
Abstract
In this study, we show that NAC-like transcription factor (TF) has 90% sequence similarity with cDNA of the OsNac5 gene present in the NCBI database. Phylogenetic analysis of the NAC gene family was performed with inclusion of the highly diverse C-terminal sequences. We report that this gene is also found to be orthologous to Glycine max NAC8, NAC2, Triticum aestivum NAC6 and paralogous to OsNAC6. mRNA was purified from five recombinant inbred lines (RILs) and parents of rice at three different stages of grain filling under aerobic conditions, with grain protein content (GPC) spanning from 4 to 14%. The NAC-like TF encoding a protein was found to be upregulated at the \(\hbox {S}_{2}\) stage in the leaf (3.9-fold) and panicle (1.84-fold) of parent HPR14 and in five RILs (1.9 to 4.51-fold in leaves and 0.47 to 3.2-fold in panicles). Expression analysis of the NAC-like TF encoding a protein for the rice gene was found to be upregulated at the \(\hbox {S}_{2}\) stage in the leaf and panicle of parental line HPR14 and RILs with high protein content.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is one of the most important staple food crops in Southeast Asia. Globally, India is the first in area of rice cultivation and second in rice production after China. India produces 21.5% of rice globally (Viraktamath et al. 2011). Enhancing grain nutrient quality is one of the most important objectives of many rice breeding programmes. Rice seeds consist of over 90% starch and protein by dry weight (Juliano 1971). However, several advances have been made in understanding the molecular and genetic basis of grain quality in rice. The molecular basis and response of signalling pathways to environmental factors, which can have strong and adverse effects on grain quality, are not completely conclusive. Seed-storage protein genes and the genes underlying starch biosynthesis have been cloned, and their functions in grain quality have been described. The regulation of these genes affecting the levels of transcription and post-transcription during development and in response to environmental conditions are not yet understood (Krishnan et al. 2009).
Scientists have been attempting to develop and improve grain quality through classical breeding as well as biotechnology. However, caution must be taken, as downregulation or upregulation of the expression of a single gene might result in unpredictable and complex biochemical and physiological changes. It has been reported that the modification of a single starch synthesis gene led to changes in all the traits related to cooking quality (Tian et al. 2009), and a reduction of one or several storage proteins could be compensated by increasing other proteins (Kawakatsu et al. 2010).
NAC is a domain name derived from first letters of three different genes NAM (no apical meristem), ATAF (Arabidopsis transcription activation factor) and CUC (cup-shaped cotyledon). Several NAM, ATAF1/2 and CUC2 (NAC) transcription factors (TFs) are involved in different cellular processes in plant species, for example, hormone signal pathways and development (Greve et al. 2003; Peng et al. 2009). NAC proteins may function as homodimers and heterodimers in plants (Xie et al. 2000; Hegedus et al. 2003; Ernst et al. 2004). Even OsNAC5 is reported to form homodimers and heterodimers with different OsNACs (Jeong et al. 2009; Takasaki et al. 2010). Genes induced by pathogen attack and wounds were found in the ATAF subfamily (Ooka et al. 2003).
To date, various NAC TFs from rice such as AtNAC072 (RD29), AtNAC019, AtNAC055 (Fujita et al. 2004; Tran et al. 2004), ANAC102 (Christianson et al. 2009) from Arabidopsis, BnNAC from Brassica napus (Hegedus et al. 2003), and SNAC1 (Hu et al. 2006), SNAC2/OsNAC6 (Nakashima et al. 2007; Hu et al. 2008), OsNAC5 (Sperotto et al. 2009; Zheng et al. 2009; Takasaki et al. 2010; Song et al. 2011) and OsNAC10 (Jeong et al. 2010) from rice have been shown to be involved in responses to various environmental stresses.
Recently, the stress-responsive gene OsNAC5 was reported in rice (Takasaki et al. 2010; Song et al. 2011). OsNAC5-overexpressing transgenic plants had increased tolerance to drought, high salinity and low temperature. Studies, however, were limited to tolerance and increasing yield (Jeong et al. 2013). A few reports determined that its function could be related to Fe, Zn and amino acid remobilization from green tissues to seed (Lim et al. 2007; Sperotto et al. 2009, 2010). To investigate the role of OsNAC5 in regulating seed-storage protein content, an expression analysis of rice NAC TFs was conducted to identify its role in improving grain protein content (GPC) in aerobic environments. More importantly, expression of this gene could significantly enhance GPC in rice.
Methods and materials
Estimation of total protein and amylose content (%)
A near-infrared reflectance spectroscopy (NIR system, FOSS, Denmark) system was used for estimation of crude protein content and amylose content in recombinant inbred lines (RILs). The NIR system was standardized using biochemical data from genotypes of \(\hbox {F}_{3}\) and \(\hbox {F}_{4}\) generations through a mathematical equation suitable for this mapping population. To measure protein content, rice was placed in a dry room with a constant humidity of 12% for a week to balance the moisture content. Samples were scanned by using a Fourier transform (FT)-NIR (Bruker MATRIX-1, Germany). Protein content was predicted from spectrum models developed with 3000 samples at the University of Agricultural Sciences (UAS), Bengaluru.
Gene expression studies for GPC
For in silico analysis, putative uncharacterized genes and known genes for seed storage protein accumulation were downloaded from the BGI database and searched for possible orthologues against the Ensembl database. To identify the most suitable candidate genes for expression studies, about 1246 putative genes with uncharacterized function expressed in the root, leaf and panicle during the grain filling stage of panicle development were downloaded and searched for their possible orthologues. Among grain filling genes investigated, a total of 49 genes qualified for possible orthologues involved in the seed storage mechanism either directly or indirectly. From these, the gene LOC_Os011g0184900 was selected for expression analysis. Further, selection was based on their protein sequence with ID percent similarity of more than 90% for query and subject.

Relative quantification in real-time quantitative PCR for NAC-like TF between parents and five selected RILs spanning 4–14% GPC in rice.
mRNA was purified from leaves and panicles of five RILs and parents (table 1) at three different stages as described by Jain et al. (2007) under aerobic conditions with GPC spanning from 4 to 14% of a population as shown in table 2.
Total RNA was extracted from leaves and panicles with the RNeasy Plant Mini kit (Qiagen) and mRNA is reverse transcribed into cDNA with SuperScript III (Invitrogen). Gene-specific primers (table 3) were designed for each candidate gene based on the cDNA sequence available in the Ensemble database. The real-time polymerase chain reaction (RT-PCR) products of 50–170 bp long were designed to span an intron, providing an internal control for the detection of contaminating genomic DNA. Quantitative RT-PCR was performed on three biological replicates with a RealPlex (Eppendorf) using SYBR Green PCR Master Mix (Qiagen) as shown in figure 1. Melt curves were examined for problems associated with genomic DNA contamination (figure 2) such as primer-dimers and multiple products. Suitable targets were then used in a template dilution series to optimize reaction efficiency. Cycle threshold values for each RIL were normalized to ubiquitin (reference gene).
In silico analysis of candidate gene
Protein and gene sequences of rice were used as queries to retrieve 36 homologous sequences of different plant genera using BLASTN and BLASTP (McGinnis and Madden 2004) of NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi). For the search, we used an E-value threshold of 0.001, query coverage \({\ge }70\%\) and sequence identity \(\ge 65\%\). Redundant sequences were removed using CD-HIT (Li and Godzik 2006).

Single melt curve for NAC-like TF.
The nonredundant homologous sequences were then aligned using the Kimura 2 parameter in MUSCLE, a multiple alignment web server (Edgar 2004). A phylogenetic tree was constructed using the maximum likelihood (ML) method with PHYLIP (Felsenstein 1989) with 100-bootstrap replicates. Further, the protein sequence of a NAC-like TF was used for multiple sequence alignment and tree building with other NAC family proteins. The resulting tree was visualized and edited using a cladogram program.
Standardization of RT-PCR condition
Primer design
All TF families have a conserved region, which is common for all the members within the family. Nonconserved regions specific to each member of the family were scanned for primer design. The primer pair specific to the TFs was designed using Primer3Plus software (http://www.bioinformatics.nl/cgibin/primer3plus/primer3plus.cgi), and the primers were synthesized at Sigma-Aldrich, USA. The following predicted parameters such as melting temperature (\(T_{\mathrm{m}}\)) of \(60\pm 2^{\circ }\hbox {C}\), primer lengths of 20–24 nucleotides, guanine–cytosine (GC) content of 45–55% and PCR amplicon length of 100–200 bp were used for designing the primer pairs of the TFs.
Primer concentration
In real-time quantitative PCR, for any gene expression, the primer concentration is one of a key factors. Primer concentrations 150, 200, 250, 300, 350 and 400 nmol were used to optimize the amplification. Primers at a concentration of 200 nmol gave a single melting curve, low Ct value, high fluorescence value and no primer dimers. Hence, this concentration was used for all experimental studies.
Annealing temperature
It is essential to determine optimal annealing temperature (\(T_{\mathrm{a}}\)) for each primer pair before its actual use. The selection of \(T_{\mathrm{a}}\) is based on the length and positions of the primers. The common view is that the \(T_{\mathrm{a}}\) should be \(5^{\circ }\hbox {C}\) below the melting temperature (\(T_{\mathrm{m}}\)) of primers. Both OsNAC and Ubiquitin were adjusted for an optimal annealing temperature of 59–60\(^{\circ }\hbox {C}\) and 61–63\(^{\circ }\hbox {C}\), respectively.
Reaction mixture
The RT-PCR master mix was freshly prepared to avoid handling errors. The reaction mixture of \(10\,\mu \hbox {L}\) contained 1.0 ng cDNA, 200 nmol of each gene-specific primer and \(5\,\mu \hbox {L}\) of \(2\times \hbox {SYBR}\) green reagents (Qiagen, USA). Individual components of the reaction mixture were standardized for a \(10\,\mu \hbox {L}\) reaction volume.
Setting baseline and threshold levels
The point of measurement (baseline and threshold levels) should be accurately determined to reflect the quantity of a particular target within the reaction. Eppendorf RealPlex detection software was used to set the default baseline from 3 to 15 cycles. The highly abundant gene ubiquitin and a few other TF genes started to amplify very early in the 20th PCR cycle. Therefore, the baseline was not changed, with default set between 3 and 15 cycles. Once the baseline has been set correctly, the software automatically sets the threshold at 10 standard deviations above the mean baseline fluorescence. It was ensured that the threshold line was placed in the exponential phase to increase the precision and quality of the experimental data, and the same baseline and threshold default setting was used for all PCR reactions.
RT-PCR condition and analysis of data
An Eppendorf RealPlex instrument (Eppendorf, India) was used for all RT-PCR amplifications. The wells defined as ‘unknown’ were used to calculate the relative quantifications. pyQPCR software (https://www.projet-plume.org/en/relier/pyqpcr) was used with an improved \(\Delta { Ct}\) method that allows reliable quantifications and error to be obtained. The confidence level is modifiable and can be calculated either using Gaussian or t-test. The programme plots results as histograms that are easy to understand. Two of the assumptions for a t-test are that both groups of \(\Delta { Ct}\) will have Gaussian distributions, and they will have equal variances (Hollander and Wolfe 1973; Yuan and Neal Stewart 2005). To generate a baseline-subtracted plot of the logarithmic increase in fluorescence signal (\(\Delta { Rn}\)) versus cycle number, baseline data were collected between cycles 3 and 15. All amplifications were analysed with the threshold automatically set by the instrument. To compare data from different PCR runs or cDNA samples, Ct values for all the TF genes were normalized to the Ct value of the ubiquitin gene, which was the most stable and had the lowest gene expression stability. Different methods are available for estimating PCR efficiency; the classical method uses Ct values obtained from a series of template dilutions.
Results
Alignment and phylogenetic analysis
The initial approach of the BLAST method for gene prediction was to identify homologues to known genes, since single nucleotide comparisons only provide an indication whether genes may or may not be involved in the seed storage protein mechanism. Therefore, phylogenetic analysis was performed to predict associated homology. In the nucleotide BLASTN study, a NAC-like TF was found to have cDNA sequence similar to the mRNA of the NAC5 gene.
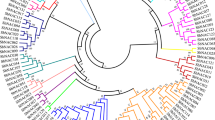
Multiple sequence alignment of the NAC protein sequence of O. sativa with different NAC proteins of other crop plants suggested that residues towards the N-terminal region appeared to be more conserved than the C-terminal region. A total of 10 clusters were generated using the neighbour-joining (NJ) method.
In eight cases, different NAC genes from other species formed a single cluster indicating that they are possible orthologues of each other. The NAC-like TF from O. sativa indica used in this study was found to be a close relative of OsNAC5 in the above phylogenetic studies and an orthologue to Glycine max NAC8, NAC2 and Triticum aestivum NAC6, whereas it was paralogous to OsNAC6, as they formed a single cluster (figure 3).
Expression analysis of OsNAC-like TF at flowering initiation and different stages of grain filling in BPT5204 and HPR14
Expression in leaves: Expression of OsNAC-like TF at flowering initiation in comparison with the baseline was 0.88-fold in BPT5204 and 1.14-fold in HPR14, whereas expression was 2.16-, 2.28- and 2.16-fold at the \(\hbox {S}_{1}, \hbox {S}_{2}\) and \(\hbox {S}_{3}\) stages of grain filling in BPT5204, respectively. HPR14 showed a high transcript abundance of 3.95-, 4.9- and 4.29-fold at the \(\hbox {S}_{1}, \hbox {S}_{2}\) and \(\hbox {S}_{3}\) stages of grain filling, respectively, summarized in table 4.
Expression in panicle: Low transcript abundance was observed in the panicle of BPT5204 at all stages. The OsNAC-like TF showed 0.75-, 0.3-, 0.34- and 0.32-fold expression at the \(\hbox {S}_{0}, \hbox {S}_{1}, \hbox {S}_{2}\) and \(\hbox {S}_{3}\) stages during grain filling in BPT5204, respectively, whereas moderate transcript abundance was observed in the panicle of HPR14, with 1.0-, 2.30-, 2.84- and 0.88-fold expression at the \(\hbox {S}_{0}, \hbox {S}_{1}, \hbox {S}_{2}\) and \(\hbox {S}_{3}\) stages, respectively, summarized in table 4.

Phylogenetic relationship among rice NAC genes, OsNAC5 and NAC-like TF. GLYMA, G. max; At, Arabidopsis; MLOC, Hordeum vulgare; GRMZM, Zea maize; Ta, Triticum aestivum; MTR, Medicago truncatula; SB, Sorghum bicolor; BNA, B. napus.

Determination of relative expression of NAC-like TF at \(\hbox {S}_{0}, \hbox {S}_{1}, \hbox {S}_{2}\) and \(\hbox {S}_{3}\) stage in (a) leaf and (b) panicle tissues of BPT 5204 and HPR14 genotype using RT-PCR.
Relatively higher transcript stage of OsNAC-like TF
OsNAC-like TF expressed relatively higher transcript at the \(\hbox {S}_{2}\) stage; HPR14 accumulated a 3.9-fold increase in leaves and a 1.84-fold increase in the panicle, whereas BPT5204 had a 1.28-fold increase in leaves and a 0.34-fold increase in the panicle (figure 4).
Expression analysis of OsNAC-like TF at S2 stage during grain filling in five selected RILs
Expression in leaves: The expression level of the NAC-like TF was found to be linear in leaves of RILs with increasing GPC. Here, RIL1 (4–5% GPC), RIL2 (6–8% GPC), RIL3 (8–10% GPC), RIL4 (10–12% GPC) and RIL5 (13–14% GPC) showed NAC-like TF expression of 1.9-, 1.9-, 2.5-, 3.04- and 4.51-fold in leaves, respectively (table 5).
Expression in panicle: A nearly linear relationship was observed in the expression level of NAC-like TF in panicles of RILs with increasing GPC (figure 5). Here, RIL1 (4–5% GPC), RIL2 (6–8% GPC), RIL3 (8–10% GPC), RIL4 (10–12% GPC) and RIL5 (13–14% GPC) showed 0.47-, 0.62-, 1.2-, 1.5- and 3.2-fold expression in the panicle, respectively, as summarized in table 5.
Discussion
Regulation of gene expression is vital for a variety of essential processes in plants, including growth, development, differentiation, metabolic regulation and adaptation to biotic and abiotic stresses. Initiation of transcription is the first step in the expression of any downstream gene; it plays a central role in the regulation of the expression of downstream genes. Transcription appears to be controlled by numerous TFs that mediate the effects of intracellular and extracellular signals (Verma and Agarwal 2010). Therefore, the functional analysis of TF genes is essential for understanding their role in grain filling.
Alignment and phylogenetic analysis
The first systematic analysis of Arabidopsis and rice NAC proteins classified them into 18 subgroups (Ooka et al. 2003). However, another phylogenetic analysis of rice NAC proteins suggested that the NAC family can be divided into five groups, and each subfamily was largely diversified (Fang et al. 2008). In a report concerning eight subfamilies (Shen et al. 2009), the main reason for the discrepancies in the reported phylogenetic trees may lie in the fact that all the previous NAC protein classifications were based on the conserved N-terminal NAC domains, either from sub-domain A to D or from A to E, which did not take the highly divergent C-terminal sequences into consideration (Ooka et al. 2003; Pinheiro et al. 2009; Shen et al. 2009).
To gain a better understanding of the phylogeny of the NAC gene family, we performed phylogenetic analysis with the inclusion of the highly diverse C-terminal sequence. Moreover, different algorithms utilized in the phylogenetic analyses may lead to the inconsistent interpretations. In the previous analyses, different algorithms, including NJ (Ooka et al. 2003; Fang et al. 2008), ML (Shen et al. 2009) and Bayesian method (Pinheiro et al. 2009) were implemented, which may make results less comparable.
In this study, NAC-like TF showed 90% sequence similarity with cDNA of the OsNac5 gene present in the NCBI database. Further, multiple sequence alignment of derived protein sequences of NAC-like TF with other NAC protein sequences of different crop plants revealed that sequences are more conservative at the N-terminal region forming a more conserved sub-domain of the NAC family, whereas the C-terminal region was found to be highly diverse among different NAC genes. In most cases, different NAC genes from different species formed a single cluster, indicating that they are possible orthologues of each other. Of the 10 clusters observed using the NJ method during phylogenetic analysis, one cluster, NAC-like TF was found to be a close relative of OsNAC5 and an orthologue to G. max NAC8, NAC2, T. aestivum NAC6, whereas it was a paralogue to OsNAC6.

Determination of relative expression of NAC-like TF transcript at \(\hbox {S}_{2}\) stage in (a) leaf and (b) panicle tissues of BPT 5204, HPR14 and low to high GPC RILs using RT-PCR. Y-axis, expression in fold. x-axis, genotypes. Ctrl, control (ubiquitin gene); TG, target gene.
Based on the available information on various TFs and their annotated functions in the model system O. sativa, important genes were selected for their expression analysis on the basis of their direct and indirect roles in regulating grain filling. TF genes showing detectable expression levels belonged to OsNAC families. The NAC genes constitute one of the largest families of plant-specific TFs and are present in a wide range of land plants. Genes in the NAC family have been shown to regulate a wide range of developmental processes including seed development, embryo development, shoot apical meristems, fibre development, leaf senescence and cell division (Souer et al. 1996; Aida et al. 1997; Sablowski and Meyerowitz 1998; Xie et al. 2000; Uauy et al. 2006).
In this study, we found a NAC-like TF encoding a protein for the rice gene to be upregulated at the \(\hbox {S}_{2}\) stage in the leaves and panicles of the parent HPR14 and also in the five RILs. A significant increase in the expression of transcript has been reported. A wheat NAC gene, NAM-B1, was reported to be involved in nutrient remobilization from leaves to developing grains (Uauy et al. 2006; Zhang et al. 2008). In rice it was also reported that OsNAC10, a closest plant protein to NAM-B1 (NAC TF), which promotes accelerated senescence and increases nutrient remobilization from leaves to developing grain was located near QTL qCP7. GPC is highly influenced by the amount of N remobilization from leaves to grain and dry matter accumulation in the seeds. The protein to starch ratio in the grain is one of the results of events occurring at both the sink (developing grains) and at the source (leaves).
Several studies suggest that source regulation plays a significant role in grain protein accumulation (Barneix and Guitman 1993; Martre et al. 2003). However, efficient remobilization of N takes place during short duration grain filling but may result in reduced total kernel weight under favourable conditions (Kade et al. 2005). OsNAC5 was demonstrated to be a senescence-associated gene that is upregulated during grain maturation in rice flag leaves (Sperotto et al. 2009). Lim et al. (2007) reported that OsNAC5 is regulated by ABA, a hormone with a known central role in senescence processes. A comparison of diverse cultivars showed a positive correlation of OsNAC5 expression in flag leaves before and during anthesis with final Fe, Zn and protein concentrations in mature grains (Sperotto et al. 2009, 2010).
In our results, there was a consistent increase in the relative abundance of transcript in leaves with increasing GPC across RILs. The result is consistent with reports by Lim et al. (2007) and Sperotto et al. (2009, 2010). This suggests that the putative OsNAC5 TF acts as a candidate gene for nutrient reservoir activity to enhance GPC during grain filling in rice.
References
Aida M., Ishida T., Fukaki H., Fujisawa H. and Tasaka M. 1997 Genes involved in organ separation in Arabidopsis: an analysis of the cup-shaped cotyledon mutant. Plant Cell 9, 841–857.
Barneix A. J. and Guitman M. R. 1993 Leaf regulation of the nitrogen concentration in the grain of wheat plants. J. Exp. Bot. 44, 1607–1612.
Christianson J. A., Wilson I. W., Llewellyn D. J. and Dennis E. S. 2009 The low oxygen induced NAC domain transcription factor ANAC102 affects viability of Arabidopsis thaliana seeds following low-oxygen treatment. Plant Physiol. 149, 1724–1738.
Edgar R. C. 2004 MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797.
Ernst H. A., Olsen A. N., Skriver K., Larsen S. and Leggio L. L. 2004 Structure of the conserved domain of ANAC, a member of the NAC family of transcription factors. EMBO Rep. 5, 297–303.
Fang Y., You J., Xie K., Xie W. and Xiong L. 2008 Systematic sequence analysis and identification of tissue-specific or stress-responsive genes of NAC transcription factor family in rice. Mol. Genet. Genom. 280, 547–563.
Felsenstein J. 1989 PHYLIP – phylogeny inference package (Version 3.2). Cladistics 5, 163–166.
Fujita M., Fujita Y., Maruyama K., Seki M., Hiratsu, K., Ohme-Takagi M. et al. 2004 A dehydration induced NAC protein, RD26, is involved in a novel ABA-dependent stress signaling pathway. Plant J. 39, 863–876.
Greve K., La Cour T., Jensen M. K., Poulsen F. M. and Skriver K. 2003 Interactions between plant RING-H2 and plant-specific NAC (NAM/ATAF1/2/CUC2) proteins: RING-H2 molecular specificity and cellular localization. Biochem. J. 371, 97–108.
Hegedus D., Yu M., Baldwin D., Gruber M., Sharpe A., Parkin I. et al. 2003 Molecular characterization of Brassica napus NAC domain transcriptional activators induced in response to biotic and abiotic stress. Plant Mol. Biol. 53, 383–397.
Hollander M. and Wolfe D. A. 1973 Nonparametric statistical methods, pp. 503. Wiley, New York.
Hu H., Dai M., Yao J., Xiao B., Li X., Zhang Q. et al. 2006 Overexpressing a NAM, ATAF and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. 103, 12987–12992.
Hu H., You J., Fang Y., Zhu X., Qi Z. and Xiong L. 2008 Characterization of transcription factor gene SNAC2 conferring cold and salt tolerance in rice. Plant Mol. Biol. 67, 169–181.
Jain M., Nijhawan A., Arora R., Agarwal P., Ray S., Sharma P. et al. 2007 F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol. 143, 1467–1483.
Jeong J. S., Park Y. T., Jung H., Park S. H. and Kim J. K. 2009 Rice NAC proteins act as homodimers and heterodimers. Plant Biotechnol. Rep. 3, 127–134.
Jeong J. S., Kim Y. S., Baek K. H., Jung H., Ha S. H., Choi Y. D. et al. 2010 Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 153, 185–197.
Jeong J. S., Kim Y. S., Redillas M. C., Jang G., Jung H., Bang S. W. et al. 2013 OsNAC5 overexpression enlarges root diameter in rice plants leading to enhanced drought tolerance and increased grain yield in the field. Plant Biotechnol. J. 11, 101–114.
Juliano B. O. 1971 A simplified assay for milled-rice amylose. Cereal Sci. Today 16, 334, 340, 360.
Kade M. A., Barneix J., Olmos S. and Dubcovsky J. 2005 Nitrogen uptake and remobilization in tetraploid Langdon durum wheat and a recombinant substitution line with the high grain protein gene GPC-B1. Plant Breed. 124, 343–349.
Kawakatsu T., Wang S., Wakasa Y. and Takaiwa F. 2010 Increased lysine content in rice grains by over-accumulation of BiP in the endosperm. Biosci. Biotechnol. Biochem. 74, 2529–2531.
Krishnan A., Guiderdoni E., An, G., Hsing Y. I., Han C. D. and Lee M. C. 2009 Mutant resources in rice for functional genomics of the grasses. Plant Physiol. 149, 165–170.
Li W. and Godzik A. 2006 Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659.
Lim P. O., Kim H. J. and Nam H. G. 2007 Leaf senescence. Annu. Rev. Plant Biol. 58, 115–136.
Martre P., Porter J. R., Jamieson P. D. and Triboi E. 2003 Modeling grain nitrogen accumulation and protein composition to understand the sink/source regulations of nitrogen remobilization for wheat. Plant Physiol. 133, 1959–1967.
McGinnis S. and Madden T. L. 2004 BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, 20–25.
Nakashima K., Tran L. S., Van Nguyen D., Fujita M., Maruyama K., Todaka, D. et al. 2007 Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-responsive gene expression in rice. Plant J. 51, 617–630.
Ooka H., Satoh K., Doi K., Nagata T., Otomo Y., Murakami K. et al. 2003 Comprehensive analysis of NAC family genes in Oryza sativa and Arabidopsis thaliana. DNA Res. 20, 239–247.
Peng H., Cheng H. Y., Chen C., Yu X. W., Yang J. N., Gao W. R. et al. 2009 A NAC transcription factor gene of chickpea (Cicer arietinum), CarNAC3, is involved in drought stress response and various developmental processes. J. Plant Physiol. 166, 1934–1945.
Pinheiro G. L., Marques C. S., Costa M. D., Reis P. A., Alves M. S., Carvalho C. M. et al. 2009 Complete inventory of soybean NAC transcription factors: sequence conservation and expression analysis uncover their distinct roles in stress response. Gene 444, 10–23.
Sablowski R. W. and Meyerowitz E. M. 1998 A homolog of NO APICAL MERISTEM is an immediate target of the floral homeotic genes APETALA3/PISTILLATA. Cell 92, 93–103.
Shen H., Yin Y. B., Chen F., Xu Y. and Dixon R. A. 2009 A bioinformatic analysis of NAC genes for plant cell wall development in relation to lignocellulosic bioenergy production. Bioenergy Res. 2, 217–232.
Song S. Y., Chen Y., Chen J., Dai X. Y. and Zhang W. H. 2011 Physiological mechanisms underlying OsNAC5-dependent tolerance of rice plants to abiotic stress. Planta 234, 331–345.
Souer E, Van Houwelingen A, Kloos D, Mol J. and Koes R. 1996 The no apical meristem gene of petunia is required for pattern formation in embryos and flowers and is expressed at meristem and primordia boundaries. Cell 85, 159–170.
Sperotto R. A., Ricachenevsky F. K., Duarte G. L., Boff T., Lopes K. L. and Sperb E. R. 2009 Identification of up-regulated genes in flag leaves during rice grain filling and characterization of OsNAC5, a new ABA-dependent transcription factor. Planta 230, 985–1002.
Sperotto R. A., Boff T., Duarte G. L., Santos L. S., Grusak M. A. and Fett J. P. 2010 Identification of putative target genes to manipulate Fe and Zn concentrations in rice grains. J. Plant Physiol. 167, 1500–1506.
Takasaki H., Maruyama K., Kidokoro S., Ito Y., Fujita Y., Shinozaki K. et al. 2010 The abiotic stress responsive NAC-type transcription factor OsNAC5 regulates stress-inducible genes and stress tolerance in rice. Mol. Genet. Genom. 284, 173–183.
Tian Z., Qian Q., Liu Q., Yan M., Liu X. and Yan C. 2009 Allelic diversities in rice starch biosynthesis lead to a diverse array of rice eating and cooking qualities. Proc. Natl. Acad. Sci. USA 106, 21760–21765.
Tran L. S., Nakashima K., Sakuma Y., Simpson S. D., Fujita Y., Maruyama K. et al. 2004 Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 16, 2481–2498.
Uauy C., Distelfeld A., Fahima T., Blechl A. and Dubcovsky, J. 2006 A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 314, 1298–1301.
Verma P. S. and Agarwal V. K. 2010 Genetics, 3rd edition. S. Chand publication.
Viraktamath B. C., Bentur J. S., Rao K. V. and Mangal S. 2011 Vision 2030, Directorate of Rice Research, Hyderabad.
Xie Q., Frugis G., Colgan D. and Chua N. H. 2000 Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 14, 3024–3036.
Yuan S. J. and Neal Stewart C. N. 2005 Real-time PCR statistics. PCR Encycl. 1, 101127–101149.
Zhang W., Bi J., Chen L., Zheng L., Ji S., Xia Y. et al. 2008 QTL mapping for crude protein and protein fraction contents in rice (Oryza sativa L.). J. Cereal Sci. 48, 539–547.
Zheng X., Chen B., Lu G. and Han B. 2009 Overexpression of a NAC transcription factor enhances rice drought and salt tolerance. Biochem. Biophys. Res. Commun. 379, 985–989.
Acknowledgements
We thank the Department of Biotechnology (DBT), New Delhi, Government of India, for funding this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: Arun Joshi
Rights and permissions
About this article

Cite this article
Sharma, G., Upadyay, A.K., Biradar, H. et al. OsNAC-like transcription factor involved in regulating seed-storage protein content at different stages of grain filling in rice under aerobic conditions. J Genet 98, 18 (2019). https://doi.org/10.1007/s12041-019-1066-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12041-019-1066-5