
Abstract
Glycogen, the largest cytosolic macromolecule, is soluble because of intricate construction generating perfect hydrophilic-surfaced spheres. Little is known about neuronal glycogen function and metabolism, though progress is accruing through the neurodegenerative epilepsy Lafora disease (LD) proteins laforin and malin. Neurons in LD exhibit Lafora bodies (LBs), large accumulations of malconstructed insoluble glycogen (polyglucosans). We demonstrated that the laforin–malin complex reduces LBs and protects neuronal cells against endoplasmic reticulum stress-induced apoptosis. We now show that stress induces polyglucosan formation in normal neurons in culture and in the brain. This is mediated by increased glucose-6-phosphate allosterically hyperactivating muscle glycogen synthase (GS1) and is followed by activation of the glycogen digesting enzyme glycogen phosphorylase. In the absence of laforin, stress-induced polyglucosans are undigested and accumulate into massive LBs, and in laforin-deficient mice, stress drastically accelerates LB accumulation and LD. The mechanism through which laforin–malin mediates polyglucosan degradation remains unclear but involves GS1 dephosphorylation by laforin. Our work uncovers the presence of rapid polyglucosan metabolism as part of the normal physiology of neuroprotection. We propose that deficiency in the degradative phase of this metabolism, leading to LB accumulation and resultant seizure predisposition and neurodegeneration, underlies LD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lafora disease (LD) is a fatal teenage-onset neurological disorder characterized by progressive myoclonic epilepsy, neurodegeneration, and neuronal accumulation of a poorly branched and insoluble amylopectin-like form of glycogen, called polyglucosan, into inclusion bodies termed Lafora bodies (LBs) or polyglucosan bodies (PBs) [1–4]. Loss-of-function mutations in either of two genes cause LD: EPM2A, which encodes the dual-specificity phosphatase laforin, and NHLRC1, which encodes the E3 ubiquitin ligase malin [5, 6]. Laforin possesses a carbohydrate-binding module critical for its binding to glycogen and plant amylopectin in vitro and to polyglucosans in vivo [7–10]. Laforin has been shown to dephosphorylate amylopectin and glycogen [10–12], and in serum-starved cells the glycogen synthase kinase GSK3β [13–15]. This effect is not seen in non-starved condition, e.g., in unchallenged Epm2a knockout mice [12, 16].
Laforin and malin form an interacting complex [17, 18]. We recently observed that in PB containing neuronal cells, the laforin–malin complex assembles on PBs, and that this is associated with degradation of PBs, and with protection of the cells against the endoplasmic reticulum (ER) stress-induced apoptosis [19]. These results raised the question: Is polyglucosan formation a normal part of the response of neurons to stress, and is the function of the laforin–malin complex to participate in the digestion of PBs?
Brain glycogen is stored almost exclusively in astrocytes, with minimal to no glycogen present in neurons. One source of energy utilized by neurons is lactate generated in astrocytes from glycogen and other sources. However, in the main, neurons rely for energy on glucose supplied by the systemic circulation [20, 21]. In this report, we show that neuronal ER stress increases cellular levels of glucose-6-phosphate (G6P), the main intracellular form of glucose. G6P is also a very potent allosteric activator of the glycogen synthesizing enzyme GS1, its action able to greatly overcome any inactivation of the enzyme by phosphorylation by GS kinases such as GSK3β. We show that the increase in G6P in response to ER stress drives GS1 to produce polyglucosans and PBs. We demonstrate that digestion of the polyglucosans requires laforin, without which accumulating PBs lead to neurodegeneration and epilepsy. Finally, the epilepsy itself provokes PB formation. Our results suggest that the undegradability of PB in the absence of laforin, and PB generation as a result of the epilepsy, combine to set into motion a mechanism that might represent the basic pathogenesis of LD.
Materials and Methods
Mice and Cells
Epm2a knockout (KO) mice (originally a 129Sv strain) used in this study have been backcrossed more than ten generations onto a C57BL/6 background [22]. Experiments were performed using wild type (WT) and Epm2a KO mice that were littermates born from homozygous breeding pairs. At the age of approximately 2 to 3 months, the mice were randomly placed into groups of five to ten mice. 2-DG, 2.0 or 2.5 g/kg, was administered intraperitoneally every other day to grouped mice for a total of eight injections. Control group mice were administered phosphate-buffered saline (PBS). Twenty-four hours after the last injection, the brain sections were prepared for immunohistochemistry, PAS, and Fluoro-Jade C staining. All experiments have been performed in accordance with the Principles of Laboratory Animal Care.
HEK293FT (HEK293) and N2A cell lines were from Invitrogen and ATCC, respectively. HEK293 cells were cultured in DMEM medium supplemented with 4.5 g glucose, 2 mM glutamine, 2 % penicillin, and 10 % fetal bovine serum (FBS). N2A cells were cultured in MEM medium supplemented with 2 mM glutamine, 2 % penicillin, and 10 % FBS.
For primary neuron culture, forebrain cortical neuron layers were microdissected from the brains of postnatal day 2 Epm2a WT or KO mice into Neurobasal medium. The tissue was then digested by 0.1 % trypsin plus 0.25 mM EDTA at 37 °C for 15 min. The resultant cells were titrated, neutralized, filtered, and pelleted by centrifugation at 800 × g for 8 min. The isolated neurons were cultured in Neurobasal medium supplemented with the nutrient B27 and antibiotics (culture medium) in poly-l-lysine-coated plates. To limit astrocyte contamination, 50 μM uridine and 20 μM 5-fluoro-2´-deoxyuridine were added to cultured neurons on the second day of culture. Three days after culture, the neurons were treated in neuron culture medium.
Antibodies and Reagents
Rabbit polyclonal antibodies of muscle GS1 (EP817Y, Abcam), phosphor-GS1 (Ser641/645) (NSB1092, Novus) and GFAP (Abcam); mouse monoclonal antibodies to Tuj-1 (Promega), Myc tag (Invitrogen), Flag tag (M2, Sigma), Flag-Cy3 (Sigma), β-actin (Sigma); and an APC-Annexin V antibody for flow cytometry (BD) were used in the present study. Monoclonal anti-glycogen IgM antibody is described in [23]. Thapsigargin (T9033), tunicamycin (T7765), 2-deoxyglucose (D3179), and Flag peptide (F3290), enzymes and substrates of glucose-6-phosphate dehydrogenase (G6378), myokinase (M3003), pyruvate kinase (P1506), amyloglucosidase (A7420), α-amylase (A6814), glycogen (G0885), NADP + (N5755), glucose-1-phosphate (G6750), UDP-glucose (U4625), phosphoenolpyruvate monopotassium (P7127 for AMP determination), and cyclohexylammonium (P3636 for GS determination) were purchased from Sigma. The PAS staining kit (Richard-Allan Scientific), Malachite Green Phosphate assay kit (10009325, Cayman), Glucose (GO) assay kit (GAGO20, Sigma), ATP assay kit (A22066, Invitrogen) were purchased from the indicated vendors. Recombinant glycogen phosphorylase GPBB (10-663-45308) was purchased from GenWay. Fluoro-Jade C was purchased from Histo-Chem (Jefferson, AR) and used to stain degenerative neurons as previously described [24, 25].
Plasmids and RT-PCR
The coding regions of mouse GS1 (Gys1) and mouse PTG cDNAs were amplified by PCR using reverse transcription (RT) of C57BL/6 J bone marrow mRNA as template and were cloned into a pcDNA vector with Flag tags at the C-terminus. Human PTG cDNA from hepatoma HepG2 cells was cloned into the same vector. Both human and mouse PTG plasmids functioned identically. Gys1 truncated at its N- or C-terminus or both, named ΔN10 (missing the first ten amino acid [aa] residues), Δ639 (missing all aa residues after residue 639), and ΔN10-639 (missing the first ten aa residues and all aa residues after residue 639) were generated by PCR using full-length Gys1 as template. Gene silencing sequences were as follows: Epm2a, 5-ttccagactgaatgggata-3; Nhlrc1, 5-attctcttcttgtgctgga-3; Pygb, 5-tccgatatgaatttgggat-3; and Gys1, 5-acagttgtcgcattcttca-3. The silencing sequence was cloned into a small hairpin RNA lentiviral vector that was created by replacing the CMV promoter of the plenti6/V5TOPO plasmid (Invitrogen) with a mouse U6 promoter and a downstream PGK-EGFP cassette.
Transfection and Western Blot
Generally, HEK293 and N2A cells were transiently transfected with a total of 0.25 μg plasmid and 0.75 μL Lipofectamine 2000 per well in a 24-well plate for 24 h in Opti-MEM medium containing 10 % FBS. The transfected cells were then lysed with 1 % Triton X-100 lysis buffer containing 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 40 mM NaF, 2 mM DTT, and a protease and phosphatase inhibitor cocktail (Sigma). Lysate supernatants were resolved on a reducing, heat-denaturing 10 % SDS-PAGE. To determine laforin phosphatase activity in vivo, isolated glycogen pellets were digested with 5 U/mL α-amylase in PBS, pH 7.4, at 37 °C for 1 h and then lysed with 5X 1 % Triton X-100 plus 0.2 % SDS lysis buffer.
Immunofluorescence and PAS Staining
Mouse brains were infused with freshly prepared 4 % PFA in PBS and maintained in 15 % sucrose for 1 day before being transferred to 30 % sucrose. Frozen brain tissues were cut into 12-μm thick sections. Prepared brain sections and cold methanol-fixed cells were permeabilized with 0.3 % Triton X-100 in 10 mM Tris–HCl buffer for 30 min. Immunofluorescence staining with primary antibody was performed in 10 mM Tris–HCl buffer containing 2 % bovine serum albumin (BSA) at 4 °C, overnight, and with secondary antibody in the BSA–Tris–HCl buffer at room temperature for 2–4 h. PAS staining was performed on permeabilized sections or cells after pretreatment with 0.5 % periodic acid for 30 min (sections) or 1 h (cells) at 37 °C. Additional procedures were performed in accordance with the PAS kit protocol.
Polyglucosan Isolation and α-Amylase Hydrolysis
Polyglucosan was isolated as described in our paper [19]. To isolate polyglucosan particles of cultured neurons, the period of the last centrifugation was extended to 2.5 h. The polyglucosan pellets were dissolved in phosphatase assay buffer containing 50 mM Hepes, pH 5.5–6.0, 50 mM NaCl, 5 mM EGTA and 5 mM mercaptoethanol, and stored at −70 °C. In an α-amylase-resistance assay, frozen–thawed cells were pretreated with 0.2 U/ml α-amylase in PBS buffer at 37 °C for 30 min, then pelleted at 18,000 g for 35 min, and completely hydrolyzed with 1 U/mL of amyloglucosidase and 5 U/mL of α-amylase at 37 °C overnight.
Glycogen Content Determination
Glycogen content in lysate was determined in lysates as described previously [26]. Briefly, 250 μL of 0.2 M sodium acetate, pH 4.8, was added to cells grown in a 24-well plate. The plate was frozen at −70 °C overnight and then subjected to two cycles of freezing with liquid nitrogen and thawing at 37 °C. 0.5 U/mL amyloglucosidase was added per each 200 μL of lysate and glycogen was hydrolyzed into glucose at 55 °C for 2–4 h depending on glycogen content, and then neutralized with 10 μL of 1 N NaOH. Glucose content in the 50 μL of supernatant after centrifugation at 12,000 × g for 10 min was determined by Glucose (GO) assay kit based on the glucose oxidase/peroxidase system. Absorbance was then measured at 540 nm.
Laforin Phosphatase Activity in Polyglucosan In Vitro
Laforin and C265S proteins were purified by Flag peptide competitive elution from anti-Flag antibody immunoprecipitates of laforin-Flag or C265S-Flag, which was transiently overexpressed in HEK293 cells. Transfected cells were lysed with 1 % Triton X-100 lysis buffer as described above and the supernatants were incubated with protein G beads and anti-Flag antibody for 24 h at 4 °C. Immunoprecipitated beads were washed three times with a total of 120 mL lysis buffer at 4 °C for 10 min. Laforin–Flag proteins were eluted with 250 mg/mL Flag peptide in phosphatase buffer at 4 °C overnight with rotation. Phosphatase activity of laforin was determined in phosphatase buffer supplemented with 10 μg GS1-ΔN10 polyglucosan and 100 ng laforin or C265S, in a 40-μL reaction volume at 37 °C for 30 min. After centrifugation, 5 μL of the solution was used for phosphate determination by the Malachite Green Phosphate Assay Kit. The remaining solution was digested with 1 U/mL of amyloglucosidase and 5 U/mL of α-amylase, and then the digestion was stopped by addition of 5X Triton X-100 lysis buffer and subjected to Western blot to detect dephosphorylation of GS1 at Ser641/645 sites.
Glucose-6-Phosphate Determination
G6P content was determined as described previously [27]. Briefly, cells grown in a 12-well plate were covered with 200 μL of 5 % (w/v) HClO4 solution and frozen at −70 °C overnight. The frozen cells were lysed by two cycles of freezing–thawing, collected, neutralized with 33 μL of 10 N NaOH, and centrifuged at 12,000 × g for 10 min. The G6P content in 50 μL of the supernatant was determined spectrophotometrically after the supernatant underwent an enzyme-coupling reaction in a buffer containing 0.1 M Tris–HCl, pH 8.0, 0.3 mM NADP+, 5 mM MgCl2, and 1 μg G6P dehydrogenase for 1 h. The absorbance was measured at 340 nm and G6P content was calculated using the standard molar absorption of NADPH.
Glycogen Synthase Activity Measurement
GS activity was measured as previously described [28, 29]. Briefly, the cells grown in a 12-well plate were trypsinized and suspended in 100 μL of glycine buffer containing 0.15 M glycine, pH 8.5, 20 mM Tris–HCl, 1.0 mM EDTA and 25 mM MgSO4, and then frozen by liquid nitrogen. The cells were then lysed via two cycles of freezing by liquid nitrogen and subsequent thawing at 37 °C. Fifty microliters of the supernatant was then mixed with 50 μL of the glycine buffer supplemented with 5 mM UDPG and 8 mg/mL glycogen. The reaction was carried out at 37 °C for 1 h and was stopped by heating at 100 °C for 5 min. The UDP product was determined by pyruvate kinase-catalyzed transfer of phosphoenolpyruvate (PEP) into pyruvate, which then reacted with 2,4 dinitrophenylhydrazine. This was accomplished by addition of 50 μL of 10 mM PEP–0.4 M KCl solution and 50 μL of 0.4 U/μL pyruvate kinase–0.1 M MgS04 solution to the heat-stopped reaction. This reaction lasted 30 min at 37 °C, colorized by 80 μL of 0.1 % dinitrophenylhydrazine-2 N HCl solution for 5 min and stopped by addition of 100 μL of 10 N NaOH and 200 μL of 95 % ethanol. The optical density of the supernatant after centrifugation at 12,000 × g for 10 min was measured at 520 nm.
AMP Determination
AMP was measured as described [30]. ADP and AMP were determined using a bioluminescent ATP detection kit after they were converted into ATP by pyruvate kinase and myokinase plus pyruvate kinase, respectively, in the presence of inorganic phosphate. Briefly, cells cultured in a 24-well plate were lysed with 100 μL of buffer containing 0.1 M Tris–HCl, pH 7.5 and 1 mM EDTA. ADP in a 10-μL sample was converted into ATP by addition of 20 U/mL pyruvate kinase and 10 μL PEP substrate buffer containing 0.3 mM PEP, 9 mM MgCl2, and 5 mM KCl. AMP in a 10-μL sample was converted to ATP by addition of 120 U/mL myokinase, 20 U/mL pyruvate kinase, and 10 μL of the PEP substrate buffer. The reactions lasted for 30 min at room temperature and were terminated by addition of 200 μL of the Tris–EDTA buffer. The amount of AMP was determined by subtracting the value of the ADP-to-ATP conversion from the value of the AMP-to-ATP conversion.
GPBB Activity Determination
GPBB activity was measured by determining free inorganic phosphate released from G1P in the reverse reaction of glycogen breakdown as described previously [31]. Briefly, cells grown in a 12-well plate were trypsinized, washed twice with 0.9 % NaCl, and frozen at −70 °C in a buffer containing 30 mM triethanolamine, pH 6.8, 0.5 mM EDTA, and 0.5 mM dithiothreitol. The frozen cells were lysed via subjection to two cycles of freezing in liquid nitrogen and thawing at 37 °C. Two hundred microliters of the resultant lysate was mixed with 0.1 mM G1P and 1 % (w/w) glycogen, and after incubation for 30 min at 37 °C, the reaction was stopped by addition of 6 μL of 70 % (w/w) HClO4 and placement on ice for 5 min. The phosphate content in 50 μL of supernatant after centrifugation at 12,000 × g for 10 min was determined using the Malachite Green Phosphate Assay Kit.
Results
ER stress induces glycogen synthesis
To determine the mechanism by which laforin protects cells from stress-induced apoptosis [32, 33], we purified laforin-associated proteins by Flag peptide competitive elution from immunoprecipitates of lysates of HEK293 cells that were transiently transfected with Flag-tagged laforin (Fig. 1a). By the same procedure, we also purified laforin-associated proteins from laforin-transfected Neuro-2a (N2A) cells after treatment (or not) with thapsigargin, an ER stressor that depletes calcium from the ER lumen [34]. As shown in Fig. 1b, in HEK293 and treated N2A cells, laforin was recruited to a glycogen synthesis complex in which GS1 and glycogenin-1 were included; the glycogenolytic enzyme AGL1 was included in the glycogenesis complex only in HEK293 cells. Unexpectedly, in the untreated N2A cells, analysis by mass spectrometry (MS) could not detect any proteins related to glycogen metabolism; this observation indicates a lack of glycogen synthesis in unstressed N2A cells. MS analysis also could not detect other proteins involved in glycogen metabolism, such as glycogen branching enzyme (GBE1), PTG and AMPK, in complex with laforin in any of the cases tested above.

ER stress induces glycogen bodies. a, b Laforin associates with the glycogen metabolic complex. HEK293 and N2A cells were transiently transfected with laforin-Flag plasmid for 24 h before N2A transfectants were treated with or without 2 μM thapsigargin for 8 h. Coomassie blue staining of laforin-Flag eluate of HEK293 cells is shown in a; MS analysis of Flag peptide-purified laforin-associated proteins is shown in b. Only proteins involved in glycogen metabolism are shown. The asterisk denotes that two thirds of the laforin band on Coomassie blue-stained gel was cut out, and the remaining gel was digested for MS analysis. c GS1-positive glycogen granules are induced by ER stressors. N2A cells were treated with or without 2.5 mM 2-DG, 2 μg/mL tunicamycin or 2 μM thapsigargin for 8 h in MEM medium containing 2.5 % FBS and 5 mM (for 2-DG) or 25 mM (for others) glucose. The cells were then fixed and stained with an antibody against GS1, doubly stained with antibodies against GS1 and glycogen or stained with the PAS reagent. Nuclei were stained with DAPI and merged images of immunostainings are shown. Scale bars, 10 μm. Increased ER stress marker (d) and glycogen content (e) in stressed cells. ER stress markers and glycogen content were determined in the lysate of N2A cells treated as in c
Immunocytochemistry of thapsigargin-treated cells revealed large inclusions, which stained intensely positive with a glycogen-specific antibody. These plaques (0.2 to 20 μm in diameter) were 10 to 100 times the size of normal glycogen (20 nm) (Fig. 1c). To confirm that these glycogen masses resulted from ER stress and not from other thapsigargin effects, we also used tunicamycin [35], and 2-deoxyglucose (2-DG) [36, 37], two other well-characterized inducers of ER stress. Immunocytochemistry revealed that tunicamycin and 2-DG also led to the formation of large glycogen granules, similar to thapsigargin. The glycogen granules stained positive for periodic acid-Schiff (PAS), a stain that detects any chains of glucose (whether glycogen or otherwise), and for GS1. Compared to thapsigargin and tunicamycin, 2-DG induced the largest number and sizes of granules (Fig. 1c); however, it increased the amounts of ER stress protein markers, GRP78 and CHOP, the least, possibly indicating that glycogen granules are largely induced at early stages of ER stress (Fig. 1d).
As mentioned, neuronal cells normally make little to no glycogen [18]. We measured the quantities of glycogen in the above cells and found them to be massively elevated (Fig. 1e). Collectively, the results in this section demonstrate that under ER stress, neuronal cells generate large amounts of aggregating glycogen.
GS1 and GPBB Activities in N2A Cells are Allosterically Activated by ER Stress
To determine whether the induced glycogenesis in stressed N2A cells resulted from alterations of the activities of GS1 and glycogen phosphorylase (GPBB, the principal glycogen digesting enzyme), we measured these activities following application of ER stress (2-DG) over a 20-h time course. We also chased the cellular levels of G6P and AMP, respectively the allosteric activators of GS1 and GPBB, and of glycogen. As seen in Fig. 2a, the stress led to a major rise in the level of G6P, GS1 activity, and glycogen, followed subsequently by a large rise in AMP and GPBB activity. Tunicamycin and thapsigargin treatment gave similar results (Fig. 2b).

ER stressors elevate allosteric regulators and enzyme activities required for glycogen synthesis and then breakdown, in that sequence. a Time course of indicated components as induced by 2-DG. N2A cells were treated with 2-DG for indicated times in 5 mM glucose DMEM medium containing 2.5 % FBS. The levels of G6P, glycogen, and AMP, the activity levels of GS1 and GPBB, and the values of untreated vehicle controls, shown at time zero, were monitored. b Same experiment as in a but using all three ER stressors and showing results at selected time points rather than the entire chase; N2A cells used; 2-DG (2.5 mM), tunicamycin (2 μg/mL), thapsigargin (2 μM); G6P at 4 h, GS at 4 h, AMP at 6 h, and GPBB at 6 h. The error bar represents the mean + SD of three separate experiments
GS1 Synthesizes α-Amylase-Resistant Polyglucosan
Normal glycogen is an intricately spherical structure which hides its hydrophobic glucan chains within the sphere and exposes only hydrophilic ends of chains at the surface, allowing solubility, and preventing precipitation and aggregation. Normal glycogen stains positively with PAS and is rapidly digested with amylase. PBs, or LBs, are histochemically defined as large PAS positive structures that have relative resistance to amylase digestion because amylase has restricted access into the densely packed insoluble polyglucosan aggregates [3]. We treated our ER stress-induced cells with α-amylase and found that their large PAS-positive glycogen plaques were indeed resistant to amylase digestion (Fig. 3a), indicating that these structures are highly similar to PBs.

GS1 synthesizes LB-like polyglucosans that resist α-amylase hydrolysis. a GS1 synthesizes α-amylase-resistant polyglucosan in stressed cells. After treatment with 2 μM thapsigargin for 8 h, N2A cells were fixed by methanol. The fixed cells were treated or not with 0.2 U/mL α-amylase for 15 min at 37 °C and then stained with PAS. b, c Overexpressed GS1 synthesizes polyglucosans. HEK293 cells were transiently transfected with Flag-tagged WT GS1, truncated forms of GS1, PTG, or GS2 for 24 h. Next, the transfected cells were fixed by methanol and stained with Cy3-Flag antibody. PAS reagent stained the fixed cells that were pretreated with or without 0.2 U/mL α-amylase for 30 min at 37 °C (b). Glycogen content in lysates of transfected HEK293 cells before or after α-amylase pretreatment was quantified (c). Scale bars, 10 μm
To corroborate our observation in Fig. 2 that increased GS1 activity underlies PB formation, we overexpressed the enzyme as well as three truncated forms which eliminate phosphorylation sites that downregulate the enzyme’s activity when phosphorylated (ΔN10, N-terminal phosphorylation sites; Δ639, C-terminal sites; and ΔN10-Δ639, both N and C-terminal sites). All four forms generated PBs, the largest and most abundant with the non-inactivatable ΔN10-Δ639 form (Fig. 3b, c), and in all cases the PBs exhibited the typical amylase resistance, to varying degrees (Fig. 3c). GS1 is the muscle and brain isoform of GS. Interestingly, when we used the liver isoform of GS, GS2, instead of PBs, normal glycogen was generated. Also, when we overexpressed the PTG protein, a protein that targets the PP1 phosphatase to activate GS, again we did not see PBs, but instead large amounts of readily amylase-digestible normal glycogen (Fig. 3b, c). Precisely why GS2 and PTG lead to normal glycogen rather than PBs is unclear, but in any case it is clear that increased GS1 is associated with PB formation. This result is consistent with the massive presence of GS1, and no other tested enzymes of glycogen metabolism in LB isolated from mouse models of LD [38–42].
GS1 in Polyglucosan Bodies is Phosphorylated, and Laforin is a GS1 Phosphatase in This Location
Abundant GS1 activated allosterically by increased G6P explains the formation of polyglucosans in the ER-stressed cells. We asked whether, in addition to allosteric activation, the GS1 in stressed cells is also activated by dephosphorylation. The converse was the case. GS1 in the PBs was phosphorylated at the GSK3 sites (Fig. 4a, b), which are the main phosphoinhibition sites and reflect the overall phosphorylation state of the enzyme [43]. This suggests an attempt by GSK3 to regulate the amount of polyglucosans being formed by GS1 under ER stress. To confirm that excess GS1 activity, despite its phosphorylation, results in polyglucosans, we overexpressed various deletion and other forms of GS1 with mutations affecting phosphorylation sites. In all cases where the GSK3 Ser 641/645 sites were preserved, these sites became phosphorylated, despite which PBs still formed (Fig. 4c–e).

GS1 is phosphorylated in synthesized polyglucosans. a, b GS1 is phosphorylated in PBs induced by stress. N2A cells treated with or without 2 μM thapsigargin for 8 h were fixed and doubly stained with antibodies to p-GS1 and glycogen (a). After treatment with thapsigargin as a, N2A cells were lysed by 0.55 % NP-40 detergent and PBs were fractioned and subjected to Western blot (b). c, d GS1 is phosphorylated in PBs at physiological conditions. HEK293 cells were transiently transfected with WT GS1, its truncated forms, PTG, or GS2. Twenty-four hours after transfection, the cells were fixed and stained with antibodies to p-GS1 and glycogen (c). Western blot detected p-GS1 in HEK293 cells transfected with GS1 or other plasmids as indicated (d). e Quantitation of glycogen content in polyglucosans isolated from HEK293 cells transfected with indicated plasmids for 24 h. Scale bars, 10 μm
To assess what effect if any laforin has on the phosphorylation state of GS1, we overexpressed laforin with GS1 under normal or ER stress condition. Firstly, laforin intensely localizes on PBs (Fig. 5a). Secondly, and surprisingly, presence of laforin leads to dephosphorylation of GS1. This dephosphorylation is dependent on laforin’s phosphatase action because laforin phosphatase-inactive mutant C265S likewise localized on PBs but did not dephosphorylate GS1 (Fig. 5b–d).

Laforin is a phosphatase of GS1 in PBs. a, b Examination of laforin phosphatase activity on GS1 present in PBs in vivo. GS1-ΔN10 was co-transfected with laforin or C265S into HEK293 cells for 24 h before the transfected cells were treated or not with 2 μM thapsigargin for 8 h. The co-localization of laforin or C265S with p-GS1-ΔN10 in PBs was identified by double staining (a), and the dephosphorylation of p-GS1-ΔN10 by laforin was detected by Western blot (b). c, d Determination of laforin phosphatase activity in vitro. Isolated GS1-ΔN10 polyglucosans, at an amount equal to 10 μg glycogen, was added to phosphatase buffer containing purified laforin or C265S. After a 30-min incubation at 37 °C, dephosphorylation of GS1-ΔN10 was detected by binding of antibody to p-GS1 (c). The released phosphate content by laforin from the GS1-ΔN10 polyglucosans was monitored (d). Scale bars, 10 μm
Stress Accelerates LD
LD is a progressive disorder. LBs are not present in neonatal mice. They appear around age 2 months and become profuse by 9 months. To assess the effect of ER stress on LB formation in LD, we treated postnatal cultured neurons from Epm2a KO mice with ER stressors. Markedly large and more numerous p-GS1 bodies were formed in the nuclei of the cultured neurons from Epm2a KO mice than that from WT mice (Fig. 6a). GS1 was phosphorylated to a much greater extent in the Epm2a KO cells than in WT neurons (Fig. 6b). While WT neurons do not make as many or as large p-GS1 bodies in the cytoplasm under ER stress as Epm2a KO cells, they do make some (Fig. 6a), and the cytoplasmic p-GS1 bodies were biochemically measurable to contain polyglucosan (Fig. 6c). This confirms that the observed PB formation under ER stress in N2A cells (Fig. 4a) occurs equally in stressed neurons.

ER stress accelerates LD. a, b, c PB induction in cultured neurons. Postnatal cortical neurons from WT and Epm2a KO mice were stimulated or not by ER stressors (0.5 mM 2-DG, 0.5 μg/mL tunicamycin, or 0.25 μg/mL thapsigargin) in neuron culture medium for 6 h. The neurons were then fixed and doubly stained with antibodies to p-GS1 and the neuronal marker Tuj-1 (a). The treated neurons were lysed in buffer containing 1 % Triton X-100 and 0.1 % SDS for Western blot (b). Quantitation of glycogen content of the PBs isolated from stressed neurons of WT mice is shown (c). The result is expressed as total glycogen content divided by total protein content in the isolated polyglucosan deposit after it was completely digested by amyloglucosidase and amylase. d PB induction in mice by 2-DG. Age-matched 8-week-old WT and Epm2a KO mice were intraperitoneally administered 2 g/kg 2-DG every other day for a period encompassing eight injections. Twenty-four hours after the last injection, the hippocampal sections of each group were stained by antibodies to GS1, p-GS1, or PAS reagent (bottom). Selected areas of WT sections are enlarged and shown on the right. e Phosphorylated GS1 is highly abundant in PBs in brain as Epm2a KO mice age. Numerous PBs staining positive for GS1, p-GS1, and PAS are observed in the hippocampal CA1 regions of 9-month-old Epm2a KO mice. Only merged photos are shown. f Phosphorylated GS1 is abundant in LBs of hippocampus CA1 region of 6-month-old malin KO mouse. Scale bars, 10 μm
We next tested the effect of ER stress on LB formation in live animals. We treated 8-week-old Epm2a KO mice and WT controls with 2-DG (which crosses the blood–brain barrier) at a dose of 2 g/kg every other day for a period encompassing eight injections. After the last injection, hippocampal sections were analyzed via staining with PAS, GS1, and p-GS1. Very small, almost insignificant amounts of LBs formed in the WT animals. In the Epm2a KO mice, massive amounts of LBs formed to an extent normally seen in 9-month-old animals (Fig. 6d, e), i.e., 16 days of sustained ER stress (eight injections) led to a 7-month equivalent disease progression.
The above results indicate that ER stress drastically accelerates LB formation in LD. They also show that animals possessing laforin resist this effect. These results and the previous section also include the observations that laforin absence leads to increased GS1 phosphorylation and laforin overexpression to decreased GS1 phosphorylation. These observations are paradoxical as decreased GS1 phosphorylation increases GS1 activity, and hence, laforin’s dephosphorylation of GS1 could not serve the purpose of reducing the synthesis of polyglucosans. This dephosphorylation must serve towards another purpose, possibly the degradation of polyglucosans after they have formed and aggregated. This dephosphorylation-led degradation of PBs likely requires malin because massive p-GS1-positive LBs also accumulate in hippocampi of 6-month-old malin KO mice (Fig. 6f).
Stress-Accelerated LD Includes Increased Seizure Susceptibility and Neurodegeneration
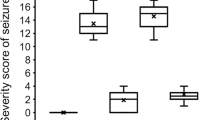
In human LD patients, seizures begin in early teenage years and are initially infrequent and treatable. Over time, they become extremely frequent and intractable. Their neurodegeneration is likewise progressive as documented by radiological and pathological studies [3], and the same applies to LD mouse models, where spontaneous seizures begin only after age 9 months. Both control and Epm2a knockout (KO) mice treated with high dose 2-DG (2.5 mg/kg, see Fig. 7a) developed Racine stage 5 [44] seizures but at a frequency of 40 % in WT animals compared to 80 % of Epm2a KO mice (Fig. 7a). Pathological analysis revealed massive and profuse PBs in Epm2a KO mice, but PBs were now, with the high-dose treatment, also seen in wild-type (WT) animals, although much smaller and fewer than in the KO mice (Fig. 7b). PBs were clearly induced in neuronal processes as we observed only limited co-localization of PBs with astrocytes (Fig. 7c). Neurodegeneration (Jade C positive neurons) was present in both stressed WT and KO mice, again to a much greater extent in the latter (Fig. 7c–e).

Stress-accelerated LD includes increased seizure susceptibility and neurodegeneration. a Tonic–clonic seizure induction by 2-DG. Ten-week-old WT and Epm2a KO mice were given 2-DG, every other day, at doses of 2 g/kg for the first four injections and 2.5 g/kg for the second four injections. The number of mice in each group with a seizure score of 5 induced at each injection was compared. b, c, d 2-DG induced PB formation and neurodegeneration in the hippocampal neurons. Twenty-four hours after the last injection with 2-DG, the hippocampal sections were prepared and stained with antibodies to glycogen only b and glycogen plus GFAP c. In d, the hippocampal sections from the mice treated in a were first stained with Fluoro-Jade C dye and then with an antibody against the astrocyte marker GFAP. Similar regions and merged photos are shown. e Fluoro-Jade C positive neurons in a similar area section of each dentate gyrus of five individual mice were numerated. Scale bars, 10 μm
Are ER Stress-Induced Polyglucosans Toxic or Physiologic?
The above series of experiments revealed associations between ER stress, polyglucosan generation, PB formation, and neurodegeneration. The latter two are vastly enhanced in laforin deficiency. The association of PB with neurodegeneration and with disease, LD, leads to the intuitive notion that polyglucosans are per se toxic. However, the question arises, why does ER stress lead to polyglucosan generation? Indeed, ER stress is frequently followed by cell demise if the stress is not relieved, but is it possible that the polyglucosan generation is an attempt at averting cell death, and are polyglucosans a rapid capture of energy, without construction of elaborate spherical glycogen for subsequent quick usage in times of stress? To begin addressing this question, we subjected to ER stress N2A cells deficient of GS1 or GPBB, the enzymes that synthesize and digest polyglucocan chains, and found that the amount of cell apoptosis with ER stress is markedly higher in the absence of either enzyme (Fig. 8a, b). Similar to GS1 or GPBB, the deficiency of laforin or malin in stressed N2A cells increased apoptosis (Fig. 8b). These results suggest that synthesis and breakdown of polyglucosans enhance the survival of ER-stressed cells and that perhaps polyglucosans, as long as they are able to be degraded, are not toxic but rather part of neuronal survival physiology.

Knockdown of key enzymes related to polyglucosan metabolism enhances cell apoptosis induced by 2-DG. a Gene knockdown efficiency. Scrambled control or each small hairpin (Sh) RNA in combination with Flag-tagged laforin, malin, GPBB, or GS1 was transiently co-transfected into HEK293 cells for 24 h. The cells were then lysed and subjected to Western blot using an antibody to Flag. b N2A cells were transfected with a plasmid expressing the reporter EGFP and Sh-RNA of indicated genes or scrambled control. Blasticidin-resistant cells were treated with 2.5 mM 2-DG in MEM medium containing 2.5 % FBS and 25 mM glucose, or left untreated, for 36 h. The cells were then stained with Annexin V and nuclear dye DAPI and analyzed by flow cytometry. The EGFP-positive population was gated to evaluate apoptosis (Annexin V-positive) and death (DAPI-positive), respectively. One representative of two separate experiments is shown
Discussion
In LD, massive accumulations of LBs, which often outgrow and destroy the very cells they form in, are clearly harmful [3]. This led to the idea that polyglucosans are pathogenic. In the present work, we obtained results that suggest otherwise, that polyglucosans might be a physiologic form of emergency short-term energy storage. Neurons are extremely sensitive to damage from diverse insults, including ischemia, hypoglycemia, energy deprivation, ER stress, oxidative stress, etc. and must therefore possess a protective mechanism activated under these conditions. Stress would increase glucose uptake and conversion to G6P, which would meet immediate energy needs, but which through its potent allosteric effect would also strongly activate GS1. The allosterically activated enzyme would synthesize polyglucosans, which would be degraded subsequently to continue promoting cell survival if stress is sustained. That randomly structured polyglucosans, and not elaborately constructed spheres of normal glycogen, are generated would be consistent with the necessity of very rapid turnover. Consistent with the above hypothesis, we show that stress increases intracellular G6P, increases GS1 activity, leads to polyglucosan formation, and several hours after the activation of GS1 leads to increased AMP and activation of the main polyglucan digesting enzyme GPBB.
One remarkable observation is that the stress-induced G6P-driven GS1 is phosphorylated. This suggests presence of downregulatory controls, without which GS1 would be excessively hyperactive and generate large excesses of polyglucosans, which, precipitating and aggregating, would escape digestion and accumulate to form LBs.
The laforin–malin complex appears to function in the degradation of polyglucosans, through mechanisms yet unknown [19]. Intermingled with polyglucosans are large amounts of p-GS1 [39, 42], and we showed here that laforin dephosphorylates this p-GS1. This dephosphorylation may serve as a primer for laforin–malin complex-mediated degradation of PBs in order to permit glycogen phosphorylase unimpeded progression down the polyglucan chain as it digests successive α1-4 interglucosidic linkages.
LD is a progressive disease. Our experiments revealed that non-LD neuronal cell lines, cultured normal neurons, and brains of live WT animals form polyglucosans under certain conditions. It is conceivable that these conditions are intermittently met in the course of normal life. The absence of laforin or malin would result in failure to digest polyglucosans, leading to their accumulation. This leads to increasing amounts of cell stress and formation of greater amounts of polyglucosans that cannot be digested. This escalating process might underlie the accelerating progression of the disease that leads to dementia and rapid death of previously completely normal children.
The present findings advance our understanding of one of the most severe forms of epilepsy. They also have wider clinical implication because, in addition to LD, PBs characterize the pathologies of certain cases of Alzheimer’s disease [45], temporal lobe epilepsy [46], and several other neurological disorders [1]. Most importantly, our results afford an initial characterization of a novel glycogen (polyglucosan) metabolic process related to cellular stress. Detailed uncovering of this process will be highly informative to the understanding of the bioenergetics and natural neuroprotective mechanisms in neurons.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- CBD:
-
Carbohydrate-binding domain
- 2-DG:
-
2-Deoxyglucose
- ER:
-
Endoplasmic reticulum
- G6P:
-
Glucose-6-phosphate
- GDE1/AGL1:
-
Glycogen debranching enzyme 1
- GSK3:
-
Glycogen synthase kinase 3
- GPBB:
-
Glycogen phosphorylase isoenzyme BB
- GS1:
-
Muscle glycogen synthase
- LB:
-
Lafora body
- LD:
-
Lafora disease
- PB:
-
Polyglucosan body
- N2A:
-
Neuro-2a
- PAS:
-
Periodic acid-Schiff
- PME:
-
Progressive myoclonic epilepsy
- PTG:
-
Protein targeting to glycogen
References
Cavanagh JB (1999) Corpora-amylacea and the family of polyglucosan diseases. Brain Res Brain Res Rev 29:265–295
Gambetti P, Di Mauro S, Hirt L, Blume RP (1971) Myoclonic epilepsy with lafora bodies. Some ultrastructural, histochemical, and biochemical aspects. Arch Neurol 25:483–493
Minassian BA (2002) Progressive myoclonus epilepsy with polyglucosan bodies: Lafora disease. Adv Neurol 89:199–210
Sakai M, Austin J, Witmer F, Trueb L (1970) Studies in myoclonus epilepsy (Lafora body form). II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology 20:160–176
Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, Avanzini G, Elia M, Ackerley CA, Jovic NJ, Bohlega S, Andermann E, Rouleau GA, Delgado-Escueta AV, Minassian BA, Scherer SW (2003) Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet 35:125–127
Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, Dunham I, Gardner R, Fong CY, Carpenter S, Jardim L, Satishchandra P, Andermann E, Snead OC 3rd, Lopes-Cendes I, Tsui LC, Delgado-Escueta AV, Rouleau GA, Scherer SW (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet 20:171–174
Chan EM, Ackerley CA, Lohi H, Ianzano L, Cortez MA, Shannon P, Scherer SW, Minassian BA (2004) Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum Mol Genet 13:1117–1129
Liu Y, Wang Y, Wu C, Liu Y, Zheng P (2006) Dimerization of Laforin is required for its optimal phosphatase activity, regulation of GSK3beta phosphorylation, and Wnt signaling. J Biol Chem 281:34768–34774
Wang J, Stuckey JA, Wishart MJ, Dixon JE (2002) A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J Biol Chem 277:2377–2380
Worby CA, Gentry MS, Dixon JE (2006) Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem 281:30412–30418
Tagliabracci VS, Heiss C, Karthik C, Contreras CJ, Glushka J, Ishihara M, Azadi P, Hurley TD, DePaoli-Roach AA, Roach PJ (2011) Phosphate incorporation during glycogen synthesis and Lafora disease. Cell Metab 13:274–282
Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, Delgado-Escueta AV, Minassian BA, Depaoli-Roach AA, Roach PJ (2007) Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A 104:19262–19266
Liu R, Wang L, Chen C, Liu Y, Zhou P, Wang Y, Wang X, Turnbull J, Minassian BA, Liu Y, Zheng P (2008) Laforin negatively regulates cell cycle progression through glycogen synthase kinase 3beta-dependent mechanisms. Mol Cell Biol 28:7236–7244
Lohi H, Ianzano L, Zhao XC, Chan EM, Turnbull J, Scherer SW, Ackerley CA, Minassian BA (2005) Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum Mol Genet 14:2727–2736
Wang Y, Liu Y, Wu C, Zhang H, Zheng X, Zheng Z, Geiger TL, Nuovo GJ, Liu Y, Zheng P (2006) Epm2a suppresses tumor growth in an immunocompromised host by inhibiting Wnt signaling. Cancer Cell 10:179–190
Puri R, Suzuki T, Yamakawa K, Ganesh S (2009) Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease. J Biol Chem 284:22657–22663
Gentry MS, Worby CA, Dixon JE (2005) Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A 102:8501–8506
Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B, Criado-Garcia O, Fernandez-Sanchez E, Medrano-Fernandez I, Dominguez J, Garcia-Rocha M, Soriano E, Rodriguez de Cordoba S, Guinovart JJ (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 10:1407–1413
Zeng L, Wang Y, Baba O, Zheng P, Liu Y (2012) Laforin is required for the functional activation of malin in endoplasmic reticulum stress resistance in neuronal cells. FEBS J 279:2467–2478
Brown AM (2004) Brain glycogen re-awakened. J Neurochem 89:537–552
Gruetter R (2003) Glycogen: the forgotten cerebral energy store. J Neurosci Res 74:179–183
Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y, Akagi T, Gomi H, Suzuki T, Amano K, Agarwala KL, Hasegawa Y, Bai DS, Ishihara T, Hashikawa T, Itohara S, Cornford EM, Niki H, Yamakawa K (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet 11:1251–1262
Baba O (1993) Production of monoclonal antibody that recognizes glycogen and its application for immunohistochemistry. Kokubyo Gakkai Zasshi 60:264–287
Bian GL, Wei LC, Shi M, Wang YQ, Cao R, Chen LW (2007) Fluoro-Jade C can specifically stain the degenerative neurons in the substantia nigra of the 1-methyl-4-phenyl-1,2,3,6-tetrahydro pyridine-treated C57BL/6 mice. Brain Res 1150:55–61
Schmued LC, Stowers CC, Scallet AC, Xu L (2005) Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res 1035:24–31
Worby CA, Gentry MS, Dixon JE (2008) Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J Biol Chem 283:4069–4076
Cervenansky C, Arias A (1984) Glucose-6-phosphate dehydrogenase deficiency in pleiotropic carbohydrate-negative mutant strains of Rhizobium meliloti. J Bacteriol 160:1027–1030
Cabib E, Leloir LF (1958) The biosynthesis of trehalose phosphate. J Biol Chem 231:259–275
Leloir LF, Goldemberg SH (1960) Synthesis of glycogen from uridine diphosphate glucose in liver. J Biol Chem 235:919–923
Spielmann H, Jacob-Muller U, Schulz P (1981) Simple assay of 0.1–1.0 pmol of ATP, ADP, and AMP in single somatic cells using purified luciferin luciferase. Anal Biochem 113:172–178
Maridakis GA, Sotiroudis TG (1996) A catalytic assay for the detection of sub-nanomolar glycogen phosphorylase concentrations. Anal Biochem 240:304–306
Liu Y, Wang Y, Wu C, Zheng P (2009) Deletions and missense mutations of EPM2A exacerbate unfolded protein response and apoptosis of neuronal cells induced by endoplasm reticulum stress. Hum Mol Genet 18:2622–2631
Wang Y, Liu Y, Wu C, McNally B, Liu Y, Zheng P (2008) Laforin confers cancer resistance to energy deprivation-induced apoptosis. Cancer Res 68:4039–4044
Treiman M, Caspersen C, Christensen SB (1998) A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol Sci 19:131–135
Helenius A (1994) How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell 5:253–265
Yu SM, Kim SJ (2010) Endoplasmic reticulum stress (ER-stress) by 2-deoxy-d-glucose (2DG) reduces cyclooxygenase-2 (COX-2) expression and N-glycosylation and induces a loss of COX-2 activity via a Src kinase-dependent pathway in rabbit articular chondrocytes. Exp Mol Med 42:777–786
Zhang K, Kaufman RJ (2006) The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66:S102–S109
DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, Roach PJ (2010) Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J Biol Chem 285:25372–25381
Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, Depaoli-Roach AA, Roach PJ (2008) Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem 283:33816–33825
Tiberia E, Turnbull J, Wang T, Ruggieri A, Zhao XC, Pencea N, Israelian J, Wang Y, Ackerley CA, Wang P, Liu Y, Minassian BA (2012) Increased laforin and laforin binding to glycogen underlie lafora body formation in malin-deficient Lafora disease. J Biol Chem 287:25650–25659
Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG, Kameka AP, Pencea N, Zhao X, Ackerley CA, Minassian BA (2010) Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol 68:925–933
Valles-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L, Serafin A, Canas X, Soriano E, Delgado-Garcia JM, Gruart A, Guinovart JJ (2011) Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol Med 3:667–681
Skurat AV, Roach P (2004) Regulation of glycogen synthesis, p. xviii, 1540 p. In: LeRoith D, Taylor SI, Olefsky JM (eds) Diabetes mellitus: a fundamental and clinical text, 3rd edn. Lippincott Williams & Wilkins, Philadelphia
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294
Inoue M, Yagishita S, Itoh Y, Amano N, Matsushita M (1996) Coexistence of paired helical filaments and polyglucosan bodies in the same neuron in an autopsy case of Alzheimer's disease. Acta Neuropathol 92:511–514
Abubakr A, Wambacq I, Donahue JE, Zappulla R (2005) The presence of polyglucosan bodies in temporal lobe epilepsy: its role and significance. J Clin Neurosci 12:911–914
Acknowledgments
This work was supported by grants from the National Institutes of Health, 1R21NS062391 and 5R21CA164469, and the Canadian Institutes of Health Research to BAM. BAM holds the University of Toronto Michael Bahen chair in epilepsy research.
Conflict of Interest
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Y., Ma, K., Wang, P. et al. Laforin Prevents Stress-Induced Polyglucosan Body Formation and Lafora Disease Progression in Neurons. Mol Neurobiol 48, 49–61 (2013). https://doi.org/10.1007/s12035-013-8438-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8438-2