
Abstract
A novel strategy for the chemoselective conversion of sulfoxides to sulfide utilizing D-camphorsulfonic acid (D-CSA) as a reducing reagent has been developed under metal and additive-free conditions. A variety of sulfoxides such as alkyl–aryl, allyl–aryl, benzyl–aryl, aryl–ethyl sulfoxides have been effectively utilized to achieve the corresponding reduced products in good to excellent yields under mild conditions without any additive. The proposed method offers a practical solution for the deoxygenation of sulfoxides, which has potential applications in various fields such as pharmaceuticals, materials science, and organic synthesis.
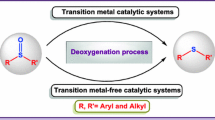
Graphical abstract
A novel strategy for the chemoselective deoxygenation of diverse sulfoxides without metal and additive has been disclosed using D-camphorsulfonic acid (D-CSA) as the reducing agent. The reaction proceeds under mild conditions to afford the sulphides in moderate to good yields with great tolerance of diverse functional groups.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Organosulfur chemistry is one of the important areas of research in organic synthesis. Numerous pharmacological compounds and bioactive natural products contain ‘S’ atom and such moieties serve as the building block for synthesizing many biologically active compounds.1 Reduction reaction is one of the most fundamental reactions in organic chemistry, which generally involves relatively spotless reagents like molecular hydrogen with extremely heavy metal salts or different phosphines.2 The deoxygenation strategies of diverse sulfoxides have been summarized in a few reviews.3 Significant progress has been achieved during the last two decades in the field of thioether synthesis through the reduction of sulfoxides using various deoxygenation reagents such as metal complexes, oxo and phosphines containing metal complexes, cyanuric chlorides, triflic anhydride, phosphines, electrophilic chlorine, and strong acids with additives, etc.4,5,6 Among various reducing agents, acids have gained profound attention in reducing sulfoxides in combination with different additives. It was extracted from the literature that various acids, such as 3-mercaptopropionic, thioacetic, ascorbic, sulfuric, hydrochloric, and Lewis acid (anhyd AlCl3), etc., have been utilized as a deoxygenating agent in combination with different additives such as I2, NBS, TMCS, NaI, H2O2, etc., for the reduction of various sulfoxides.7,8,9,10,11,12 However, these reactions are generally conducted under harsh conditions utilizing toxic and expensive reagents where chemoselective conversion can’t be carried out always due to the reduction of a few functional groups (C=O, NO2, C≡N, C=C) during the course of reactions. In addition, using phosphine-based ligands affords phosphonium oxide as the side product, which creates difficulties in the purification of the products. To address the above hurdles, herein, we report a novel and chemoselective deoxygenation strategy of diverse sulfoxides using common organic reagent D-camphorsulfonic acid (D-CSA) as the reducing agent without any additive. This reagent is well known as a promising organocatalyst with low sensitivity to air and moisture.13
2 Experimental procedure
2.1 General procedure for deoxygenation of sulfoxides
A mixture of corresponding sulfoxide derivative (1) (0.72 mmol) and D-Camphorsulfonic acid (1.8 mmol) in dry acetonitrile (2 mL) was stirred at 90°C for 12 h. After completion of the reaction (monitored by TLC), the solvent was removed under reduced pressure, and the residue was extracted with EtOAc (2 × 40 mL) and washed with H2O (2 × 20 mL). The combined organic layer was washed with brine (2 × 15 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography to obtain the desired sulfide (3).
3 Results and discussion
Initially, 1-methoxy-4-[(phenylsulfinyl)methyl]benzene (1a) was considered as the model substrate, and it was treated with 1 equiv. of various acids such as benzoic acid, trifluoromethane sulfonic acids, trifluoro acetic acid, pivalic acid, and D-camphorsulfonic acid (D-CSA) in dichloroethane at 90°C for 24 h (Table 1, Entries 1–5). It was found that D-camphorsulfonic acid afforded the best result to obtain the deoxygenated product (3a) in 41% yield (Table 1, Entry 5). On increasing the loading of D-CSA up to 2 equiv. and subsequently, up to 2.5 equiv., enhanced yield of the corresponding sulfide (3a) could be attained (53% and 62%, respectively) (Table 1, Entries 6 and 7). Further enhancement of D-CSA loading up to 3 equiv. did not increase the yield of the product. Sequentially, the reaction was optimized by screening various solvents such as DMF, DMSO, ethanol, toluene, and acetonitrile (Table 1, Entries 8–12). It was observed that CH3CN afforded the best yield (85%) within 12 h, probably due to the lowering of the activation energy through coordination with the polar sulfoxide (Table 1, Entry 12). The product yields decreased when the deoxygenations were carried out at 50°C and 60°C instead of 90°C (Table 1, Entries 13 and 14). Subsequently, the conduction of the reaction at room temperature afforded the deoxygenation product (3a) in only 22% yield (Table 1, Entry 15). Thus, the optimal reaction conditions were established as follows: 2.5 equiv. of D-CSA in CH3CN (2 mL) at 90°C for 12 h. With the optimized conditions in hand, we attempted the deoxygenation reaction of several other benzylphenyl sulfoxides under the standardized reaction conditions (Scheme 1). Different electron-donating and -withdrawing substituents (OMe, Me, OPh, CO2Me, CN, NO2, F, Br, Cl) on the benzyl ring of benzylphenyl sulfoxides were well tolerated under the optimized reaction conditions and provided moderate to good yields of corresponding sulfides (66–85%). Benzylphenyl sulfoxide (1e) without substituents afforded the desired product (3e) in 64% yield. Notably, the presence of ortho-substituents (F, Cl, Br, Me, CN, CO2Me) on the benzyl ring did not hamper the reaction yields (3h-3m). Meta-substituted sulfoxides (1n-1p) also afforded good yields of corresponding thioethers (69–74%, 3n-3p). In addition, chloro and methyl groups at the para-position on the phenyl ring of benzylphenyl sulfoxides also produced target thioethers (3q-3s) in good yields (Scheme 1).

Substrates scope.a
Next, we explored the substrate scopes for aryl–ethyl (1t–1v), aryl–alkyl (1w, 1x), aryl–allyl (1y) sulfoxides under optimal conditions. Notably, 1-chloro-4-(methylsulfinyl)benzene (1w), 1-methoxy-3-(methylsulfinyl)benzene (1x), and 1-(allylsulfinyl)-4-methylbenzene (1y), afforded the related thioethers (3w, 3x, 3y) in 62%, 42%, and 73% yields, respectively (Scheme 1). Whereas, in the case of (phenethylsulfinyl)benzenes (1t–1v), the reduction reactions were found to be very sluggish and provided the corresponding sulphides (3t–3v) in 31%, 16%, and 28% yields under standard conditions even after treatment for 24 h. In all cases, starting materials remained, which were recovered. Surprisingly, these reactions almost proceeded to completion (58–80%) within just 5 h upon the addition of a catalytic amount of NBS under the standardized conditions (Scheme 1, compounds 3t–3v). The remaining starting materials were recovered without any side product in all cases. The presence of NBS in this reaction was examined in a few cases. The addition of a catalytic amount of NBS under standard conditions enhanced the reaction rate to complete within 4 h with a slight increase in the yields of the products along with remaining starting materials (3a, 3e, and 3f). Unfortunately, the established protocol did not work with diaryl sulfoxides (1z and 1za).
The scalability of this protocol was verified by a gram-scale reaction where 84% yield of the target product (3a) was obtained (Scheme 2).

Gram scale reaction of 1a.
A competition experiment was carried out to determine the electronic preference of this transformation. The reaction between equimolar mixtures of 1-methoxy-4-[(phenylsulfinyl)methyl]benzene (1a) and methyl 4-[(phenylsulfinyl)methyl]benzoate (1f) favored both the electron-rich (1a) and electron-deficient (1f) sulfoxides in an equal (1:1) ratio under optimal conditions (Scheme 3).

Electronic effect on the reaction.
A mechanism for this deoxygenation reaction is proposed in Scheme 4. Benzylphenyl sulfoxide (1e) reacts with D-CSA to form intermediate A, which, after intramolecular rearrangement followed by S–O bond cleavage, generates benzylphenyl sulfide (3e) along with dioxirane species (B).

Proposed pathway of deoxygenation.
4 Conclusions
In conclusion, we have successfully developed a strategy for the deoxygenation reaction of diverse sulfoxides under mild reaction conditions. This strategy using D-camphorsulfonic acid is an efficient reducing agent for deoxygenating diverse sulfoxides under metal and additive-free conditions. This protocol might have broad synthetic applications in organic synthesis because of the mild reaction conditions, high chemoselectivity, simplicity in operation, and broad substrate scope for synthesizing drug intermediates and biologically active products.
References
(a) Feng Q, Li Y, Wang N, Hao Y, Chang J, Wang Z, Zhang X, Zhang Z and Wang L 2020 A biomimetic nanogenerator of reactive nitrogen species based on battlefield transfer strategy for enhanced immunotherapy Nano. Micro. Small. 16 25; (b) Liu L, Li J, Yue F, Yan X, Wang F, Bloszies S and Wang Y 2018 Effects of arbuscular mycorrhizal inoculation and Biochar Amendment on maize growth, cadmium uptake and soil cadmium speciation in CD-contaminated soil Chemosphere 194 495; (c) Song K, Zhu W, Li X and Yu Z 2020 A novel mechanical robust, self-healing and shape memory hydrogel based on PVA reinforced by cellulose nanocrystal Mater. Lett. 260 126884; (d) Shamsi G A, Mortazavi Z, Mirshekar M A, Mohammadi M, Moradi K N, Jafari M S and Shahraki M 2020 Comparative effects of curcumin versus nano-curcumin on insulin resistance, serum levels of Apelin and lipid profile in type 2 diabetic rats Diabetes Metab. Syndr. Obes. 13 2337; (e) Zhu W, Deng M, Chen D, Zhang Z, Chai W, Chen D, Xi H, Zhang J, Zhang C and Hao Y 2020 Dual-phase CsPbCl3–Cs4PbCl6 perovskite films for self-powered, visible-blind UV photodetectors with fast response ACS Appl. Mater. Interfaces 12 32961
(a) Kunieda N, Nokami J and Kinoshita M 1974 Stereoselective reactions of (+)-(r)-α(p-tolylsulfinyl)acetophenone with alkyl Grignard reagents in ethyl ether Chem. Lett. 3 369; (b) Solladié G, Demailly G and Greck C 1985 Reduction of β-hydroxysulfoxides: Application to the synthesis of optically active epoxides Tetrahedron Lett. 26 435
(a) Li W, Chen X, Zheng T and Zou Q 2019 Research Progress on Reduction of Sulfoxides to Thiothers Chin. J. Org. Chem. 39 2443; (b) Lin H, Wu L and Kazemi M 2021 A decade updates (2011–2020): Reduction of sulfoxides to sulphides Synth. Commun. 11 1609
(a) Castrillón J P and Szmant H H 1965 Reduction of sulfoxides by triphenylphosphine and carbon tetrachloride J. Org. Chem. 30 1338; (b) Kikuchi S, Konishi H and Hashimoto Y 2005 The deoxygenation of sulfoxide mediated by the PH3P/Lewis acid combination and the application to the kinetic resolution of racemic phosphines using optically active sulfoxide Tetrahedron 61 3587; (c) Bahrami K, Khodaei M M and Khedri M 2007 A novel method for the deoxygenation of sulfoxides with the PPH3/Br2/CuBr system Chem. Lett. 36 1324; (d) Jang Y, Kim K T and Jeon H B 2013 Deoxygenation of sulfoxides to sulfides with thionyl chloride and triphenylphosphine: Competition with the Pummerer reaction J. Org. Chem. 78 6328; (e) Ghorbani V R, Shiri L and Ghorbani C A 2014 A novel method for the reduction of sulfoxides with the N, N, N, N-tetrabromobenzene-1,3-disulfonamide (TBBDA)/PPh3 system C. R. Chimie. 17 1002
(a) Clarke A K, Parkin A, Taylor R J, Unsworth W P and Rossi A J A 2020 Photocatalytic deoxygenation of sulfoxides using visible light: Mechanistic investigations and synthetic applications ACS Catal. 10 5814; (b) Sousa S C A and Fernandes A C 2009 Highly efficient and chemoselective reduction of sulfoxides using the system silane/oxo-rhenium complexes Tetrahedron Lett. 50 6872; (c) Fernandes A C, Fernandes J A, Romão C C, Veiros L F and Calhorda M J 2010 Highly efficient reduction of sulfoxides with the system borane/oxo-rhenium complexes Organometallics 29 5517; (d) Sousa S C, Bernardo J R, Wolff M, Machura B and Fernandes A C 2014 Oxo-rhenium(V) complexes containing heterocyclic ligands as catalysts for the reduction of sulfoxides Chem. Eur. J. 2014 1855
(a) García N, García G P, Fernández R M A, Rubio R, Pedrosa M R, Arnáiz F J and Sanz R 2012 Pinacol as a new green reducing agent: Molybdenum-catalyzed chemoselective reduction of sulfoxides and Nitroaromatics Adv. Synth. Catal. 354 321; (b) Garcia N, Garcia G P, Fernandez R M A, Garcia D, Pedros M R, Arnaiz F J and Sanz R 2013 An unprecedented use for glycerol: Chemoselective reducing agent for sulfoxides Green Chem. 15 999; (c) Fernandes A C and Romão C C 2006 A novel method for the reduction of sulfoxides and pyridine N-oxides with the system silane/MOO2CL2 Tetrahedron 62 9650; (d) Fernandes A C and Romão C C 2007 Reduction of sulfoxides with Boranes catalyzed by MOO2Cl2 Tetrahedron Lett. 48 9176; (e) Sakai N, Shimada R and Ogiwara Y 2021 Indium-catalyzed deoxygenation of sulfoxides with hydrosilanes Asian J. Org. Chem. 10 845; (f) Cabrita I, Sousa S C A and Fernandes A C 2010 Reduction of sulfoxides catalyzed by Oxo-complexes Tetrahedron Lett. 51 6132; (g) Enthaler S 2012 Zinc-catalyzed deoxygenation of sulfoxides to sulfides applying [b(pin)]2 as deoxygenation reagents Catal. Lett. 142 1306; (h) Joshi A, Iqbal Z and De S R 2022 1,2-bis(diphenylphosphino)ethane (DPPE)/NBS: An unprecedented combination for deoxygenation of sulfoxides under mild conditions ChemistrySelect 7 e202202924; (i) Acosta G P, Mahecha M C and Gamba S D 2020 Electrophilic chlorine from chlorosulfonium salts; A highly chemoselective reduction of sulfoxides Chem. Eur. J. 26 10348
Drabowicz J and Mikolajczyk M 1978 An iodine-catalysed reduction of sulphoxides by formamidinesulphinic acid Synthesis 7 542
Karimi B and Zareyee D 2003 n-bromosuccinimide and iodine catalyzed highly efficient deoxygenation of sulfoxides to thioethers using 3-mercaptopropionic acid under mild reaction conditions Synthesis 12 1875
Jabbari A, Zarei M and Jamaleddini A 2012 I2 as a mild and efficient catalyst in deoxygenation of sulfoxides with thioacetic acid J. Sulphur Chem. 33 413
Abbasi M, Mohammadizadeh M R and Moradi Z 2016 Reduction of sulfoxides with ascorbic acid catalyzed by NBS Chem. Soc. Jpn. 89 405
Wang J, Shi F, Dai D, Xiong L and Yang Y 2021 n-bromosuccinimide/HCl mediated reduction of sulfoxides to sulfides Sulfur Silicon Relat. Elem. 196 43
Kong Z, Pan C, Li M, Wen L and Guo W 2021 Scalable electrochemical reduction of sulfoxides to sulfides Green Chem. 23 2773
Brahmachari G, Nurjamal K, Karmakar I and Mandal M 2019 Camphor-10-sulfonic acid (CSA): A water compatible organocatalyst in organic transformations Curr. Organocatal. 5 165
Acknowledgements
Joshi A and Sandeep Singh are thankful to NIT Uttarakhand and CSIR, New Delhi, respectively, for their fellowships. The authors are thankful to NIT Uttarakhand and TEQIP-III for financial support. The funding from DST-SERB, New Delhi (103/2014 dt. 18.07.2015) is also greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Joshi, A., Singh, S. & De, S.R. Deoxygenation of sulfoxides using D-camphorsulfonic acid as an efficient reducing agent under metal and additive-free conditions. J Chem Sci 136, 33 (2024). https://doi.org/10.1007/s12039-024-02261-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-024-02261-x