTL for ACE and AC
For QTL with no pleiotropic effects, the direction of their average additive effects (Table 5) permits easy determination whether the target genotype should have the allele from Asominori or IR24. The alleles from IR24 on segments M23 and M56 have positive effects on ACE. Therefore, the segments from Asominiro should be present in the target genotype so as to reduce ACE. In contrast, the allele from IR24 on segments M35 has negative effect on ACE, and the segment from IR24 should be present. Based on the same principle, the IR24 segments of M4 and M60, and the Asominori segment of M14 should be present in the target genotype to increase AC (Table 5).
The two QTL on M57 and M59 have pleiotropic effects on both traits and so we gave the four possible genotypes, denoted as DG1 to DG4 (Table 5). As expected, DG1 has the lowest-predicted genetic values on ACE, and DG4 has the highest-predicted genetic values on AC. Compared with Asominori, DG1 has lower ACE but higher AC, which can be considered as an improvement of Asominori. DG2, DG3, and DG4 have high values on both ACE and AC. When maximizing AC is the breeding objective while ACE is less important, DG3 and DG4 may be used as target genotypes.
Achievement of the target genotype by optimum crossing strategies
This section illustrates the most efficient methodology in selecting the four designed genotypes (Table 5) using QuLine.
From their genotypic constitution, we can see DG1 and DG2 can be achieved from a topcross among the three lines CSSL4, CSSL28, and CSSL49. There are three options to conduct the topcross, based on which line is used in the second crossing, i.e., TC1: (CSSL4 × CSSL28) × CSSL49. TC2: (CSSL4 × CSSL49) × CSSL28, and TC3: (CSSL28 × CSSL49) × CSSL4. Under the assumption of the absence of linkage, the parent with the largest number of favorable alleles should be used as the third parent to maximize the frequency of favorable alleles, as the third parent has the largest contribution to their progeny (Wang et al. 2007). In our study, M56 and M57, and M59 and M60 are linked on chromosomes 8 and 9, respectively (Table 1). Hence, there is no simple answer to the question which topcross will yield in the highest frequency of the target genotype. Via 1000 simulation runs, we established that TC2 and TC3 resulted in a higher DG1 frequency in their DH populations, whereas TC1 resulted in the highest DG2 frequency (Table 6). Consequently, if the target genotype is only selected from DH lines, TC2 or TC3 should be made for selecting DG1, but TC1 should be made for selecting DG2.
Four lines are required to select DG3 and DG4, i.e., CSSL4, CSSL28, CSSL49, and CSSL52 (Table 5). The order of parents in a double cross also affects the frequency of the target genotype to be selected. Simulation results revealed that the double cross between the two single crosses (CSSL4 × CSSL28) and (CSSL49 × CSSL52) resulted in the highest frequency of both DG3 and DG4 (Table 6) in the derived doubled haploid population. The reason here for the difference in the target genotype frequency is the linkage between M59 and M60 (recombination frequency is r = 0.2789).
Letting \(\frac{00}{00}, \frac{00}{00}, \frac{02}{02},\) and \(\frac{20}{20}\) represent the four genotypes of CSSL4, CSSL28, CSSL49, and CSSL52 at M59 and M60, respectively (Table 5), the genotypes of the two F1 hybrids between CSSL4 and CSSL28, and between CSSL49 and CSSL52 are, \(\frac{00}{00},\) and \(\frac{02}{20},\) respectively. The four genotypes \(\frac{00}{00}, \frac{00}{02}, \frac{00}{20},\) and \(\frac{00}{22}\) have frequencies \(\frac{1}{2}r, \frac{1}{2}(1 - r), \frac{1}{2}(1 - r),\) and \(\frac{1}{2}r\) in the F2 of DC1 (Table 6), respectively, and \(\frac{00}{22}\) is the only one to derive the target genotype of \(\frac{22}{22}.\) Thus the frequency of the target genotype in its derived DH lines is \(\frac{1}{2}r \times \frac{1}{2}(1 - r) = \frac{1}{4}r(1 - r).\) For DC2 (Table 6), two genotypes of the two F1 hybrids are \(\frac{00}{02},\) and \(\frac{00}{20},\) respectively. Individuals in its F2 generation to derive the target genotype is \(\frac{02}{20}\) and have the frequency of \(\frac{1}{4}.\) Thus the frequency of the target genotype in its derived DH lines is \(\frac{1}{4} \times \frac{1}{2}r = \frac{1}{8}r.\) As \(0 < r < \frac{1}{2},\) it can be easily seen that \(\frac{1}{4}r(1 - r) \geqslant \frac{1}{8}r,\) which explains the higher target frequency in DC1 (Table 6).
We want to make two points here. First, people may normally ignore the importance of the order in selecting parents to make a double cross when considering each parent has the same contribution of 25%. When target genes are linked, the order of parents in a double cross affects the frequency of target genotype, as shown in Table 6. Therefore simulation can help correct some intuitive misleading and help identify the optimum crossing strategy. Second, we gave an explicit explanation in our double cross example, where only one couple of two linked QTL affect the difference in target genotype frequency. When two or more couples of linked QTL are included, there may be no theoretical way to select the best crossing strategy. Simulation is not limited by the number of linked QTL, and therefore is of great value in plant breeding when more and more QTL information becomes available.
Achievement of the target genotype by optimum selection strategies
Considering the low frequency of the target genotype in the breeding population, a multi-stage selection scheme (Wang et al. 2007) can be adopted, i.e., schemes 2 and 3 in Fig. 2. Taking DG2 as an example to illustrate the enhancement selection process. In the F1 generation of TC1, the genes on M4 and M35 are segregating (i.e., multiple genotypes for each locus), so enhancement selection can be applied to select heterozygous individuals on M4 and M35. The genes on M56, M57, and M60 are in the heterozygous stage, hence no enhancement selection can be applied. In the F1 generation of TC2, the genes on M4, M56, M57, and M60 are segregating, and enhancement selection can be applied to selected individuals which are heterozygous on these segments. For TC3, enhancement selection can be applied to select heterozygous individuals on M35, M56, M57, and M60. A three-stage selection scheme is commonly used in breeding (i.e., scheme 3 in Fig. 2). To do this, the selected TCF1 individuals from scheme 2 were self pollinated to have the TCF2 generation with a population size of 200 (Fig. 2). Genes on all the five segments are segregating in TCF2, and therefore enhancement selection for individuals carrying one or two target alleles at the five segments M4, M34, M56, M57, and M60 was applied.
For Scheme 1 where the target genotype is selected only with DH lines, on average 12.72, 20.77, and 20.71 DH lines of DG1 can be selected from TC1, TC2, and TC3, respectively (Table 7). After once enhancement selection (i.e., scheme 2 in Fig. 2), these numbers increase to 40.48, 66.40, and 66.48, and to 100.41, 141.75, and 141.95 after two times of enhancement selection (i.e., scheme 3 in Fig. 2; Table 7). The similar trend can be seen for DG2 (Table 7). The frequency of the number of target genotypes for each crossing strategy and selection scheme can be obtained from simulation (Figs. 4, 5). For Scheme 1, distinct peaks appear around 10 for TC1, and around 18 for TC2 and TC3 (Fig. 4). Scheme 2 can shift the peak of the distribution to 38 for TC1, and to 66 for TC2 as well as TC3. Scheme 3 can shift the peak of the distribution to 94 for TC1, and to 138 for TC2 and TC3. Similar results were observed for DG2 (Fig. 5). Despite the large variation in the number of selected target genotype (Figs. 4, 5), there is a high probability that selection schemes 2 and 3 will select a much higher number of target genotypes and give breeders greater chance to improve other value-added traits.


Discussion
QTL mapping with non-idealized CSS lines
CSS lines can be very useful in QTL fine mapping and cloning. In a preliminary CSS population, typically, each line carries a few segments from the donor parent rather than just one segment each, which impedes locating QTL on a single chromosome segment through the comparison of trait performance between individual CSS lines and the background parent. In previous studies (Wan et al. 2004, 2005), the composite interval mapping was used for QTL mapping to the CSS population due to the lack of an appropriate mapping method. The RSTEP-LRT method employed in this study identified the same major QTL on M57 and M59. Based on the high-density linkage maps (Harushima et al. 1998; McCouch et al. 2002), the QTL on M57 with the largest effects on ACE and AC was also detected in other mapping populations (He et al. 1999), but RSTEP-LRT in addition identified QTL that were not detected by composite interval mapping.
Interval and composite interval methods were developed for mapping populations derived from biparental crosses under the condition that there was no segregation distortion. Hence the RSTEP-LRT method may be more suitable for a non-idealized CSS population where there is severe segregation distortion due to the high background selection during population development. Extensive simulation studies revealed that RSTEP-LRT greatly increased the power detecting QTL compared to single marker analysis (Wang et al. 2006). The methodology used in RSTEP-LRT has also been applied to improve the popular composite interval mapping (Li et al. 2007).
Due to the significance of genotype by environment interaction on ACE and AC, QTL mapping in each environment was conducted separately and the stability of QTL was assessed from its effects in different environments. It has been suggested that future research should apply a QTL-mapping method capable of analyzing QTL major effects and QTL by environment interactions (Piepho 2000; van Eeuwijk et al. 2002), and follow-on research is undergoing.
Use of identified marker-QTL associations assisted by simulation tools
A vast number of studies on QTL mapping have been conducted for various traits in plants and animals in the last decade. As the number of published genes and QTL for various traits continues to increase, plant breeders are challenged on how to best utilize this multitude of information in applied crop improvement (Bernardo and Charcosset 2006; Wang et al. 2007). This study demonstrated the simulation tool QuLine facilitated use of QTL mapping results from a non-idealized CSS population in designing a breeding program. Results showed that this approach can greatly assist breeders in using identified QTL to improve breeding efficiency and effectiveness. Even though only additive and pleiotropic QTL were considered in this study, the simulation platform QU-GENE and its modules allow the definition of more complicated genetic models including epistasis, and genotype by environment interaction (Podlich and Cooper 1998; Wang et al. 2004; Cooper et al. 2005), which suggests that the potential of applying simulation tools in genetics and breeding is far greater compared to what has been demonstrated in this paper.
In a CSS population, each chromosome segment was represented by a marker. The true length of introgressed segments is rarely known, and in practice a double crossover may happen between two adjacent markers especially for a large interval. Hence a gap may occur between two neighboring segments. These factors were not considered in this study. We made three assumptions in QTL mapping and marker-assisted selection using the identified QTL-marker associations, (1) the length of the same segment in different CSS lines does not change, (2) at most one QTL exists on each segment, and (3) the QTL is closely linked with the marker representing the chromosome segment. Thus, if one QTL associated with a marker was identified, every CSS line possessing the marker will also have the QTL. The recombination between genes and their markers, and false positive QTL can all reduce the efficiency of marker-assisted selection. As a consequence, the incomplete linkage between QTL and marker will affect the simulation output given in this paper. The QuLine simulation tool can investigate the effects of different QTL-marker distances if such information becomes known (Wang et al. 2007).
The genetic models and selection schemes presented in this study still do not represent all complexities which exist in applied breeding. Our objective was to demonstrate the application of simulation tools using real data from genetic experiments and breeding programs to compare realistic breeding scenarios. Hence, the examples given in this study represent a realization of the so-called design breeding or breeding by design (Peleman and Voort 2003). The marker-QTL associations identified with the CSS population need to be validated in other populations, such as recombinant inbred lines and secondary populations by crossing CSS lines with the background parent, although several major QTL we reported in this paper have been confirmed in other mapping populations. In addition, the tentative results from this simulation study need to be further tested in field experiments. The identification of discrepancies between simulation and field evaluation will provide the basis for modifying and further improving the genetic models used in simulation. As the information on genes and QTL for important breeding traits dramatically increases, computer simulation may likely become an essential tool guiding breeders in designing effective crossing and selection strategies through testing the range of possible scenarios in silico.
References
Belknap JK (2003) Chromosome substitution strains: some quantitative considerations for genome scans and fine mapping. Mamm Genome 14:723–732
Bernardo R, Charcosset A (2006) Usefulness of gene information in marker-assisted recurrent selection: a simulation appraisal. Crop Sci 46:614–621
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Cooper M, Podlich DW, Smith OS (2005) Gene-to-phenotype and complex trait genetics. Aust J Agric Sci 56:895–918
Cowley AW Jr, Roman RJ, Jacob HJ (2003) Application of chromosome substitution techniques in gene-function discovery. J Physiol 554:46–55
Dekkers JCM, Hospital F (2002) The use of molecular genetics in the improvement of agricultural populations. Nat Rev Genet 3:22–32
van Eeuwijk FA, Crossa J, Vargas M, Ribaut JM (2002) Analysing QTL-environment interaction by factorial regression, with an application to the CIMMYT drought and low-nitrogen stress programme in maize. In: Kang MS (ed) Quantitative genetics, genomics and plant breeding. CABI Publishing, Wallingford
Eshed Y, Zamir D (1995) An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141:1147–1162
Harushima Y, Yano M, Shomura A, Sato M, Shimano T, Kuboki Y, Yamamoto T, Lin SY, Antonio BA, Parco A, Kajiya H, Huang N, Yamamoto K, Nagamura Y, Kurata N, Khush GS, Sasaki T (1998) A high-density rice genetic linkage map with 2275 markers using a single F2 Population. Genetics 148:479–494
He P, Li SG, Qian Q, Ma YQ, Li JZ, Wang WM, Chen Y, Zhu LH (1999) Genetic analysis of rice grain quality. Theor Appl Genet 98:502–508
Huang FS, Sun ZX, Hu PS, Tang SQ (1998) Present situations and prospects for the research on rice grain quality forming. Chin J Rice Sci 12(3):172–176
Kubo T, Aida Y, Nakamura K, Tsunematsu H, Doi K, Yoshimura A (2002) Reciprocal chromosome segment substitution series derived from Japonica and Indica cross of rice (Oryza sativa L.). Breed Sci 52:319–325
Lander ES, Botstein D (1989) Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Li H, Ye G, Wang J (2007) A modified algorithm for the improvement of composite interval mapping. Genetics 175:361–374
Mackill DJ, Ni JJ (2001) Molecular mapping and marker-assisted selection for major-gene traits in rice. In: Rice genetics, vol IV. Science Publishers, New Delhi, pp 137–151
McCouch SR, Teytelman L, Xu Y, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Myers RH (1990) Classical and modern regression with applications, 2nd edn. Duxbury Thomson Learning, Pacific Grove
Nadeau JH, Singer JB, Martin A, Lander ES (2000) Analysis complex genetics traits with chromosome substitution strains. Nat Genet 24:221–225
Peleman JD, Voort JR (2003) Breeding by design. Trends Plant Sci 8:330–334
Piepho HP (2000) A mixed-model approach to mapping quantitative trait loci in barley on the basis of multiple environment data. Genetics 156:2043–2050
Podlich DW, Cooper M (1998) QU-GENE: a platform for quantitative analysis of genetic models. Bioinformatics 14:632–653
Tanksley SD, Nelson JC (1996) Advanced backcross QTL analysis: a method for the simultaneous discovery and transfer of valuable QTL from unadpated germplasm into elite breeding lines. Theor Appl Genet 92:191–203
Tsunematsu H, Yoshimura A, Harushima Y, Nagamura Y, Kurata N, Yano M, Sadaki T, Iwata N (1996) RFLP framework map using recombination inbred lines in rice. Breed Sci 46:279–284
Wan X-Y, Wan J-M, Su C-C, Wang C-M, Shen W-B, Li J-M, Wang H-L, Jiang L, Liu S-J, Chen L-M, Yasui H, Yoshimura A (2004) QTL detection for eating quality of cooked rice in a population of chromosome segment substitution lines. Theor Appl Genet 110:71–79
Wan X-Y, Wan J-M, Weng J-F, Jiang L, Bi J-C, Wang C-M, Zhai H-Q (2005) Stability of QTLs for rice grain dimension and endosperm chalkiness characteristics across eight environments. Theor Appl Genet 110:1334–1346
Wang J, van Ginkel M, Podlich D, Ye G, Trethowan R, Pfeiffer W, DeLacy IH, Cooper M, Rajaram S (2003) Comparison of two breeding strategies by computer simulation. Crop Sci 43:1764–1773
Wang J, van Ginkel M, Trethowan R, Ye G, DeLacy I, Podlich D, Cooper M (2004) Simulating the effects of dominance and epistasis on selection response in the CIMMYT Wheat Breeding Program using QuCim. Crop Sci 44:2006–2018
Wang J, Eagles HA, Trethowan R, van Ginkel M (2005) Using computer simulation of the selection process and known gene information to assist in parental selection in wheat quality breeding. Aust J Agric Sci 56:465–473
Wang J, Wan X, Crossa J, Crouch J, Weng J, Zhai H, Wan J (2006) QTL mapping of grain length in rice (Oryza sativa L.) using chromosome segment substitution lines. Genet Res 88:93–104
Wang J, Chapman SC, Bonnett DB, Rebetzke GJ, Crouch J (2007) Application of population genetic theory and simulation models to efficiently pyramid multiple genes via marker-assisted selection. Crop Sci 47:580–588
Young ND (1999) A cautiously optimistic vision for marker-assited selection. Mol Breed 5:505–510
Zeng Z-B (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
The authors wish to thank Professor A. Yoshimura, Kyushu University, Japan for providing the CSS lines. This work was supported by the National 973 and 863 Programs of China (No. 2006CB101700 and 2006AA10Z1B1), and the Generation and HarvestPlus Challenge Programs of the Consultative Group for International Agricultural Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. van Eeuwijk.
Rights and permissions
About this article
Cite this article
Wang, J., Wan, X., Li, H. et al. Application of identified QTL-marker associations in rice quality improvement through a design-breeding approach. Theor Appl Genet 115, 87–100 (2007). https://doi.org/10.1007/s00122-007-0545-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-007-0545-x