
Abstract
The progressive myoclonic epilepsy of Lafora or Lafora disease (LD) is a neurodegenerative disorder characterized by recurrent seizures and cognitive deficits. With typical onset in the late childhood or early adolescence, the patients show progressive worsening of the disease symptoms, leading to death in about 10 years. It is an autosomal recessive disorder caused by the loss-of-function mutations in the EPM2A gene, coding for a protein phosphatase (laforin) or the NHLRC1 gene coding for an E3 ubiquitin ligase (malin). LD is characterized by the presence of abnormally branched water insoluble glycogen inclusions known as Lafora bodies in the neurons and other tissues, suggesting a role for laforin and malin in glycogen metabolic pathways. Mouse models of LD, developed by targeted disruption of the Epm2a or Nhlrc1 gene, recapitulated most of the symptoms and pathological features as seen in humans, and have offered insight into the pathomechanisms. Besides the formation of Lafora bodies in the neurons in the presymptomatic stage, the animal models have also demonstrated perturbations in the proteolytic pathways, such as ubiquitin-proteasome system and autophagy, and inflammatory response. This review attempts to provide a comprehensive coverage on the genetic defects leading to the LD in humans, on the functional properties of the laforin and malin proteins, and on how defects in any one of these two proteins result in a clinically similar phenotype. We also discuss the disease pathologies as revealed by the studies on the animal models and, finally, on the progress with therapeutic attempts albeit in the animal models.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Much of our understanding on the neurodegenerative processes and the players involved in the neuronal survival pathways have come from the discoveries made in the pathobiology of rare forms of neurodegenerative disorders. For example, the very concept of mitophagy—a quality control mechanism that regulates the mitochondrial homeostasis (Corti and Brice 2013)—and its causal role in neurodegeneration have come from the study on parkin, a protein found to be defective in a small subset of Parkinson’s disease patients (Kitada et al. 1998). Similarly, the discovery of the infectious agent of the rare disorder known as Creutzfeldt–Jakob disease (Field et al. 1969)—the agent later identified to be protein and hence named as prion (Collinge et al. 1996)—led to a revolutionary change in our understanding on a nonRNA/DNA mode transmission of disease which we believe could underlie the pathogenesis of several neurodegenerative disorders (Goedert 2015). Yet another example of a rare form of a disorder offering novel insights into the neuropathophysiology is the subset of amyotrophic lateral sclerosis (ALS), patients with mutations in the FUS or TDP-43 gene—accounting to less than 3% of the ALS cases (Lattante et al. 2013). Functional studies on the mutant forms of FUS and TDP-43 lead to our current understanding on their regulatory role post-transcriptional regulation, and as to how defects in this process could underlie neurodegeneration (Lagier-Tourenne et al. 2010). These discoveries have led to our current understanding on the role of noncoding RNA in neurological disorders (Lourenco et al. 2015). Studies on yet another rare disorder known as Lafora disease (LD) and the topic of the current review, likewise offered novel insights into the role of glycogen metabolism in the neuronal survival. Dissecting the function of laforin and malin proteins (the two proteins defective in LD) in diverse cellular pathways, especially on the glycogen metabolism and autophagy, extended our understanding on the common pathways connecting diverse set of neurodegenerative disorders. This review aims to summarize the findings on LD and attempts to functionally link the disease genotype with the disease phenotype by providing a comprehensive overview on the LD biology.
Introduction to LD
LD is an adolescent onset neurodegenerative disorders, with the disease-defining symptoms of epileptic attacks, including myoclonus, tonic–clonic and absence seizures (Minassian 2001; Ganesh et al. 2006). The other symptoms include ataxia, dementia, hallucination, and dysarthria, and all those, including the seizures, show progressive worsening. Hence, LD is classified as one of the five forms of progressive myoclonus epilepsies (Delgado-Escueta et al. 2001). With typical age at onset around 12–15 years, the affected patients die around 25 years, often due to respiratory failure (Ganesh et al. 2006; Striano et al. 2008). Thus, LD is considered to be one of the most severe forms of epilepsies (Minassian 2001). LD shows an autosomal recessive inheritance with 100% penetrance and fatality. LD is named after Gonzalo Rodríguez Lafora (1886–1971), a Spanish neurologist who first described the presence of intracellular inclusions, which he referred to as ‘amyloid bodies,’ in the condition progressive myoclonic epilepsy (Nanduri et al. 2008). These inclusions were later referred to as ‘Lafora bodies’ in honor of the contributions of Gonzalo Lafora. LD, though a rare disorder, is relatively frequent in the Mediterranean region, the Middle East, eastern Europe and the South Asian populations (Singh and Ganesh 2009). There are a few reports of LD in the Japanese and Chinese population as well (Ganesh et al. 2001; Singh et al. 2005; Yildiz et al. 2017).

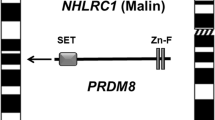
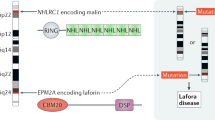
Schematic diagram showing the position of LD genes EPM2A and NHLRC1 on chromosome 6 and PRDM8 gene on the chromosome 4. The domain organization of the encoded proteins are also shown (drawn not to scale). SET, SET domain; Zn-F, Zinc-finger domain; RING, RING domain; NHL, NHL repeat domain; CBD, carbohydrate-binding domain; DSPD, dual-specificity phosphatase domain.
Genetic heterogeneity in LD
A genetic locus for LD was mapped on chromosome 6q24 (Serratosa et al. 1995; Sainz et al. 1997), and a causative gene, named EPM2A, was identified in the year 1998 (Minassian et al. 1998; Serratosa et al. 1999) (see figure 1). Subsequently, the full gene sequence was discovered and characterized (Ganesh et al. 2000). The coding sequence of the EPM2A gene spans 4 exons, and codes for a dual-specificity protein phosphatase named laforin (Ganesh et al. 2000). Besides the phosphatase domain at the carboxyl terminal, the laforin protein also harbors a carbohydrate-binding domain at the amino terminal (Wang et al. 2002; Ganesh et al. 2004) (see figure 1). Several loss-of-function mutations were identified in the gene, confirming that the EPM2A gene is indeed the causative gene for LD (see below). Some of the LD families did not show mutations in the EPM2A gene or mapped to the 6q24 locus, suggesting the presence of at least one more locus for LD (Minassian et al. 1999). Subsequent studies, specifically by using the nonEPM2A LD families, identified a second locus for LD named as EPM2B and mapping at 6p23 (Chan et al. 2003a, b). Sequence analyses of genes mapped in this region lead to the identification of NHLRC1 gene as the second causative gene for LD (Chan et al. 2003a, b). The NHLRC1 is a single exon gene (Chan et al. 2003a, b), coding for an E3 ubiquitin ligase protein named malin (Gentry et al. 2005) (see figure 1). A number of mutations have been identified in the NHLRC1 gene and all of them appear to be loss-of-function mutations (see below), as expected for an autosomal recessive mode of inheritance of LD. It is of interest to note that the two genes showed population specific distributions of the mutations; e.g., mutations in the EPM2A gene is far more frequent in Spanish, French and the US population whereas the NHLRC1 gene appears to be the major causative gene for LD in the Italian, Canadian, Arab, Indian and Brazilian populations (Singh and Ganesh 2009; Turnbull et al. 2016). Among the reported families, defects in the EPM2A accounts for nearly 50% of them, and the rest for the NHLRC1 gene. There appears to be a third locus for LD since not all LD families show mutations in the EPM2A or the NHLRC1 gene (Chan et al. 2004a, b). Indeed, a novel locus for LD was mapped on chromosome 4q21 in a family that showed early onset LD phenotype (Turnbull et al. 2012). Homozygosity mapping leads to the identification of a novel variant of the PRDM8 gene in a family of Pakistani origin (Turnbull et al. 2012) (see figure 1). Although recent studies indicate a role for this gene in neuronal differentiation during development (Inoue et al. 2015; Jung et al. 2015; Iwai et al. 2018), the possible role for this gene in LD is yet to be established (see below). Nevertheless, the LD families that do not map to EPM2A and NHLRC1 loci strongly indicate the presence of multiple genetic loci for LD.
Allelic heterogeneity in LD
Genetic screen for more than 250 LD families are available in the literature and more than 150 distinct mutations have been reported in the two genes (Singh and Ganesh 2009; Turnbull et al. 2016). These include large and small deletions, insertions, point mutations and mutations in the splice junctions (Singh and Ganesh 2009; Turnbull et al. 2016). This extreme allelic heterogeneity, with orphan mutations spreading across the gene region poses an obvious challenge for the genetic diagnostics. Haplotype analyses indicate that only a minor fraction of these mutations are due to the founder effect and a large number represents mutational hot spots (Singh and Ganesh 2009; Turnbull et al. 2016). For example, the p.R241X mutation in the EPM2A gene is often seen in the Spanish population and their decent seem to be due to the founder effect (Ganesh et al. 2002a). Similarly, the p.C26S mutation in the NHLRC1 gene in the French Canadian appears to be due to a founder effect (Chan et al. 2003a, b; Singh et al. 2006). Similarly, quite a few discrete spots of high recurrent mutations have also been reported in both genes (Singh and Ganesh 2009; Turnbull et al. 2016). These include, but not limited to, the large deletions and p.G279S mutation in the EPM2A gene, and the p.P69A and p.G158fs16 mutations in the NHLRC1 gene (Ganesh et al. 2002a). Heterozygous mutations were reported in a few families, suggesting the possible presence of undetected mutations in the other allele (Gómez-Garre et al. 2000; Ganesh et al. 2002a; Chan et al. 2003a, b; Singh et al. 2005). Some of these might carry deletions, since hemizygous deletions may go undetected in a conventional PCR. Indeed such instances of compound heterozygous mutations complicating the genetic diagnosis have been recorded and reported (Minassian et al. 2000).
Genotype–phenotype correlations in LD
LD is generally considered to be a clinically homogenous disorder. It is clinically difficult to differentiate LD patients who have mutations in EPM2A gene from those who have in NHLRC1. Few reports did suggest that the NHLRC1 defective patients may show a slower disease progression (Singh et al. 2006). However, such slow progressive forms have also been identified for a few patients with EPM2A defects (Jara-Prado et al. 2014), suggesting that the observed difference could be mutation specific (Ferlazzo et al. 2014). Similarly, subsyndromes of LD have also been reported; an atypical form of LD, with childhood onset learning disabilities was shown to be associated with the mutations in the first exon of the EPM2A gene (Ganesh et al. 2002a; Annesi et al. 2004). Since the first exon of the EPM2A gene codes for the carbohydrate binding domain (CBD), a positive correlation for the CBD and atypical form was also proposed (Ganesh et al. 2002a). Later, the possible differential effect of mutations on the alternatively spliced transcript of the EPM2A gene was suggested (Dubey et al. 2012). However, such correlations could not be extended to other patients with exon 1 mutations (Ganesh et al. 2002a), and more importantly, a founder effect mutation in an Arab family showed variable expression, suggesting the possible presence of ‘modifier’ genes for LD (Lesca et al. 2010; Singh and Ganesh 2012b). A strong support to this notion comes from a study where the same mutation on the EPM2A gene showed variable pathologies, and a rare sequence variant in the PPP1R3C gene coding for the protein targeting to glycogen (PTG)—a protein that interacts with laforin—caused slower progression of the disease (Guerrero et al. 2011). Given that laforin and malin are shown to interact with a number of proteins, a possible disease modifying role for these interacting proteins are suspected (Singh and Ganesh 2012b). Recently, several variants of LD clinical course are reported. For example, LD with obsessive compulsive symptoms (Nasri et al. 2017) and a late onset LD with Parkinsonism (Lynch et al. 2016) are observed. Another example is the early onset of LD associating with the PRDM8 gene mutation, thus highlighting the primary defect—in one or more genes—associating with subsyndromes of LD. Clearly further work is needed to correlate genetic defects in other genes with the variation in clinical symptoms.
Functional properties of laforin phosphatase and malin ubiquitin ligase
The cellular functions of the two LD associated gene products—laforin and malin—are reasonably well characterized (table 1). The suggested functions for the LD proteins include a critical role in the glycogen metabolism (Roach 2015), ubiquitin-proteasome pathway (Mittal et al. 2007; Garyali et al. 2009; Vernia et al. 2009; Rao et al. 2010a), autophagy (Knecht et al. 2010; Puri and Ganesh 2010, 2012), heat shock response (Sengupta et al. 2011; Jain et al. 2017), ER stress response (Vernia et al. 2009; Sharma et al. 2013), oxidative stress response (Romá-Mateo et al. 2014, 2015; Sánchez-Elexpuru et al. 2017a, b), translational regulation (Ganesh et al. 2000), RNA metabolism (Singh et al. 2012a), cell death pathway (Upadhyay et al. 2015), and mitochondrial homeostasis (Upadhyay et al. 2017) (see table 1) (also discussed below). The EPM2A gene product laforin is a dual-specificity protein phosphatase (Ganesh et al. 2000; Minassian et al. 2000; Girard et al. 2006). Although the phosphatase activity of this protein is well established, and a number of interacting proteins are identified for laforin, it is yet not clear as how many of them are substrates of laforin (table 2). In vitro studies have identified Tau (Puri et al. 2009), HIRIP5 (Ganesh et al. 2003), malin (Gentry et al. 2005; Lohi et al. 2005a), AMPK (Moreno et al. 2010) to be a few of the potential substrates of laforin. Similarly, for malin, the potential substrates are laforin (Gentry et al. 2005; Lohi et al. 2005a; Solaz-Fuster et al. 2008), PTG (Worby et al. 2008), MGS (Valles-Ortega et al. 2011), Dishevelled (Sharma et al. 2012), Neuronatin (Sharma et al. 2011), and abnormally misfolded proteins (Garyali et al. 2009; Rao et al. 2010a; Jain et al. 2017). It may be noted here that a majority of these studies relied upon cell models, and overexpressed laforin or malin since no reliable antibodies were available for both the proteins for further characterization of the interactions.

Representative images showing the exclusively cytoplasmic localization pattern of transiently expressed laforin (red) and malin (green) in normal condition (top row), their import to nucleus on exposure of COS7 cells on heat shock (second row), their perinuclear aggresomal localization upon proteasomal blockade (third row) or their recruitment to processing bodies upon puromycin treatment, as indicated (scale \(10\,\mu \hbox {m}\)).
Studies using overexpression constructs have shown exclusively cytoplasmic localization for laforin (Ganesh et al. 2000, 2002b; Wang et al. 2002), while malin appears to localize both in cytoplasm and nucleus (Chan et al. 2003b; Mittal et al. 2007). Moreover, their localization pattern appears to be dynamic and aligned to the physiological state of the cell. For example, a heat shock or glucose starvation induces the nuclear localization of laforin (Sengupta et al. 2011; Singh et al. 2012a) (see figure 2) (also see below). Studies have also documented the subcellular compartment in which laforin and malin might localize (see figure 2). Both laforin and malin appear to localize in the endoplasmic reticulum and in the polysomal fraction (Ganesh et al. 2000; Minassian et al. 2001; Chan et al. 2003b). In situ localization revealed that laforin colocalizing with glycogen particles in the cell (Wang et al. 2002; Tiberia et al. 2012) and has been shown to dephosphorylate glycogen (Ganesh et al. 2004; Worby et al. 2006). The transgenically overexpressed laforin was also found to localize with the Lafora inclusion bodies in the LD mice (Chan et al. 2004a, b). Malin was shown to localize with processing bodies in the cells (Singh et al. 2012a; Singh and Ganesh 2012b).
Laforin harbours two functional domains—the amino terminal CBD which enables laforin to bind to glycogen, and the carboxyl terminal dual-specificity phosphatase domain (DSPD) which confers the phosphatase activity to laforin (figure 1). Intriguingly, laforin functions as a phosphatase in dimeric form (Liu et al. 2006; Dubey and Ganesh 2008; Dubey et al. 2012; Sankhala et al. 2015) while it binds to glycogen as monomer (Dubey and Ganesh 2008). At least in humans, the EPM2A gene codes for multiple isoforms, each with varying localization and possible function (Ganesh et al. 2002b; Ianzano et al. 2004; Dubey and Ganesh 2008; Dubey et al. 2012). For example, one of the minor isoforms harbor both CBD and DSPD, yet inactive as a phosphatase due to a sequence variation at the carboxyl terminal, and it localizes to the nuclear compartment as well (Dubey et al. 2012). In addition, this isoform can potentially interact with the major form, to form a heterodimer and block its phosphatase activity (Dubey et al. 2012). The other isoforms, resulting from the alternative mRNA splicing harbor one of the functional domains or a novel protein sequence with unknown function (Dubey et al. 2012). The biological significance of these potential isoforms of laforin is not known although a majority of them interact among each other, and with malin. Malin, however, do not appear to function as a dimer although malin–malin interaction has been established (Mittal et al. 2015). This self-interaction perhaps promotes the autoubiquitination of malin when the cellular levels of its substrates are low (Mittal et al. 2015). Laforin could possibly minimize the autoubiquitination of malin by presenting itself as a substrate (Mittal et al. 2015). Intriguingly, malin appears to have a higher affinity for the monomeric form of laforin, suggesting a regulatory role for malin in laforin’s phosphatase activity (Mittal et al. 2015). Conversely, laforin seems to phosphorylate malin although its biological significance is not clear (Gentry et al. 2005).
Animal models of LD
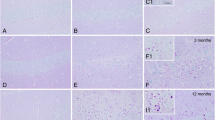
The mouse lines deficient for laforin or malin, created by targeted disruption of the Epm2a or the Nhlrc1 gene, offer attractive models for the LD (Ganesh et al. 2002c; DePaoli-Roach et al. 2010; Criado et al. 2012). Each of the models replicated most of the symptoms and pathological features of the LD. These include the formation of polyglucosan bodies (figure 3), epileptic attacks, abnormal motor coordination, behavioural and cognitive deficits (Ganesh et al. 2002c; DePaoli-Roach et al. 2010; Criado et al. 2012; Berthier et al. 2015; Sánchez-Elexpuru et al. 2017a). Unlike the LD in humans, the animal models however do not die early possibly due to the species specific difference in the disease progression. Detailed studies on these models has led us to discover the following: (i) mutation in both the genes and thus both the animal models develop similar pathological and clinical course (García-Cabrero et al. 2012; ii) formation of Lafora bodies predates the epileptic symptoms (Ganesh et al. 2002c; Sánchez-Elexpuru et al. 2017a; iii) Lafora bodies develop first in the neurons (as early as in one-month-old animals) followed by other tissues such as liver and muscle (from 3–4 months onwards) (Ganesh et al. 2002c; (iv) the Lafora bodies increase in size and dimension with age of the mice (Ganesh et al. 2002c; v) the Lafora bodies are lesser branched than normal glycogen, rich in phospho content and are insoluble in water (Tagliabracci et al. 2008; Roach 2015; vi) an unique form of cell death—called dark cell death—predate the formation of Lafora bodies in the neurons (Ganesh et al. 2002c; Machado-Salas et al. 2012; vii) several of such degenerating neurons do not show visible Lafora bodies (Ganesh et al. 2002c; Machado-Salas et al. 2012), suggesting multiple causes for the neuronal death; (viii) the Lafora bodies are positive for ubiquitin, p62—an adaptor protein for autophagy targets, and advanced glycation end-products, hereby suggesting a higher amount of protein content in the glycogen-rich inclusions (Ganesh et al. 2002c; Puri et al. 2012; Criado et al. 2012; Duran et al. 2014); (ix) the affected neurons show hyperphosphorylated neurofibrillary tangles, intraneuronal abeta deposits,

Representative images showing the periodic acid-schiff (PAS)-positive Lafora inclusion bodies (identified by arrows) in the brain and the skeletal muscle tissues of the laforin-deficient mouse and their absence in the wild-type littermate, as indicated (scale \(10\,\mu \hbox {m}\)).
senile plaques (Puri et al. 2009; Machado-Salas et al. 2012) and abnormally large lysosomes (Puri and Ganesh 2012; x) the neurites showed shunted projections, abnormally structured endoplasmic reticulum and the Gogli networks (Ganesh et al. 2002c; Puri and Ganesh 2012) and fragmented mitochondria (Ganesh et al. 2002c; Upadhyay et al. 2017); (xi) the brain showed increased gliosis indicating persistent insult to the neurons (Puri et al. 2012; Pederson et al. 2013; Turnbull et al. 2014; Sánchez-Elexpuru et al. 2017a; Rai et al. 2017; xii) brain tissue showed increased levels of long-lived proteins (Aguado et al. 2010; Criado et al. 2012), and insoluble ubiquitinated proteins (Puri et al. 2012; xiii) autophagic defects in the brain and other tissues occurring prior to the formation of Lafora bodies (Aguado et al. 2010; Puri and Ganesh 2012; Criado et al. 2012); (xiv) increased levels of neuroinflammatory markers revealing increased innate inflammatory responses in the LD model (López-González et al. 2017; xv) increased susceptibility to drug-induced seizures (Sánchez-Elexpuru et al. 2017a, b; xvi) cardiomyopathy characterized by hypertrophy and systolic dysfunction, suggesting pathologies beyond the neurological defects (Villalba-Orero et al. 2017). In addition to the transgenic mouse models for LD, a selected breed of dogs displayed myoclonic seizures and Lafora bodies in the brain, showed an expanded repeat mutation in the Nhlrc1 gene confirming the natural occurrence of LD in the canine species (Lohi et al. 2005b).
Pathomechanisms in LD
The locus heterogeneity and clinical homogeneity in LD suggest that the products of the two LD genes are nonredundant partners in physiological pathways that are defective in the LD. Thus, defect in any one of them will likely to affect the same set of pathways, leading to clinically identical symptoms and pathologies. Consistent with this model, a number of studies showed that both laforin and malin as a functional complex and are involved in multiple pathways, and loss of anyone of them would affect the cellular physiology (see tables 1 and 2). Notwithstanding the substantial progress made in deciphering the functions of the laforin and malin in cellular context, whether or not they contribute to the disease defining symptoms—epilepsy and other neurological defects as seen in LD patients—is yet to be unequivocally established. Grossly, the possible functions of the LD proteins (laforin/malin) can be grouped in to the following three generic cellular processes: (i) cellular stress response mechanisms, (ii) proteolytic pathways and (iii) glycogen metabolic pathways. And each one of them can contribute to the defect in the other. For example, the polyglucosan bodies can trap ubiquitin, proteasome, chaperones and other players of the protein quality control pathways, thus leading to a compromise in their function (Rao et al. 2010a, b; Criado et al. 2012; Puri et al. 2012). Similarly, the increased glycogen level in the LD tissues can block autophagy through the AMPK-mTOR axis (Singh et al. 2013). Conversely, the oxidative stress, for example, can lead to increased glycogen in the neuronal cells (Wang et al. 2013; Saez et al. 2014; Rai et al. 2018), possibly, as a transient protective response to the cellular stress (Rai et al. 2018). Thus, each of these three axes can potentially feed-in to the other, and it would therefore be difficult to dissect the cause-consequence relationship. The narrative below provides a brief summary of the each of these pathways in the context of LD.
Lafora bodies—the disease defining pathology of LD represent phospho-rich, lesser branched form of glycogen. These conspicuous inclusions in the neurons led to the suggestion that LD could be possibly a metabolic disorder, and these inclusions are causative for the symptoms seen in LD (Criado et al. 2012; Singh et al. 2013; Duran et al. 2014). Consistent with this hypothesis, the laforin phosphatase was found to bind to the glycogen and dephosphorylate the same (Irimia et al. 2015; Raththagala et al. 2015). The findings that the Lafora bodies are rich in phosphate moieties, and that they tend to form water insoluble, ‘sticky’ aggregates support this notion (Tagliabracci et al. 2008). Thus, the addition of phosphate group was thought to be an error in the glycogen synthetic process, and that laforin is an error fixing enzyme (Roach 2011). Thus, the ‘hyperphosphorylated’ abnormal glycogen, resulting from the loss of laforin function was considered to be the primary cause for LD (Tagliabracci et al. 2008). However, the recent reports that the abnormal chain length pattern and not the hyperphosphorylation could be the cause for the genesis of Lafora bodies (Nitschke et al. 2017). More recently, a transgenic mouse expressing a phosphatase inactive mutant of laforin was shown to correct the LD pathology in the laforin-deficient mouse (Gayarre et al. 2014), suggesting defects in functions other than the phosphatase activity of laforin is critical for pathology. An alternate model on the role for laforin/malin in glycogen metabolism was that these proteins regulate the glucose uptake in the cells (Singh et al. 2012a; Singh and Ganesh 2012b; Singh et al. 2013). It is long known that in addition to the Lafora bodies the LD tissues including the brain showed abnormally higher levels of glycogen. Studies have shown that laforin, and possibly malin, functions as energy sensors in the cell, and that they negatively regulate the cellular glucose uptake by regulating cell surface localization of glucose transporters by modulating the activity of the serum/glucocorticoid-induced kinase 1 (SGK1) (Singh et al. 2012a; Singh and Ganesh 2012b; Singh et al. 2012c, 2014). Thus, loss of either malin or laforin is expected to promote excessive glucose uptake and its conversion into glycogen. These observations explain as to why the LD tissues show higher levels of intracellular glycogen, and that the increased glycogen may play a role in the aetiology. Consistent with this notion, a recent report demonstrates an increased level of glycogen in the astrocytes in a LD mouse model, and that astrocyte glycogen could contribute to the LD pathology (Rubio-Villena et al. 2018).
Studies have shown that the Lafora bodies are positive for ubiquitin, proteasome, p62 and other markers of proteolytic pathways (Ganesh et al. 2002c; Criado et al. 2012; Duran et al. 2014), suggesting that Lafora bodies could contribute defects in the proteolytic pathways. Subsequently, a few reports suggested a direct involvement of laforin and malin in the ubiquitin-proteasome pathway (Mittal et al. 2007; Garyali et al. 2009), and autophagy–lysosome pathway (Aguado et al. 2010; Puri et al. 2012; Criado et al. 2012; Knecht et al. 2012). Intriguingly, studies have shown that the defects in autophagy predate the formation of visible Lafora bodies in the LD mouse models (Criado et al. 2012), suggesting that the proteolytic defects could be one of the primary triggers in LD. However, the defects in glycogen metabolism contributing to the compromised autophagy process cannot be ruled out. For example, a link between intracellular glucose level and the mTOR activation was shown in cellular LD models (Singh et al. 2013), and reducing the glycogen build-up in the LD animals restored the autophagy defects (Duran et al. 2014). Thus, metabolic changes in the LD tissues can modulate the other processes, including the proteolytic, stress response and inflammatory pathways. The findings that the LD tissues show elevated ROS level (Romá-Mateo et al. 2014), ER stress (Vernia et al. 2009; Zeng et al. 2012), compromised chaperone activity (Sengupta et al. 2011; Rao et al. 2010a, b) and the elevated inflammatory response (López-González et al. 2017) point to the possibility of defective cellular processes. Thus restoring the glycogen metabolic flux might fix most of the other pathologies (also see below).
Therapy for LD
Despite the advances made in identifying the genes involved in LD and elucidating their cellular functions, the treatment for LD remains at best symptomatic (Goldsmith and Minassian 2016; Michelucci et al. 2016). The current treatments for LD include the use of channel blockers such as valproic acid and recently the AMPA antagonist perampanel was shown to be more efficient in controlling the seizures (Goldsmith and Minassian 2016). Given the glycogen load in the LD tissues, a few studies have also attempted ketogenic diet for LD but did not show an appreciable effect (Cardinali et al. 2006; Kossoff et al. 2014). Therefore, the LD animal models are being used to identify and test ‘druggable’ targets for effective therapy. Obviously, the best way would be to block the glycogen synthesis since genetic inactivation of the glycogen synthase or its positive regulators led to the suppression of Lafora bodies and seizure susceptibility in the mouse models (Pederson et al. 2013; Turnbull et al. 2014; Sánchez-Elexpuru et al. 2017a, b; Rai et al. 2017). Thus attempts are being made to identify small molecules that can block glycogen synthase activity (Solmesky et al. 2017). Similarly, gene therapy is also being tested in the animal models (Cornford et al. 2016) to restore the function of laforin or malin. As a pharmacological approach, treatment with metformin, an activator of AMP-activated protein kinase (AMPK), was shown to reduce the Lafora body load, neurodegeneration and seizure susceptibility in LD animals (Berthier et al. 2015; Sánchez-Elexpuru et al. 2017a, b), although its effect on epilepsy or seizure susceptibility was not documented. Similarly, sodium selenite administration was shown to decrease the seizure susceptibility possibly by protecting neurons from oxidative stress (Sánchez-Elexpuru et al. 2017a, b). Recently, a study from our own group has demonstrated that blocking the leptin signalling in the brain could reduce the glycogen level, Lafora body load and seizure susceptibility possibly by lowering the neuronal glucose uptake (Rai et al. 2017). Nonetheless how the loss of laforin or malin results in the epileptic episodes, and whether epilepsy is primary symptom or secondary to the Lafora bodies or neuroinflammation are not studied well. Similarly, the one-to-one correlation on the specific pathologies and the disease symptoms (such as epilepsy, dementia, psychosis and ataxia) have not been established.
Concluding statement
The first 20 years since the discovery of the first LD gene, EPM2A, has seen a remarkable progress in DNA diagnostics, understanding the gene function, developing models and deciphering the pathomechanisms. However not much has changed with regard to the prognosis or the treatment; LD continues to be a debilitating condition affecting hundreds of families worldwide. Often termed as a ‘rare orphan disorder’, work on LD genes and LD models continue to shape our understanding on the biology of glycogen metabolism, and its relevance to neuronal function. With the availability of LD models, the next two decades is likely to witness a remarkable progress in our understanding behind the epilepsy and other symptoms of LD and contribute to the management and treatment of LD in humans.
References
Aguado C., Sarkar S., Korolchuk V. I., Criado O., Vernia S., Boya P. et al. 2010 Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet. 19, 2867–2876.
Annesi G., Sofia V., Gambardella A., Candiano I. C., Spadafora P., Annesi F. et al. 2004 A novel exon 1 mutation in a patient with atypical lafora progressive myoclonus epilepsy seen as childhood-onset cognitive deficit. Epilepsia 45, 294–295.
Berthier A., Payá M., García-Cabrero A. M., Ballester M. I., Heredia M., Serratosa J. M. et al. 2015 Pharmacological interventions to ameliorate neuropathological symptoms in a mouse model of Lafora disease. Mol. Neurobiol. 53, 1296–1309.
Cardinali S., Canafoglia L., Bertoli S., Franceschetti S., Lanzi G., Tagliabue A. et al. 2006 A pilot study of a ketogenic diet in patients with Lafora body disease. Epilepsy Res. 69, 129–134.
Chan E. M., Bulman D. E., Paterson A. D., Turnbull J., Andermann E., Andermann F. et al. 2003a Genetic mapping of a new Lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22. J. Med. Genet. 40, 671–675.
Chan E. M., Young E. J., Ianzano L., Munteanu I., Zhao X., Christopoulos C. C. et al. 2003b Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 35, 125–127.
Chan E. M., Ackerley C. A., Lohi H., Ianzano L., Cortez M. A., Shannon P. et al. 2004a Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum. Mol. Genet. 13, 1117–1129.
Chan E. M., Omer S., Ahmed M., Bridges L. R., Bennett C., Scherer S. W. et al. 2004b Progressive myoclonus epilepsy with polyglucosans (Lafora disease): evidence for a third locus. Neurology 63, 565–567.
Cheng A., Zhang M., Gentry M. S., Worby C. A., Dixon J. E. and Saltiel A. R. 2007 A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori’s disease. Genes Dev. 21, 2399–2409.
Collinge J., Sidle K. C., Meads J., Ironside J. and Hill A. F. 1996 Molecular analysis of prion strain variation and the aetiology of ’new variant’ CJD. Nature 383, 685–690.
Cornford E. M., Hyman S., Cornford M. E., Chytrova G., Rhee J., Suzuki T. et al. 2016 Non-invasive gene targeting to the fetal brain after intravenous administration and transplacental transfer of plasmid DNA using PEGylated immunoliposomes. J. Drug Target 24, 58–67.
Corti O. and Brice A. 2013 Mitochondrial quality control turns out to be the principal suspect in parkin and PINK1-related autosomal recessive Parkinson’s disease. Curr. Opin. Neurobiol. 23, 100–108.
Criado O., Aguado C., Gayarre J., Duran-Trio L., Garcia-Cabrero A. M., Vernia S. et al. 2012 Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 21, 1521–1533.
Delgado-Escueta A. V., Ganesh S. and Yamakawa K. 2001 Advances in the genetics of progressive myoclonus epilepsy. Am. J. Med. Genet. 106, 129–138.
DePaoli-Roach A. A., Tagliabracci V. S., Segvich D. M., Meyer C. M., Irimia J. M. and Roach P. J. 2010 Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J. Biol. Chem. 285, 25372–25381.
Dubey D. and Ganesh S. 2008 Modulation of functional properties of laforin phosphatase by alternative splicing reveals a novel mechanism for the EPM2A gene in lafora progressive myoclonus epilepsy. Hum. Mol. Genet. 17, 3010–3020.
Dubey D., Parihar R. and Ganesh S. 2012 Identification and characterization of novel splice variants of the human EPM2A gene mutated in Lafora progressive myoclonus epilepsy. Genomics 99, 36–43.
Duran J., Gruart A., García-Rocha M., Delgado-García J. M. and Guinovart J. J. 2014 Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum. Mol. Genet. 23, 3147–3156.
Ferlazzo E., Canafoglia L., Michelucci R., Gambardella A., Gennaro E., Pasini E. et al. 2014 Mild Lafora disease: clinical, neurophysiologic, and genetic findings. Epilepsia 55, e129–e133.
Fernández-Sánchez M. E., Criado-García O., Heath K. E., García-Fojeda B., Medraño-Fernández I., Gomez-Garre P. et al. 2003 Laforin, the dual-phosphatase responsible for Lafora disease, interacts with R5 (PTG), a regulatory subunit of protein phosphatase-1 that enhances glycogen accumulation. Hum. Mol. Genet. 12, 3161–3171.
Field E. J., Farmer F., Caspary E. A. and Joyce G. 1969 Susceptibility of scrapie agent to ionizing radiation. Nature 222, 90–91.
Ganesh S., Agarwala K. L., Ueda K., Akagi T., Shoda K., Usui T. et al. 2000 Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum. Mol. Genet. 9, 2251–2261.
Ganesh S., Shoda K., Amano K., Uchiyama A., Kumada S., Moriyama N. et al. 2001 Mutation screening for Japanese Lafora’s disease patients: identification of novel sequence variants in the coding and upstream regulatory regions of EPM2A gene. Mol. Cell Probes. 15, 281–289.
Ganesh S., Delgado-Escueta A. V., Suzuki T., Francheschetti S., Riggio C., Avanzini G. et al. 2002a Genotype-phenotype correlations for EPM2A mutations in Lafora’s progressive myoclonus epilepsy: exon 1 mutations associate with an early-onset cognitive deficit subphenotype. Hum. Mol. Genet. 11, 1263–1271.
Ganesh S., Suzuki T. and Yamakawa K. 2002b Alternative splicing modulates subcellular localization of laforin. Biochem. Biophys. Res. Commun. 291, 1134–1137.
Ganesh S., Delgado-Escueta A. V., Sakamoto T., Avila M. R., Machado-Salas J., Hoshii Y. et al. 2002c Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 11, 1251–1262.
Ganesh S., Tsurutani N., Suzuki T., Ueda K., Agarwala K. L., Osada H. et al. 2003 The Lafora disease gene product laforin interacts with HIRIP5., a phylogenetically conserved protein containing a NifU-like domain. Hum. Mol. Genet. 12, 2359–2368.
Ganesh S., Tsurutani N., Suzuki T., Hoshii Y., Ishihara T., Delgado-Escueta A. V. et al. 2004 The carbohydrate-binding domain of Lafora disease protein targets Lafora polyglucosan bodies. Biochem. Biophys. Res. Commun. 313, 1101–1109.
Ganesh S., Puri R., Singh S., Mittal S. and Dubey D. 2006 Recent advances in the molecular basis of Lafora’s progressive myoclonus epilepsy. J. Hum. Genet. 51, 1–8.
García-Cabrero A. M., Marinas A., Guerrero R., de Córdoba S. R., Serratosa J. M. and Sánchez M. P. 2012 Laforin and malin deletions in mice produce similar neurologic impairments. J. Neuropathol. Exp. Neurol. 71, 413–421.
Garyali P., Segvich D. M., DePaoli-Roach A. A. and Roach P. J. 2014 Protein degradation and quality control in cells from laforin and malin knockout mice. J. Biol. Chem. 289, 20606–20614.
Garyali P., Siwach P., Singh P. K., Puri R., Mittal S., Sengupta S. et al. 2009 The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum. Mol. Genet. 18, 688–700.
Gayarre J., Duran-Trío L., Criado Garcia O., Aguado C., Juana-López L., Crespo I. et al. 2014 The phosphatase activity of laforin is dispensable to rescue Epm2a-/- mice from Lafora disease. Brain 137, 806–818.
Gentry M. S., Worby C. A. and Dixon J. E. 2005 Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. USA 102, 8501–8506.
Girard J. M., Lê K. H. and Lederer F. 2006 A dual-specificity protein phosphatase implicated in Lafora disease. Biochimie 88, 1961–1971.
Goedert M. 2015 Neurodegeneration. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled A\(\upbeta \), tau, and \(\upalpha \)-synuclein. Science 349, 1255555.
Goldsmith D. and Minassian B. A. 2016 Efficacy and tolerability of perampanel in ten patients with Lafora disease. Epilepsy Behav. 62, 132–135.
Gómez-Garre P., Sanz Y., Rodríguez De Córdoba S. R. and Serratosa J. M. 2000 Mutational spectrum of the EPM2A gene in progressive myoclonus epilepsy of Lafora: high degree of allelic heterogeneity and prevalence of deletions. Eur. J. Hum. Genet. 8, 946–954.
Guerrero R., Vernia S., Sanz R., Abreu-Rodríguez I., Almaraz C., García-Hoyos M. et al. 2011 A PTG variant contributes to a milder phenotype in Lafora disease. PLoS One 6, e21294.
Ianzano L., Zhao X. C., Minassian B. A. and Scherer S. W. 2003 Identification of a novel protein interacting with laforin., the EPM2a progressive myoclonus epilepsy gene product. Genomics 81, 579–587.
Ianzano L., Young E. J., Zhao X. C., Chan E. M., Rodriguez M. T., Torrado M. V. et al. 2004 Loss of function of the cytoplasmic isoform of the protein laforin (EPM2A) causes Lafora progressive myoclonus epilepsy. Hum. Mutat. 23, 170–176.
Inoue M., Iwai R., Yamanishi E., Yamagata K., Komabayashi-Suzuki M., Honda A. et al. 2015 Deletion of Prdm8 impairs development of upper-layer neocortical neurons. Genes Cells 20, 758–770.
Irimia J. M., Tagliabracci V. S., Meyer C. M., Segvich D. M., DePaoli-Roach A. A., and Roach P. J. 2015 Muscle glycogen remodeling and glycogen phosphate metabolism following exhaustive exercise of wild type and laforin knockout mice. J. Biol. Chem. 290, 22686–22698.
Iwai R., Tabata H., Inoue M., Nomura K. I., Okamoto T., Ichihashi M. et al. 2018 A Prdm8 target gene Ebf3 regulates multipolar-to-bipolar transition in migrating neocortical cells. Biochem. Biophys. Res. Commun. 495, 388–394.
Jain N., Mishra R. and Ganesh S. 2016 FoxO3a-mediated autophagy is down-regulated in the laforin deficient mice, an animal model for Lafora progressive myoclonus epilepsy. Biochem. Biophys. Res. Commun. 474, 321–327.
Jain N., Rai A., Mishra R. and Ganesh S. 2017 Loss of malin, but not laforin, results in compromised autophagic flux and proteasomal dysfunction in cells exposed to heat shock. Cell Stress Chaperones. 22, 307–315.
Jara-Prado A., Ochoa A., Alonso M. E., Lima Villeda G. A., Fernández-Valverde F., Ruano-Calderón L. et al. 2014 Late onset Lafora disease and novel EPM2A mutations: breaking paradigms. Epilepsy Res. 108, 1501–1510.
Jung C. C., Atan D., Ng D., Ploder L., Ross S. E., Klein M. et al. 2015 Transcription factor PRDM8 is required for rod bipolar and type 2 OFF-cone bipolar cell survival and amacrine subtype identity. Proc. Natl. Acad. Sci. USA 112, E3010–E30109.
Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S. et al. 1998 Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608.
Knecht E., Aguado C., Sarkar S., Korolchuk V. I., Criado-García O., Vernia S. et al. 2010 Impaired autophagy in Lafora disease. Autophagy 6, 991–993.
Knecht E., Criado-García O., Aguado C., Gayarre J., Duran-Trio L., Garcia-Cabrero A. M. et al. 2012 Malin knockout mice support a primary role of autophagy in the pathogenesis of Lafora disease. Autophagy 8, 701–703.
Kossoff E. H., Veggiotti P., Genton P. and Desguerre I. 2014 Transition for patients with epilepsy due to metabolic and mitochondrial disorders. Epilepsia 3, 37–40.
Lagier-Tourenne C., Polymenidou M. and Cleveland D. W. 2010 TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–64.
Lattante S., Rouleau G. A. and Kabashi E. 2013 TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum. Mutat. 34, 812–826.
Lesca G., Boutry-Kryza N., de Toffol B., Milh M., Steschenko D., Lemesle-Martin M. et al. 2010 Novel mutations in EPM2A and NHLRC1 widen the spectrum of Lafora disease. Epilepsia. 51, 1691–1698.
Liu Y., Wang Y., Wu C., Liu Y. and Zheng P. 2006 Dimerization of Laforin is required for its optimal phosphatase activity, regulation of GSK3beta phosphorylation, and Wnt signaling. J. Biol. Chem. 281, 34768–34774.
Liu Y., Zeng L., Ma K., Baba O., Zheng P., Liu Y. et al. 2013 Laforin-malin complex degrades polyglucosan bodies in concert with glycogen debranching enzyme and brain isoform glycogen phosphorylase. Mol. Neurobiol. 49, 645–657.
Lohi H., Ianzano L., Zhao X. C., Chan E. M., Turnbull J., Scherer S. W. et al. 2005a Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 14, 2727–2736.
Lohi H., Young E. J., Fitzmaurice S. N., Rusbridge C., Chan E. M., Vervoort M. et al. 2005b Expanded repeat in canine epilepsy. Science 307, 81.
López-González I., Viana R., Sanz P. and Ferrer I. 2017 Inflammation in Lafora disease: Evolution with disease progression in Laforin and Malin knock-out mouse models. Mol. Neurobiol. 54, 3119–3130.
Lourenco G. F., Janitz M., Huang Y. and Halliday G. M. 2015 Long noncoding RNAs in TDP-43 and FUS/TLS-related frontotemporal lobar degeneration (FTLD). Neurobiol. Dis. 82, 445–454.
Lynch D. S., Wood N. W. and Houlden H. 2016 Late-onset Lafora disease with prominent parkinsonism due to a rare mutation in EPM2A. Neurol. Genet. 16, e101.
Machado-Salas J., Avila-Costa M. R., Guevara P., Guevara J., Durón R. M., Bai D. et al. 2012 Ontogeny of lafora bodies and neurocytoskeleton changes in laforin-deficient mice. Exp. Neurol. 236, 131–140.
Michelucci R., Pasini E., Riguzzi P., Andermann E., Kälviäinen R. and Genton P. 2016 Myoclonus and seizures in progressive myoclonus epilepsies: pharmacology and therapeutic trials. Epileptic Disord. 18, 145–153.
Minassian B. A., Lee J. R., Herbrick J. A., Huizenga J., Soder S., Mungall A. J. et al. 1998 Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 20, 171–174.
Minassian B. A., Sainz J., Serratosa J. M., Gee M., Sakamoto L. M., Bohlega S. et al. 1999 Genetic locus heterogeneity in Lafora’s progressive myoclonus epilepsy. Ann. Neurol. 45, 262–265.
Minassian B. A., Ianzano L., Meloche M., Andermann E., Rouleau G. A., Delgado-Escueta A. V. et al. 2000 Mutation spectrum and predicted function of laforin in Lafora’s progressive myoclonus epilepsy. Neurology 55, 341–346.
Minassian B. A. 2001 Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 25, 21–29.
Minassian B. A., Andrade D. M., Ianzano L., Young E. J., Chan E., Ackerley C. A. et al. 2001 Laforin is a cell membrane and endoplasmic reticulum-associated protein tyrosine phosphatase. Ann. Neurol. 49, 271–275.
Mittal S., Dubey D., Yamakawa K. and Ganesh S. 2007 Lafora disease proteins malin and laforin are recruited to aggresomes in response to proteasomal impairment. Hum. Mol. Genet. 16, 753–762.
Mittal S., Upadhyay M., Singh P. K., Parihar R. and Ganesh S. 2015 Interdependence of laforin and malin proteins for their stability and functions could underlie the molecular basis of locus heterogeneity in Lafora disease. J. Biosci. 40, 863–871.
Moreno D., Towler M. C., Hardie D. G., Knecht E. and Sanz P. 2010 The laforin-malin complex, involved in Lafora disease, promotes the incorporation of K63-linked ubiquitin chains into AMP-activated protein kinase beta subunits. Mol. Biol. Cell. 21, 2578–2588.
Nanduri A. S., Kaushal N., Clusmann H. and Binder D. K. 2008 The maestro don Gonzalo Rodríguez-Lafora. Epilepsia 49, 943–947.
Navarro-Sastre A., Tort F., Stehling O., Uzarska M. A., Arranz J. A., Del Toro M. et al. 2011 A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am. J. Hum. Genet. 89, 656–667.
Nasri A., Mansour M., Kacem A., Derbali H., Yahya M., Riahi A. et al. 2017 Pediatric obsessive compulsive disorder: an unusual form of Lafora disease. Encephale 43, 90–94.
Nitschke F., Sullivan M. A., Wang P., Zhao X., Chown E. E., Perri A. M. et al. 2017 Abnormal glycogen chain length pattern., not hyperphosphorylation, is critical in Lafora disease. EMBO Mol. Med. 9, 906–917
Pederson B. A., Turnbull J., Epp J. R., Weaver S. A., Zhao X., Pencea N. et al. 2013 Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann. Neurol. 74, 297–300.
Puri R. and Ganesh S. 2010 Laforin in autophagy: a possible link between carbohydrate and protein in Lafora disease? Autophagy 6, 1229–1231.
Puri R. and Ganesh S. 2012 Autophagy defects in lafora disease: cause or consequence? Autophagy 8, 289–290.
Puri R., Suzuki T., Yamakawa K. and Ganesh S. 2009 Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease. J. Biol. Chem. 284, 22657–22663.
Puri R., Suzuki T., Yamakawa K. and Ganesh S. 2012 Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum. Mol. Genet. 21, 175–184.
Rai A., Mishra R. and Ganesh S. 2017 Suppression of leptin signaling reduces polyglucosan inclusions and seizure susceptibility in a mouse model for Lafora disease. Hum. Mol. Genet. 26, 4778–4785.
Rai A., Singh P. K., Singh V., Kumar V., Mishra R., Thakur A. K. et al. 2018 Glycogen synthase protects neurons from cytotoxicity of mutant huntingtin by enhancing the autophagy flux. Cell Death Dis. 9, 201.
Rao S. N., Maity R., Sharma J., Dey P., Shankar S. K., Satishchandra P. et al. 2010a Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum. Mol. Genet. 19, 4726–4734.
Rao S. N., Sharma J., Maity R. and Jana N. R. 2010b Co-chaperone CHIP stabilizes aggregate-prone malin, a ubiquitin ligase mutated in Lafora disease. J. Biol. Chem. 285, 1404–1413.
Raththagala M., Brewer M. K., Parker M. W., Sherwood A. R., Wong B. K., Hsu S. et al. 2015 Structural mechanism of laforin function in glycogen dephosphorylation and lafora disease. Mol. Cell 57, 261–272.
Roach P. J. 2011 Are there errors in glycogen biosynthesis and is laforin a repair enzyme? FEBS Lett. 585, 3216–3218.
Roach P. J. 2015 Glycogen phosphorylation and Lafora disease. Mol. Aspects Med. 46, 78–84.
Roemer M. I. 1990 Public and private sectors in health system development. Asia Pac. J. Public Health 4, 164–168.
Romá-Mateo C., Aguado C., García-Giménez J. L., Ibáñez-Cabellos J. S., Seco-Cervera M., Pallardó F. V. et al. 2014 Increased oxidative stress and impaired antioxidant response in Lafora disease. Mol. Neurobiol. 51, 932–946.
Romá-Mateo C., Aguado C., García-Giménez J. L., Knecht E., Sanz P. and Pallardó F. V. 2015 Oxidative stress, a new hallmark in the pathophysiology of Lafora progressive myoclonus epilepsy. Free Radic. Biol. Med. 88, 30–41.
Rubio-Villena C., Garcia-Gimeno M. A. and Sanz P. 2013 Glycogenic activity of R6, a protein phosphatase 1 regulatory subunit, is modulated by the laforin-malin complex. Int. J. Biochem. Cell Biol. 45, 1479–1488.
Rubio-Villena C., Viana R., Bonet J., Garcia-Gimeno M. A., Casado M., Heredia M. et al. 2018 Astrocytes: new players in progressive myoclonus epilepsy of Lafora type. Hum. Mol. Genet. 27, 1290–1300.
Sainz J., Minassian B. A., Serratosa J. M., Gee M. N., Sakamoto L. M., Iranmanesh R. et al. 1997 Lafora progressive myoclonus epilepsy: narrowing the chromosome 6q24 locus by recombinations and homozygosities. Am. J. Hum. Genet. 61, 1205–1209.
Saez I., Duran J., Sinadinos C., Beltran A., Yanes O., Tevy M. F. et al. 2014 Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J. Cereb. Blood Flow Metab. 34, 945–955.
Sánchez-Elexpuru G., Serratosa J. M. and Sánchez M. P. 2017a Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of Lafora disease. Epilepsia 58, 467–475.
Sánchez-Elexpuru G., Serratosa J. M., Sanz P. and Sánchez M. P. 2017b 4-Phenylbutyric acid and metformin decrease sensitivity to pentylenetetrazol-induced seizures in a malin knockout model of Lafora disease. Neuroreport. 28, 268–271.
Sánchez-Martín P., Romá-Mateo C., Viana R. and Sanz P. 2015 Ubiquitin conjugating enzyme E2-N and sequestosome-1 (p62) are components of the ubiquitination process mediated by the malin-laforin E3-ubiquitin ligase complex. Int. J. Biochem. Cell Biol. 69, 204–214.
Sankhala R. S., Koksal A. C., Ho L., Nitschke F., Minassian B. A. and Cingolani G. 2015 Dimeric quaternary structure of human laforin. J. Biol. Chem. 290, 4552–4559.
Sengupta S., Badhwar I., Upadhyay M., Singh S. and Ganesh S. 2011 Malin and laforin are essential components of a protein complex that protects cells from thermal stress. J. Cell Sci. 124, 2277–2286.
Serratosa J. M., Delgado-Escueta A. V., Posada I., Shih S., Drury I., Berciano J. et al. 1995 The gene for progressive myoclonus epilepsy of the Lafora type maps to chromosome 6q. Hum. Mol. Genet. 4, 1657–1663.
Serratosa J. M., Gómez-Garre P., Gallardo M. E., Anta B., de Bernabé D. B., Lindhout D. et al. 1999 A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum. Mol. Genet. 8, 345–352.
Sharma J., Rao S. N., Shankar S. K., Satishchandra P. and Jana N. R. 2011 Lafora disease ubiquitin ligase malin promotes proteasomal degradation of neuronatin and regulates glycogen synthesis. Neurobiol. Dis. 44, 133–141.
Sharma J., Mulherkar S., Mukherjee D. and Jana N. R. 2012 Malin regulates Wnt signaling pathway through degradation of dishevelled2. J. Biol. Chem. 287, 6830–6839.
Sharma J., Mukherjee D., Rao S. N., Iyengar S., Shankar S. K., Satishchandra P. et al. 2013 Neuronatin-mediated aberrant calcium signaling and endoplasmic reticulum stress underlie neuropathology in Lafora disease. J. Biol. Chem. 288, 9482–9490.
Singh S., Sethi I., Francheschetti S., Riggio C., Avanzini G., Yamakawa K. et al. 2006 Novel NHLRC1 mutations and genotype-phenotype correlations in patients with Lafora’s progressive myoclonic epilepsy. J. Med. Genet. 43, e48.
Singh S. and Ganesh S. 2009 Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum. Mutat. 30, 715–723.
Singh P. K., Singh S. and Ganesh S. 2012a The laforin-malin complex negatively regulates glycogen synthesis by modulating cellular glucose uptake via glucose transporters. Mol. Cell Biol. 32, 652–663.
Singh S. and Ganesh S. 2012b Phenotype variations in Lafora progressive myoclonus epilepsy: possible involvement of genetic modifiers? J. Hum. Genet. 57, 283–285.
Singh S., Singh P. K., Bhadauriya P. and Ganesh S. 2012c Lafora disease E3 ubiquitin ligase malin is recruited to the processing bodies and regulates the microRNA-mediated gene silencing process via the decapping enzyme Dcp1a. RNA Biol. 9, 1440–1449.
Singh P. K., Singh S. and Ganesh S. 2013 Activation of serum/glucocorticoid-induced kinase 1 (SGK1) underlies increased glycogen levels, mTOR activation, and autophagy defects in Lafora disease. Mol. Biol. Cell. 24, 3776–3786.
Singh S., Suzuki T., Uchiyama A., Kumada S., Moriyama N., Hirose S. et al. 2005 Mutations in the NHLRC1 gene are the common cause for Lafora disease in the Japanese population. J. Hum. Genet. 50, 347–352.
Solaz-Fuster M. C., Gimeno-Alcañiz J. V., Ros S., Fernandez-Sanchez M. E., Garcia-Fojeda B., Criado Garcia O. et al. 2007 Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 17, 667–678.
Solmesky L. J., Khazanov N., Senderowitz H., Wang P., Minassian B. A., Ferreira I. M. et al. 2017 A novel image-based high-throughput screening assay discovers therapeutic candidates for adult polyglucosan body disease. Biochem. J. 474, 3403–3420.
Striano P., Zara F., Turnbull J., Girard J. M., Ackerley C. A., Cervasio M. et al. 2008 Typical progression of myoclonic epilepsy of the Lafora type: a case report. Nat. Clin. Pract. Neurol. 4, 106–111.
Sun T., Yi H., Yang C., Kishnani P. S. and Sun B. 2016 Starch binding domain-containing protein 1 plays a dominant role in glycogen transport to lysosomes in Liver. J. Biol. Chem. 291, 16479–16484.
Tagliabracci V. S., Girard J. M., Segvich D., Meyer C., Turnbull J., Zhao X. et al. 2008 Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J. Biol. Chem. 283, 33816–33825.
Tiberia E., Turnbull J., Wang T., Ruggieri A., Zhao X. C., Pencea N. et al. 2012 Increased laforin and laforin binding to glycogen underlie Lafora body formation in malin-deficient Lafora disease. J. Biol. Chem. 287, 25650–25659.
Turnbull J., Girard J. M., Lohi H., Chan E. M., Wang P., Tiberia E. et al. 2012 Early-onset Lafora body disease. Brain. 135, 2684–2698.
Turnbull J., Tiberia E., Pereira S., Zhao X., Pencea N., Wheeler A. L. et al. 2013 Deficiency of a glycogen synthase-associated protein, Epm2aip1, causes decreased glycogen synthesis and hepatic insulin resistance. J. Biol. Chem. 288, 34627–34637.
Turnbull J., Epp J. R., Goldsmith D., Zhao X., Pencea N., Wang P. et al. 2014 PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann. Neurol. 75, 442–446.
Turnbull J., Tiberia E., Striano P., Genton P., Carpenter S., Ackerley C. A. et al. 2016 Lafora disease. Epileptic Disord. 18, 38–62.
Upadhyay M., Gupta S., Bhadauriya P. and Ganesh S. 2015 Lafora disease proteins laforin and malin negatively regulate the HIPK2-p53 cell death pathway. Biochem. Biophys. Res. Commun. 464, 106–111.
Upadhyay M., Agarwal S., Bhadauriya P. and Ganesh S. 2017 Loss of laforin or malin results in increased Drp1 level and concomitant mitochondrial fragmentation in Lafora disease mouse models. Neurobiol. Dis. 100, 39–51.
Valles-Ortega J., Duran J., Garcia-Rocha M., Bosch C., Saez I., Pujadas L. et al. 2011 Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol. Med. 3, 667–681.
Vernia S., Rubio T., Heredia M., Rodríguez de Córdoba S. and Sanz P. 2009 Increased endoplasmic reticulum stress and decreased proteasomal function in lafora disease models lacking the phosphatase laforin. PLoS One 4, e5907.
Viana R., Lujan P. and Sanz P. 2015 The laforin/malin E3-ubiquitin ligase complex ubiquitinates pyruvate kinase M1/M2. BMC Biochem. 16, 24.
Vilchez D., Ros S., Cifuentes D., Pujadas L., Vallès J., García-Fojeda B. et al. 2007 Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 10, 1407–1413.
Villalba-Orero M., Sánchez-Elexpuru G., López-Olañeta M., Campuzano O., Bello-Arroyo E., García-Pavía P. et al. 2017 Lafora Disease is an inherited metabolic cardiomyopathy. J. Am. Coll. Cardiol. 69, 3007–3009.
Wang J., Stuckey J. A., Wishart M. J. and Dixon J. E. 2002 A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J. Biol. Chem. 277, 2377–2380.
Wang Y., Liu Y., Wu C., Zhang H., Zheng X. and Zheng Z. 2006 Epm2a suppresses tumor growth in an immunocompromised host by inhibiting Wnt signaling. Cancer Cell 10, 179–190.
Wang Y., Ma K., Wang P., Baba O., Zhang H., Parent J. M. et al. 2013 Laforin prevents stress-induced polyglucosan body formation and Lafora disease progression in neurons. Mol. Neurobiol. 48, 49–61.
Worby C. A., Gentry M. S. and Dixon J. E. 2006 Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 281, 30412–30418.
Worby C. A., Gentry M. S. and Dixon J. E. 2008 Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 283, 4069–4076.
Yildiz E. P., Yesil G., Ozkan M. U., Bektas G., Caliskan M. and Ozmen M. 2017 A novel EPM2A mutation in a patient with Lafora disease presenting with early parkinsonism symptoms in childhood. Seizure 51, 77–79.
Zeng L., Wang Y., Baba O., Zheng P., Liu Y. and Liu Y. 2012 Laforin is required for the functional activation of malin in endoplasmic reticulum stress resistance in neuronal cells. FEBS J. 279, 2467–2478.
Zhu Y., Zhang M., Kelly A. R. and Cheng A. 2014 The carbohydrate-binding domain of overexpressed STBD1 is important for its stability and protein-protein interactions. Biosci. Rep. 34, pii: e00117.
Acknowledgements
We apologize to the authors whose papers we could not cite in this review due to space limitations. We thank the group members (past and present) for their immense contributions towards the LD projects in the laboratory. The work on LD is currently supported by a research grant from the Science and Engineering Research Board, Department of Science and Technology, Government of India, to SG (SB/S5/AB/05/2016) and to RP (YSS/2015/001818). SG is also supported by the Tata Innovation Fellowship of the Department of Biotechnology, Government of India (BT/HRD/35/01/01/2017), and the P. K. Kelkar Endowed Chair at IIT Kanpur.
Author information
Authors and Affiliations
Corresponding author
Additional information
We dedicate this review article to our mentor and teacher Professor Rajiva Raman as a tribute to his constant encouragements and immense contributions to the field of genetics in the country.
Rights and permissions
About this article

Cite this article
Parihar, R., Rai, A. & Ganesh, S. Lafora disease: from genotype to phenotype. J Genet 97, 611–624 (2018). https://doi.org/10.1007/s12041-018-0949-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-018-0949-1