
Abstract
The hallmarks of the adaptive immune response are specificity and memory. The cellular response is mediated by T cells which express cell surface T cell receptors (TCRs) that recognize peptide antigens in complex with major histocompatibility complex (MHC) molecules on antigen presenting cells (APCs). However, binding of cognate TCRs with MHC-peptide complexes alone (signal 1) does not trigger optimal T cell activation. In addition to signal 1, the binding of positive and negative costimulatory receptors to their ligands modulates T cell activation. This complex signaling network prevents aberrant activation of T cells. CD28 is the main positive costimulatory receptor on naïve T cells; upon activation, CTLA4 is induced but reduces T cell activation. Further studies led to the identification of additional negative costimulatory receptors known as checkpoints, e.g. PD1. This review chronicles the basic studies in T cell costimulation that led to the discovery of checkpoint inhibitors, i.e. antibodies to negative costimulatory receptors (e.g. CTLA4 and PD1) which reduce tumor growth. This discovery has been recognized with the award of the 2018 Nobel prize in Physiology/Medicine. This review highlights the structural and functional roles of costimulatory receptors, the mechanisms by which checkpoint inhibitors work, the challenges encountered and future prospects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
T cells are key players in the adaptive immune response. They perform a wide range of activities: secreting cytokines that affect B cell and macrophage responses, kill infected/tumor cells etc. T cell recognize peptide antigens that are presented on Major Histocompatibility Complex (MHC) encoded molecules on antigen presenting cells (APC) and regulate immune responses (figure 1). Hyperactive T cells are observed in several autoimmune diseases such as multiple sclerosis, insulin dependent diabetes mellitus etc. On the other hand, reduced T cell function makes individuals susceptible to pathogens and tumors as observed in patients suffering from acquired immunodeficiency syndrome. The multifarious activities of T cells make them a target for immunotherapy, wherein their function is modulated to suppress the response during autoimmunity, hypersensitivity and transplantation (Buckley 2000).

Specificity during T cell activation involves the binding of cognate T cell receptors to peptide-loaded MHC molecules. Antigen presenting cells like dendritic cells and macrophages process cellular protein into peptides. MHC molecules bind and present some of these peptides on the cell surface. Optimal T cell activation involves the binding of cognate TCRs to MHC molecules (signal 1) and the binding of positive costimulatory receptors, e.g.: CD28 to their ligands on the Antigen Presenting Cells (e.g.CD80/CD86).
The development and differentiation of T cells occurs in the thymus, which is located above the heart. During this process, thymocytes are selected for their ability to express a cell surface T cell receptor (TCR) that recognizes self MHC molecules, termed as positive selection. Thymocytes that cannot express TCRs or express TCRs that bind to self MHC molecules with high affinity are deleted by the process of negative selection. Consequently, less than 5% of thymocytes survive this rigorous selection process to enter the peripheral circulation (Majumdar et al. 2018). Subsequently, the initiation of the cellular immune response occurs in the secondary lymphoid organs. TCRs present on T cells interact with antigen-bound MHC on dendritic cells (DC) to initiate the T cell activation process (figure 2). The interactions between TCRs and their cognate antigens, under appropriate conditions, lead to clonal proliferation and differentiation of naïve T cells into effector T cells. These effector cells migrate to different tissues based on their surface homing receptors, following a chemokine gradient to perform their functions. A crucial determinant of the differentiation process of naïve T cells is the strength of signal. The strength of activation signal received by the T cells following TCR-MHC-peptide complex interaction depends on the affinity/avidity and duration of this interaction, the amount of antigen present and the presence of costimulatory signals (Ahmed and Nandi 2011).

The life history of T cells. Pluripotent stem cells from the bone marrow reach the thymus via the circulatory system. T cell differentiation and maturation occurs in the thymus. Naïve T cells egress out of the thymus and reach secondary lymphoid organs, such as lymph nodes where they interact with Antigen Presenting cells, e.g. dendritic cells. The binding of cognate TCRs to their antigens leads to activation and proliferation of T cells. Once the antigen is cleared, the majority of T cells undergo cell death; however, a subset remains as memory T cells.
2 Basic principle of T cell costimulation
T cell specificity is via the cognate binding of TCRs to their MHC ligands; however, this binding alone is insufficient to activate T cells. In fact, T lymphocytes require two principal signals for complete activation: a specific signal through the TCR or signal 1 along with ‘costimulation’ or signal 2 (figure 3). Consequently, the lack of costimulation after signal 1 activation results in the development of peripheral immune tolerance, an aspect important to prevent aberrant activation of T cells. The primary positive costimulatory receptor in naïve T cells is CD28, which is constitutively expressed. The upregulation of costimulatory ligands, e.g. CD80 and CD86, is important in delivering the costimulatory signal to T cells activated by the TCR, i.e. signal 1 (Jenkins et al. 1991; Gross et al. 1992; Harding et al. 1992). Costimulation is crucial for the regulation of T cell activation to occur under immunologically relevant conditions. Once T cells are activated, Cytotoxic T lymphocyte associated antigen 4 (CTLA4 or CD152) is upregulated which binds to CD80 and CD86 with higher affinity compared to CD28 and downregulates the activation process (Brunet et al. 1987; Walunas et al. 1994; Krummel and Allison 1995). In fact, T cell activation can be compared to running an automobile. Turning on the ignition (signaling by TCR-CD3 or signal 1) is important but to get the car to move, one has to step on the accelerator (positive costimulatory receptor signaling or signal 2). Once the car has started to move, one may need to step on the brake (negative costimulatory receptor) to reduce the speed or stop. This analogy between running a car and T cell activation is often referred to as the ignition-accelerator-brake model of T cell activation (figure 3). Interestingly, CD28 and CTLA4 are closely linked in the human chromosome 2 on the q33–34 band (Lafage-Pochitaloff et al. 1990). This basic principle of costimulation is important as it led to the concept of “checkpoints” for T cells, i.e. negative costimulatory receptors such as CTLA4 and PD1 (CD279) that regulate T cell activation. In fact, the development of “checkpoint inhibitors” or antibodies to negative costimulatory receptors led to therapeutic strategies to generate higher anti-tumor responses by T cells, the primary focus of this review. There are several laboratories that contributed to the area of T cell costimulation, reinforcing that science is a group effort. Table 1 lists the major events in the field that led to the development of checkpoint inhibitors and the award of the 2018 Nobel prize in Physiology and Medicine.

Costimulatory receptors, CD28 and CTLA-4, regulate naïve T cell activation. The initial interaction of naïve T cells occurs via the binding of cognate TCRs to peptide-loaded MHC molecules (signal 1 or ignition). Robust T cell activation requires the interaction of CD28 with its ligands CD80/CD86 (signal 2) along with signal 1 (signal 1+2 or acceleration). Post activation, CTLA-4 is induced which binds with greater avidity to CD80/CD86 and lowers T cell activation (brake). This model is often referred to as the ignition, accelerator and brake model of T cell activation.
3 CD28 and CTLA4
As mentioned previously, the regulation of T cell activation is important to prevent aberrant activation as seen in autoimmunity. CD28 is a 44 kDa glycoprotein, which is homodimeric in Nature It is expressed on almost all T cells in rodents, most human CD4+ T cells and half of circulating human CD8+ T cells (Beyersdorf et al. 2015). CD28 encodes a leader sequence, an extracellular domain consisting of three complementarity determining regions (CDRs), a transmembrane region and an intracellular domain. CD28 interacts with CD80/CD86 present on APCs. CD86 is constitutively expressed on APCs but CD80 is almost absent on resting cells and is upregulated during inflammation. The interaction between CD28 and its ligands promotes production of high levels of interleukin (IL)-2 and survival factors, leading to initiation of T cell responses (Thompson et al. 1989; Boise et al. 1995; Michel et al. 2001; Boomer and Green 2010). Antibodies to some cell surface proteins, e.g. CD2, CD5, CD9, CD44, or cytokines, e.g. IL1 plus IL6, increase the in vitro proliferation of T cells; however, the amounts of IL2 produced by the CD28 pathway is much higher and lead to greater and sustained proliferation of T cells (Holsti et al. 1994; Yashiro et al. 1998). Unlike the TCR-CD3 signaling pathway, the CD28 pathway leading to high amounts of IL2 and greater T cell proliferation cum survival (Rudd et al. 2009) is resistant to the immunosuppressive drug, cyclosporine (June et al. 1987). Not surprisingly, the proliferation of Cd28-/- T cells is severely reduced upon activation and the formation of germinal centers, which are the hallmarks of an active adaptive immune response, is compromised (Green et al. 1994; Lucas et al. 1995; Ferguson et al. 1996).
CTLA4, a 33–45 kDa transmembrane glycoprotein, is present in intracellular vesicles but expressed on surface of activated T cells (Walunas et al. 1994; Walker and Sansom 2011). CTLA4 is a 223 amino acid protein and the molecule consists of a 126 amino acid variable (V)-like domain following the cleavage of 35 amino acid long signal peptide, 21 amino acid transmembrane domain and a 41 amino acid cytoplasmic domain. CTLA4 has multiple isoforms: full length, one lacking the ligand binding domain, the soluble form without the transmembrane domain which are present on activated T cells, resting T cells and in autoimmune individuals respectively (Oaks & Hallett 2000; Rudd et al. 2009). Consistent with its role as a negative regulator of T cell activation, Ctla4-/- mice display hyperactivated CD4+ T cells and the mice die within 3–4 weeks of age (Waterhouse et al. 1995; Tivol et al. 1995; Chambers et al. 1997). Blocking or deletion experiments revealed that the autoimmune phenotype of Ctla4-/- mice is dependent on interactions of CD28 with CD80/CD86 (Tivol et al. 1997; Mandelbrot et al. 1999; Tai et al. 2007). Expression of full length Ctla4 or Ctla4-Tyr201Val lowers the lymphoproliferative and autoimmune phenotype of Ctla4-/- mice. However, Ctla4 lacking the cytoplasmic tail is unable to fully rescue the phenotype of Ctla4-/- mice. These mice are long lived but display lymphoadenopathy and a Th2 bias, demonstrating the importance of the tail for full function of Ctla4 (Masteller et al. 2000). Although Ctla4+/- mice show no phenotype, CTLA4+/- in humans display adverse phenotypic effects: dysregulation of Foxp3+ regulatory T (Treg) cells with hyperactivation of effector T cells followed by infiltration of lymphocytes in various organs. In addition, there is a loss of circulating B cells and progressive increase in autoreactive CD21lo B cells. Mutations in CTLA4 also lead to impaired suppressive functions of Treg cells which suppress autoimmunity (Kuehn et al. 2014; Schubert et al. 2014).
3.1 Additional costimulatory receptors
The identification and roles of CD28 and CTLA4 led to a search for other costimulatory receptors (table 2). Besides CD28, there are other positive costimulatory receptors like Inducible T cell Costimulator (ICOS or CD278), OX40, 4–1BB, CD40L etc (figure 4). The ICOS molecule is a homodimeric protein and expressed on activated CD4+ and CD8+ T cells. It binds to ICOS ligand expressed on B cells, macrophages, DC and some non-lymphoid cells, resulting in production of effector cytokines such as IL4, IL10 and Interferon γ (IFNγ). Compared to CD28, the IL2 production induced by ICOS-ICOSL binding is lower suggesting a distinct pathway of costimulation (Arimura et al. 2002; Wikenheiser and Stumhofer 2016). ICOS is located on human chromosome 2 along with CTLA4 and CD28. Interestingly, multiple sequence alignment (figure 5) and dendrogram (figure 6) studies demonstrate that ICOS and CD28 are more closely related to each other compared to CTLA4.

T cells express a galaxy of stimulatory and inhibitory receptors that regulate the extent of activation. The discovery of CD28 and CTLA4 led to the identification of additional receptors and their respective ligands that can either stimulate or inhibit T cell activation. The expression of these molecules depends on the kinetics post activation (e.g.: naïve, activated, etc.), various types of differentiated T cells (T helper, CTL, etc.), specificity of ligand-receptor interactions (HVEM binding to LIGHT is positive whereas HVEM binding to BTLA is negative), etc. Studies are in progress to identify the basic mechanisms and clinical efficacy of several of these proteins in eliciting anti-tumor responses.

Multiple sequence alignment comparing the different secondary structures in protein sequences of T cell costimulatory receptors. Multiple sequence alignment was performed using ClustalW for the different costimulatory receptors in humans. Information on the secondary structures of PDCD1 (Acc No: Q9NZQ7), CD28 (AccNo: P10747) and CTLA4 (Acc No: P16410) was obtained using the UniProt secondary structure prediction tool (The UniProt Consortium 2019). For ICOS (Acc No: Q9Y6W8), secondary structure was predicted using the Chou Fasman algorithm (Chou and Fasman 1974). β sheets and α helices are highlighted in green and red respectively.

Multiple sequence alignment depicting the key motifs that are important for the function of the costimulatory receptors of T cell activation. (a) Multiple sequence alignment was performed using ClustalW for the different human costimulatory receptors. Seven motifs have been highlighted in the MSA. The MYPPPY motif is present in both CD28 (AccNo: P10747) and CTLA-4 (Acc No: P16410) is responsible for binding to CD80/CD86.The YMNM motif present in CD28 is phosphorylated at the tyrosine residues by the p85 subunit of Phosphatidyl-inositol 3-kinase. Out of the two PXXP motifs, the proximal one is present in both CD28 and CTLA4. The PRRP motif in CD28 binds to Itk tyrosine kinase. The distal PYAP is present only in CD28 and is required for binding to Lck, another tyrosine kinase. The YVKM motif present in CTLA4 is important in endocytosis. In PD1 (Acc No: Q9NZQ7), the VDYGEL and TEYATI encode the ITIM and ITSM motifs that regulate PD1 function. (b) The phylogenetic tree was constructed using the maximum likelihood statistical method based on the Jones-Taylor-Thornton (JTT) model (Tamura et al. 2011). High sequence similarity exists between CD28 and ICOS (Acc No: Q9Y6W8) (distance = 6.93) followed by CTLA4 (distance = ~10). The least degree of similarity was found between PDCD1 and the other three molecules (distance = ~35).
There are a large number of costimulatory molecules that are members of the tumor necrosis factor (TNF) α -TNF receptor superfamily. OX40 belongs to the TNF receptor superfamily. Its interaction with OX40 ligand leads to proliferation of T cells, increase in IL-2 and IL-2Rα production (Lathrop et al. 2004; Redmond et al. 2009). CD137 (4–1BB) is a costimulatory glycoprotein and its crosslinking by its ligand 4–1BBL aids in activation of CD8+ T cells. In addition, it can regulate activities of CD4+ T cells, Natural Killer cells and APCs. It is important for generating sustained T cell responses and immunological memory, brought about by upregulation of anti-apoptotic factors like Bcl-XL and Bfl-1 (Lee et al. 2002, Bartkowiak and Curran 2015). CD40, a TNF superfamily receptor, is expressed on APCs and interacts with its ligand CD40L, which is induced on activated T cells. This interaction results in upregulation of costimulatory cytokines for T cell activation and plays important roles during anti-tumor immunity (Laman et al. 2017). The Herpes virus entry mediator (HVEM) is another member of the TNF receptor superfamily, which is constitutively expressed on naïve T cells. Its expression decreases with activation of T cells but is restored on memory and effector T cells. The interaction between HVEM and its receptor LIGHT (CD258) sends positive costimulatory signals to T cells; however, binding of HVEM to another ligand, i.e. BTLA (CD272), sends an inhibitor signal to T cells (Cai and Freeman 2009). Among the multitude of costimulatory receptors known now, the roles of CD28, CTLA4 and Programmed Death receptor 1 (PD1 or PDCD1 or CD279) are discussed in greater detail in this review.
3.2 Structural and signaling aspects of CD28
The crystal structure of CD28 with the Fab fragment of a mitogenic antibody has revealed several structural details (Evans et al. 2005). The extracellular domain of CD28 is made up of a single anti-parallel β-barrel consisting of two layered β sheet structure whose topology resembles that of the variable domains of antigen receptors. CD28 interacts with its cognate ligands, CD80 and CD86, through the MYPPPY motif (figure 6) which is present in Complementarity Determining Region (CDR) 3. The loop structure is mainly stabilized by a water molecule that forms hydrogen bonds with the carbonyl oxygen of Pro-101 and Pro-103 residues (Evans et al. 2005). CD28 is monovalent in terms of binding with CD80 and CD86 because the dimeric structure of CD28 is more compact and binding of one CD80 or CD86 ligand creates steric hindrance for binding of the other ligand (Chattopadhyay et al. 2009). CD28 possesses a unique G-strand pocket in close proximity of its ligand binding site. This pocket may be useful for investigation of small potentially therapeutic compounds against organ graft rejection and autoimmune diseases (Esensten et al. 2016).
CD28 sustains T cell activation by consolidating immunological synapse formation with co-stimulatory signaling. CD28 bears a highly conserved short cytoplasmic tail with no intrinsic enzymatic activity. The tyrosine residues in the cytoplasmic tail are phosphorylated by Lymphocyte-specific protein tyrosine kinase (Lck) and Fyn tyrosine kinases and act as docking sites of Src homology 2 (SH2) domain containing proteins. Upon phosphorylation, the YMNM motif binds to the p85 subunit of phosphatidyl-inositol 3-kinase (PI3K). Surface expression of CD28 is regulated by endocytosis brought about by binding of PI3K to the YMNM motif, leading to clathrin-dependent receptor internalization (Céfaï et al. 1998). The two PxxP motifs in the cytoplasmic tail are sites for binding with Src homology 3 (SH3) domain containing proteins (Rudd et al. 2009; Boomer and Green 2010). The proximal PxxP motif of CD28, located adjacent to the YMNM motif, binds to the IL2 inducible T cell kinase known as Itk whereas the distal proline rich motif (PYAP) binds to Lck (Boomer and Green 2010; Ogawa et al. 2013). Adaptor proteins like Grb2 bind to the proximal YMNM motif through SH2 domain and distal PYAP motif through its SH3 domain, interact with Vav to activate PKCƟ and MAPK pathways, respectively. Some of these proteins have been found to interact with human CD28 (figure 7) using string analysis (Szklarczyk et al. 2019). The PI3K pathway leads to production of phosphatidylinositol (3,4)-bisphosphate and phosphatidylinositol (3,4,5)-trisphosphate, recruitment of pleckstrin homology domain containing protein PDK1 which activates the Protein Kinase B (PKB)/Akt. Activated PKB phosphorylates mTOR, glycogen synthase kinase 3, Bcl-XL, Bcl-2 antagonist of cell death, and inhibitor of nuclear factor-κB, activating NFκB and NFAT. This pathway regulates cellular metabolism, apoptosis and IL2 production (Boomer and Green 2010). The generation of knock in mice with mutations in CD28, i.e. Y170F, AYAA and the double mutant, has delineated the roles of distinct motifs in CD28 signaling (Dodson et al. 2009; Boomer et al. 2014). The distal PYAP motif plays a dominant role but the YMNM motif is also involved in regulating CD28 mediated proliferation and Bcl-XL induction. However, CD28-mediated cytokine responses and germinal center formation is mediated primarily via the distal PYAP motif (Boomer et al. 2014). Interestingly, the distal PYAP is essential for generation of the autoimmune phenotype in Ctla4-/-mice (Tai et al. 2007). PKB is important in mediating the effector functions of CD28: overexpression of PKB suppresses the Fas-mediated cell death of Cd28-/- T cells (Jones et al. 2002). Also, PKB is important in regulating CD28 mediated increase in glycolysis, increased glucose uptake and higher proliferation (Frauwirth et al. 2002). Finally, minor differences in the amino acid sequences of human and mouse CD28 may be responsible for differential signaling effects: cytokine induction versus association with cytoskeletal proteins (Porciello et al. 2018).

T cell costimulatory receptors possess distinct interacting partners. The STRING software was used to identify the different interacting protein molecules with respect to CD28, CTLA4, PD1 in humans with a minimum required interaction score of 0.4 (medium confidence). The maximum number of interacting partners shown is 10. STRING obtains information from experimentally-derived protein-protein interactions identified using literature curation. All the three costimulatory receptors have distinct and different interacting partners, suggesting that the regulation of T cell activation occurs through different signaling pathways.
3.3 CTLA4: Structural perspectives
Structural analysis of CTLA4 shows similarity between human and mouse soluble CTLA4 monomers, but the homodimeric organization differs. The interchain disulfide bond is formed more readily in human CTLA4 compared to mouse CTLA4. CTLA4 comprises a single V-set domain which contacts the paired V-set domain of CD80. Three interacting surfaces are involved in the binding of CTLA4 and CD80: the surface mediating CD80 homodimerization, the CTLA4 homodimer interface and the receptor-ligand binding interface driving the association without any significant conformational rearrangements of the monomers (Stamper et al. 2001). The CTLA4 and CD86 monomers are both two-layer β-sandwiches that display the chain topology characteristic of the immunoglobulin (Ig) variable domains, found in TCR β-chains and antibody VL and VH domains. The dimerization of CTLA4 and CD86 is considered to be unusual, where the ligand binding site stays distal to the dimer interface (Schwartz et al. 2001). CTLA4 shares primary amino acid sequence features with CD28 where the striking features include the presence of three consecutive proline residues in the MYPPPY sequence and a unique cysteine residue at the stalk region for the formation of biologically functional disulfide-linked homodimers which is important for their molecular organization and function (Chattopadhyay et al. 2009). The three proline residues in the MYPPPY sequence adopt a high-energy cis-trans-cis main chain configuration that provides geometric complementarity for specific recognition of the concave surface on the front sheet of CD80/CD86 ligands (Ostrov et al. 2000; Evans et al. 2005). In terms of evolution, the MYPPPY ligand binding domain of CD28/CTLA4 is conserved from fish to humans. Fishes and amphibians possess a single CD80/CD86-like molecule whereas birds and mammals have both the paralogues. The cytoplasmic domain with the YVKM motif in CTLA4 which helps in endocytosis is present in birds and mammals (figure 6). It is possible that CTLA4 evolved from a cell surface molecule to one that can undergo rapid endocytosis (Bernard et al. 2006, Hansen et al. 2009, Walker and Sansom 2011).
3.4 Mechanisms involved during immune suppression by CTLA4
There are multiple mechanisms by which CTLA4 lowers T cell activation (figure 8): First, CTLA4 binds with greater affinity to CD80 and CD86 compared to CD28. The values of the monovalent dissociation constant have been reported to be as high as 20 µM with respect to CD86-CD28 interactions and as low as 0.2 µM with respect to CD80-CTLA4 interaction (Collins et al. 2002). CTLA4 dimers can establish bivalent biophysical interactions with CD80/CD86 ligands where the interaction with CD80 dimer imparts overall higher avidity than monomeric CD86. Signaling through CTLA4 and CD80/CD86 interactions are predicted to take place through the assembly of CTLA4 dimers or an extended network of multiple CTLA4 with CD80 dimers or CD86 monomers (Schwartz et al. 2001; Walker and Sansom 2011). The stoichiometry of interaction and binding affinity between CD28/CTLA4 with CD80/CD86 were determined using equilibrium binding analyses by surface plasmon resonance technique. This analysis led to the conclusion that the CD28 homodimer is functionally monovalent whereas the CTLA4 homodimer is bivalent. Simultaneous binding of the ligand molecules on the ‘U’ shaped structure, made up of Ig domains of CD28, is prevented due to physical clash of the C-set domains. However, this does not occur in case of the ‘V’ shaped structure made by Ig domains of CTLA4 because each arm becomes accessible to bind to separate ligand molecules (Dennehy et al. 2006). Second, CTLA4 is present predominantly in intracellular vesicles in activated T cells, Treg cells and promotes a dominant negative signaling, resulting in T cell tolerance and anergy (Rowshanravan et al. 2018). In the absence of activation, CTLA4 is present mainly in intracellular vesicles and is endocytosed rapidly following interaction between its YVKM motif present on cytoplasmic domain and adaptor proteins of clathrin. The binding of the YVKM motif to adaptor protein-1 results in CTLA4 being trafficked to lysosomes for degradation (Linsley et al. 1996; Shiratori et al. 1997). Other proteins can also modulate the expression of CTLA4. Patients with lipopolysaccharide-responsive and beige-like anchor protein deficiency display autoimmunity. This protein is important increasing CTLA4 expression in activated T cells and Tregs ((Lo et al. 2015; Burnett et al. 2017). Upon T cell activation, the phosphorylation of tyrosine residue in YVKM motif leads to release of adaptor protein-2 upon T cell activation leading to higher cell surface amounts of CTLA4 (Shiratori et al. 1997; Zhang and Allison 1997; Chuang et al. 1999; Rowshanravan et al. 2018). Third, several phosphatases are thought to be associated with CTLA4. Protein phosphatase 2A associates with CTLA4 upon activation and has important role in CTLA4 mediated suppression of T cell activation as it can target Akt phosphorylation (Parry et al. 2005). SHP-2 is another phosphatase which can dephosphorylate CD3ζ following recruitment of CTLA4 and inhibiting the leukocyte-specific protein tyrosine kinase or Lck (Lee et al. 1998). Fourth, the strength of signal together with CTLA4-CD80/CD86 interactions is important in modulating T cell activation. The amount of cell surface CTLA4 directly correlates with the strength of signal (Egen and Allison 2002). Blocking CTLA4 using Fab fragments of a monoclonal antibody generated against CTLA4 can augment or inhibit clonal expansion of different T cell clones when challenged with the same antigen. This response depends on the activation state of T cells as well as the strength of signal. Blockade of CTLA4 in stimulated CD4 T cells hamper Th1 cytokines production but increases Th2 cytokines amounts. CTLA4 blockade in presence of T cells with low TCR signaling leads to enhanced levels of Th1 and Th2 cytokines, whereas blocking CTLA4 during T cell priming with high TCR signal strength causes an expansion of Th1 cells (Anderson et al. 2000). Also, in vitro CD4+ T cell activation is either increased or decreased depending on the strength of the activating signal and blockade of CTLA4-CD80/CD86 interactions (Mukherjee et al. 2002; Ahmed et al. 2009). Fifth, the lymphoproliferation phenotype display by Ctla4-/- mice is not T cell autonomous as shown by reconstitution experiments containing a mixture of wild type and Ctla4-/- cells (Bachmann et al. 1999). These experiments clearly demonstrated the involvement of extrinsic factors in the function of CTLA4 and two distinct processes may be involved. CTLA4 can remove the ligands CD80/CD86 directly from APCs by the process of transendocytosis which are then degraded inside CTLA4 expressing cells (Qureshi et al. 2011; Walker and Sansom 2011). Also, maximal cell surface expression of CTLA4 is found constitutively on Treg cells which are important for immune tolerance and prevention of auto immunity deletion of CTLA4 in Treg cells causes spontaneous development of systemic lymphoproliferation and lethal T cell mediated autoimmune disease while increasing tumor immunity (Wing et al. 2008). CTLA4 is required for the accumulation of Tregs in the intestine but not in the thymus, spleen and lymph node (Barnes et al. 2013). CTLA4-dependent suppression is shown by wild type Treg but Treg cells deficient in CTLA4 are also suppressive due to the production of high amounts of Transforming growth factor beta (TGFβ) and IL10 as a compensatory mechanism (Tang et al. 2004). CTLA4 has been shown to associate with protein kinase isoform C-η (PKC-η) in Treg cells which is important for enhancing their suppressive activity (Kong et al. 2014). Despite years of research, it is clear that further studies are required to better understand the intrinsic as well as extrinsic factors involved in immune suppression by CTLA4.

CTLA4 lowers T cell responses using multiple pathways. T cell activation induces expression of CTLA4 which has a greater binding ability to CD80/CD86 which outcompetes CD28 and lowers T cell activation (intrinsic pathway). In addition, CTLA4 is highly expressed on Treg cells, which suppresses T cell activation. CTLA4 can also bind to CD80/CD86 and cause their degradation by transendocytosis, thereby lowering the surface expression of the ligands (extrinsic pathways).
3.5 The ligands, CD80 and CD86
The ligand molecules, CD80 and CD86 share almost 25% sequence identity at the level of amino acid residues and also bind to CD28 and CTLA4 receptors with different affinities as mentioned above. CD80 and CD86 consist of two extracellular domains, which are membrane distal IgV-like domain and membrane proximal IgC-like domain; along with one transmembrane part and an intracellular domain (Bajorath et al. 1994, Stamper et al. 2001). Both the ligand molecules form a shallow concave surface for the accommodation of the MYPPPY loop of CD28 and CTLA4 through van der Waals interactions (Peach et al. 1995). Two β-bulges are present at Leu-38 and Arg-97 of CD86; and at Met-38 and Arg-94 of CD80 in the membrane distal IgV-type domain (Ikemizu et al. 2000). The dimeric interface of CD80 and CD86 bear similar total solvent accessible areas, which are 1220 and 1405 Å2, respectively (Zhang et al. 2003).
Despite having a number of structural similarities, both of these molecules have some striking structural differences, discrete spatio-temporal distribution with distinct functional properties. CD80 forms dimers but not CD86. The CDR1 of CD80 contains two α-helical turns but CD86 has a single α-helical turn. This additional turn in CD80 forms a protrusion of several residues, among which, Val-22 makes multiple contacts with another CD80 monomer, stabilizing the dimer (Sansom et al. 2003; Zhang et al. 2003). The dimer interface in CD80 is made up of eleven hydrophobic residues out of thirteen contacting residues. Crystallographic data and flow cytometry based fluorescence resonance energy transfer also indicate that formation of a stable dimer between CD86 monomers is an unlikely event. CD86 has fifteen hydrophilic residues out of twenty contacting residues between the monomers, including seven charged residues, which results in chemically distinctive nature of the dimer interface. These features also explain why heterodimerisation between CD80 and CD86 does not take place (Zhang et al. 2003). Functional differences are evident as crosslinking with monoclonal antibodies against CD86 promotes the phosphorylation of its cytoplasmic tail, followed by B cell proliferation. On the other hand, crosslinking of CD80 with monoclonal antibodies against it blocks B cell proliferation (Suvas et al. 2002). Mice lacking both Cd80 and Cd86 display lower Ig class switching and germinal center formation, although this phenotype is not observed in the single deletion, testifying to the redundancy of CD80 and CD86 (Borriello et al. 1997). Also, transgenic mice expressing higher amounts of CD86 possess lower number of B cells due to their elimination in a CD28-dependent manner (Fournier et al. 1997).
4 PD1 and its ligands, PDL1 and PDL2
PD1 is a molecule that is expressed on the surface of T cells post activation and acts as an immune checkpoint. PD1 was discovered by Tasuku Honjo’s laboratory in Kyoto University while screening for a number of genes involved in apoptosis (Ishida et al. 1992). Upon activation of T cells, high amounts of Ca2+ stimulates the transcription factor, NFAT, which increases PD1 expression (Oestreich et al. 2008). High amounts of PD1 are also observed on exhausted T cells, which display low expression of CD122 (IL2 receptor beta chain), IL15 receptor with lower ability to produce cytokines and display cytolytic activity. Exhausted T cells are often observed during chronic viral infections, resulting in persistence (Wherry 2011; Wykes and Lewin 2018). PD1 lowers T cell activation; however, CD28 signaling, but not ICOS, attenuates this effect (Mizuno et al. 2019). Interestingly, treatment with anti-PDL1 restores proliferation of hepatitis C virus specific CD8+ T cells (Urbani et al. 2006). During viral infections, Type I Interferon is produced which induces IRF9 that binds to the promoter and induces PD1 expression (Terawaki et al. 2011). In addition, demethylation (Youngblood et al. 2011) and binding of other transcription factors such as FoxO (Staron et al. 2014) may also increase PD1 expression. IFNγ, an inflammatory cytokine, is known to induce both PDL1 and PDL2 and this may be part of the host response to tumors (Brown et al. 2003).
PD1 is composed of 268 amino acids and is a Type 1 transmembrane protein consisting of a IgV extracellular domain, a transmembrane domain and a cytoplasmic tail (Ishida et al. 1992). PD1 is encoded by PDCD1 present in human chromosome 2 (Shinohara et al. 1994) and mouse chromosome 1. PD1 has two ligands, Programmed death ligand 1 (PDL1) (Dong et al. 1999; Freeman et al. 2000) and Programmed death ligand 2 (PDL2) (Latchman et al. 2001; Tseng et al. 2001), belonging to the B7 family of proteins (table 2). In general, PDL1 expression is found on lymphoid and non-lymphoid cells whereas PDL2 expression is mainly present on lymphoid cells, e.g. DCs (Latchman et al. 2001). PDL1 expression is upregulated on DCs and macrophages with GM-CSF and lipopolysaccharide whereas upregulation of PDL1 on T cells and B cells occur via TCR and B cell receptor signaling (Yamazaki et al. 2002). PDL1 expression is also upregulated in most tumors in mice and virally-infected tissues (Iwai et al. 2002). It is likely that PDL1 and PDL2 bind to other cell surface molecules, apart from PD1; however, the functional relevance of these interactions need to be better studied in the future (Wang et al. 2003).
PD1 and PDL1/PDL2 have canonical Ig-like extracellular domains for the receptor-ligand interaction interface. The extracellular domain of PD1 folds into β-strand sandwich structure where several β-strands are organized into two disulfide linked sheets, resembling the fold of the greek key (Zak et al. 2017). PD1 lacks the nearly invariant extracellular Cys which is present in the stalk region of both CD28 and CTLA4, making it incapable of forming covalent dimers and constraining it to exist as a monomer on the cell surface (Zhang et al. 2004). Its cytoplasmic domain harbors two tyrosine-based signaling motifs: Immunoreceptor tyrosine-based inhibitory motif (ITIM) and Immunoreceptor tyrosine based switch motif (ITSM) (Lázár-Molnár et al. 2017). PD1 contains the ITIM (VDYGEL) and ITSM (TEYATI) motifs which get phosphorylated at the tyrosine residues and is a docking site for SHP1 and SHP2 phosphatases which attach to the cytoplasmic tail of PD1 (Blank and Mackensen 2007). This leads to dephosphorylation of ZAP70 (figure 9) which causes upregulation of Cbl-b ubiquitin E3 ligase and blocks the subsequent cascade of T cell activation (Karwacz et al. 2011).

The binding of PD1 to its ligands, PDL1 and PDL2, lowers T cell responses. The interaction of PD1 with its ligands PDL1 and PDL2 causes the recruitment of phosphatases, SHP1/SHP2, which causes dephosphorylation of ZAP70, lowering T cell activation and, eventually, leading to T cell exhaustion.
PDL1 has two compact Ig-like folds (N-terminal Ig-V followed by Ig-C) which are connected by a short linker. Homodimeric PDL1 is organized to expose its hydrophobic N-terminal PD1 interacting domain. The C-terminal domain of PDL1 may act as a spacer to separate the binding site from the cell membrane. The interaction between PD1 and PDL1 occurs due to structural reorganization on the PD1 interacting surface. Although the broad organization of PDL2 is similar to that observed in PDL1, PDL2 displays a greater affinity for PD1 compared to PDL1 (Zak et al. 2017).
Stimulation of T cells with anti-CD3 along with PD1-Ig in vitro, causes a significant reduction in IL2 and IFNγ production and cellular proliferation (Freeman et al. 2000; Carter et al. 2002). Co-culture of DCs expressing PDL1 along with transgenic PD1 expressing total T cells demonstrates that CD8+ T cells are more affected (Barber et al. 2006). Surprisingly, Pd1-/- mice display distinct splenomegaly compared to C57BL/6 wild type mice. In vitro studies suggested that these are due to elevated B cell responses upon IgM stimulation whereas the T cell responses are the same upon anti-CD3 stimulation in Pd1-/- mice (Nishimura et al. 1998). Strain specific effects are observed in mice lacking Pd1: In the C57BL/6 background, these mice are predisposed to autoimmune diseases such as lupus-like glomerulonephritis (Nishimura et al. 1999) whereas autoimmune disorders such as gastritis and dilated cardiomyopathy are observed in the BALB/c background (Okazaki et al.,2005). Pdl1-/- mice display an autoimmune prone phenotype with accumulation of large number of CD8+, but not CD4+, T cells in the liver. There is lower apoptosis in these cells during the contraction phase during experimental autoimmune hepatitis (Dong et al. 2004). Higher CD4+ and CD8+ T cell responses are observed post in vitro activation as well as antigen specific in vivo responses in Pdl2-/- mice, demonstrating its role as a negative costimulator (Zhang et al. 2006).
5 Modulation of T cell differentiation by CTLA4 and PD1
Costimulatory receptors modulate peripheral T cell differentiation which plays important roles in determining the types of cellular immune responses. For example, Th1 responses are known to cause delayed type hypersensitivity leading to diseases such as psoriasis. Th2 responses, on the other hand, cause excessive production of IgE during allergies. In general, CTLA4 reduces Th2, whereas PD1 reduces Th1 differentiation: In vitro polarization of naïve T cells with a Th1 bias is observed upon activating with immobilized anti-CTLA4 or CD80 transfectants in a partly TGFβ dependent manner (Kato and Nariuchi 2000). Ctla4-/- mice produce higher amounts of IL-4 and IL-5 cytokines and these mice display a more Th2 profile (Khattri et al. 1999; Oosterwegel et al. 1999). Also, Ctla4 is important for lowering activation-induced cell death by reducing the expression of Fas and FasL (Pandiyan et al. 2004). Anti-PD1 increases Th1 cytokines and impairs Treg maturation after intratracheal administration of lipopolysaccharide causing lung injury in C57BL/6 mice (Gibbs et al. 2018). Most likely, PD1 binding to its corresponding ligands result in a significant drop in Th1 cytokine profile in a SHP2-dependent manner (Li and Ferris 2014).
Th17 cells play a major role in the adaptive immune system by eliminating pathogens (Weaver et al. 2013). Interactions between CTLA4 and CD80/CD86 regulate the extent of differentiation of Th17 cells (Ying et al. 2010). One of the ways this happens is through Foxp3+ Treg cells which inhibit the differentiation of Th17 cells (Lee 2018). Pd1-deficient mice are also very prone to infections by M. tuberculosis with high amounts of Th1 and Th17 responses. The excessive production of pro-inflammatory cytokines results in excessive tissue damage and lower survival of infected Pd1-/- mice (Lázár-Molnár et al. 2010).
Follicular T cells are extremely important in the generation of germinal center-dependent antigen-specific B cell responses. Ctla4-/- mice display higher number of follicular helper T cells and development of germinal centers; also injection of anti-CTLA4 in wild type elicits these cells (Wang et al. 2015). Follicular T cells express high amounts of PD1 and the interaction with its ligands lowers the generation and recruitment of these cells to follicles (Shi et al. 2018). Consequently, anti-PD1 treatment increases the numbers of follicular T cells by downregulating the KLF2 transcription factor and upregulating IL4 and IL21 (Mizuno et al. 2019). Overall, it appears that negative costimulation by CTLA4 and PD1 regulates peripheral T cell differentiation by controlling TCR based signaling along with production of various cytokines that may result in immunopathology (Wei et al. 2017; Wei et al. 2019a, b).
6 Crosstalk and regulation by costimulatory receptors
The activation of T cells is regulated by various cell surface molecules during an antigenic response (figure 4). Although these cell surface molecules impart their function through diverse mechanisms, there is a crosstalk between these molecules. CTLA4 can be thought of as the gatekeeper to T cell activation and regulates ICOS function. Ligation of CTLA4 blocks ICOS mediated production of IL-4, IL-10 and IL-13 in mouse CD4+ T cells, which can be overcome by the addition of exogenous IL-2 (Riley et al. 2001). Also, blocking the ICOS ligand (ICOSL) interaction using ICOS-Ig results in formation of CD4hi Tregs with decreased exocytosis and higher expression of CTLA4 (Zheng et al. 2013). Anti-CTLA4-elicited anti-tumor responses are lower in the absence of ICOS (Fu et al. 2011). PD1 and ICOS also have an interesting relationship with respect to Follicular T helper cells. It is well known that ICOS-ICOS-L signalling increases contacts between T cells & B cells, resulting in the generation of high affinity B cells (Liu et al. 2015a). In general, the expression of PD1 lowers the recruitment of T cells to the follicles; however, those T cells which express high amounts of ICOS can overwhelm this, resulting in the recruitment of high ICOS bearing cells to germinal centers (Shi et al. 2018). In Type 1 diabetes there is an increase in the number of CXCR5+PD1+ICOS+ follicular T cells that promotes selection of autoreactive B cells, resulting in higher titers of autoantibodies (Viisanen et al. 2017).
CTLA4 and PD1 also modulate immune responses during viral infections. Higher expression of CTLA4, PD1 and CD28 is observed on CD4+ T cells with higher Human Immunodeficiency Virus (HIV) load. Treatment with retroviral drugs leads to a downregulation of these markers on CD4+ T cells. In vitro blockade of PD1 and simultaneous stimulation via CD28 caused HIV-specific CD4+ T cell proliferation (Kassu et al. 2010). Although the temporal expression of different costimulatory receptors and their ligands is cell type dependent, they appear to cross talk with each other in regulating T cell function.
7 How do checkpoint inhibitors mediate anti-tumor effects?
The common strategies to treat tumor growth are well known: surgery, radiation (especially for bone marrow derived tumors), hormone therapy (for hormone-dependent tumors) and chemotherapy. Among these, the latter is widely used for a wide variety of tumors. Chemotherapy extends the life of patients; unfortunately, resistant tumor cells often arise, resulting in treatment regimes with alternate drugs which may be toxic. Immunotherapy is also an option as it is known that the activation of the host immune system lowers tumor burden (Rakshit et al. 2012; Podder et al. 2016); however, there are issues with respect to efficacy, ethical issues etc.
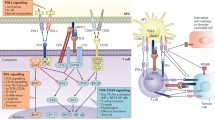
Although T cells capable of recognizing peptide antigens on tumors cells are present, they are unable to unleash their responses due to the immunosuppressive environment: expression of negative costimulatory molecules on tumors cells, altered macrophages present in tumors, higher number of Treg cells, etc. Studies are in progress to understand the conversion of “cold tumors” (without infiltrating T cells) to “hot tumors” (with infiltration of T cells). The tumor immunosuppressive environment consists of different types of molecules, e.g. PDL1, TGFβ, IL10, indoleamine 2,3-dioxygenase, and cells, e.g. Tregs and myeloid-derived suppressor cells, that lower immune responses etc (Balkwill et al. 2012; Kalathil et al. 2013; Wang et al. 2016; Syn et al. 2017). The assumption is that T cells capable of recognizing tumor antigens are present; however, their activation is sub-optimal due to the presence of immunosuppressive molecules, including negative costimulatory receptors and their ligands, e.g. CTLA4, PD1, PDL1, etc. (figure 10). These are induced as a host driven response to restrict inflammation and immunopathology; however, in the tumor environment, these responses further facilitate the growth of tumors. Therefore, by blocking the negative costimulators (checkpoints or brakes), the equilibrium is shifted to enhance anti-tumor T cell responses (figure 11). Blocking these receptors is achieved using checkpoint inhibitors or antibodies against CTLA4 (e.g. Ipilimumab) or PD1 (e.g. Nivolumab or Pembrolizumab) or PDL1 (Avelumab or Durvalumab) which enhances T cell responses against tumors (figure 11). Anti-CTLA4 was first approved by the FDA in 2011 for the treatment of late stage melanoma. Although checkpoint therapy works in a subset of patients, it is important to appreciate that some patients are “cured” due to the activation of the host immune system. Treatment of patients with stage III melanoma with anti-CTLA4 (Ipilimumab) demonstrated higher survival and less recurrence of cancers compared to placebo treated controls (Eggermont et al. 2016). Anti-PD1 was first approved for the treatment of melanoma in 2014; subsequently, anti-PD1 and anti-PDL1 have been widely used for the treatment of several types of tumors, including head and neck cancers in 2019. Readers are encouraged to find out the detailed listing of checkpoint inhibitors and other therapies targeting different tumors by visiting the following web site: https://www.cancerresearch.org/scientists/clinical-accelerator/landscape-of-immuno-oncology-drug-development

Both costimulatory ligands and their receptors are expressed across several cancer types. The data was obtained from the human protein atlas (Uhlén et al. 2015) and the transcripts levels of the different ligands and receptors are represented as median values of fragments per kilobase of transcript per million mapped reads (FPKM). The Human protein atlas comprises six parts and its pathology atlas was used to generate the above data. The pathology atlas consists of mRNA and protein expression data from 17 types of human cancers. The transcriptomics data represented above has been obtained from the cancer genome atlas.

Checkpoint inhibitors greatly stimulate anti-tumor responses. During tumor growth, the immunosuppressive environment prevents effective anti-tumor T cell responses. The blockade of the negative regulators of T cell activation (checkpoints), using specific antibodies (checkpoint inhibitors) e.g. anti-CTLA4 or anti-PD1, greatly enhances anti-tumor T cell responses. This strategy may reduce tumor growth and lead to anti-tumor immunity
The commercially available monoclonal antibody Ipilimumab binds CTLA4 at the MYPPPY site and blocks its interaction with CD80/CD86. Although CD28 and CTLA4 are structurally similar, Ipilimumab discriminates between these by recognizing Leu-39 and Ile-93 of CTLA4, which are replaced in CD28 by His and Phe residues respectively (Ramagopal et al. 2017). Treatment with anti-CTLA4 increases the numbers of anti-tumor effector T cells and lowers the numbes of intra-tumoral Tregs (Kavanagh et al. 2008; Tang et al. 2008). Interestingly, the binding of anti-CTLA4 (IgG2a) to Fc-gamma receptors in mice is essential for its ability to elicit anti-tumor responses, most likely by reducing the number of Tregs and increasing the number of anti-tumor effector T cells (Selby et al. 2013; Ingram et al. 2018). Ipilimumab which binds to human CTLA4 is IgG1 and binds well to Fc-gamma receptors also reduces the number of Tregs in bladder cancer (Liakou et al. 2008). On the other hand, Tremelimumab another antibody (IgG2) that recognizes CTLA4, does not bind to Fc-gamma receptors but mediates anti-tumor responses by increasing the tumor infiltrating CD8+ T cells (Ribas et al. 2009).
ICOS has a definite role in anti-tumor effects as studies show that anti-CTLA4 leads to upregulation of T cells expressing higher amounts of ICOS and are specific towards tumor antigens. In addition, this therapy leads to an increase in effector to Treg cell ratio (Liakou et al. 2008). In fact, Icos-/- mice are also unable to combat tumors after anti-CTLA4 therapy, demonstrating an important role for ICOS-ICOSL interactions (Fu et al. 2011). It is also interesting that concomitant ICOS activation followed by CTLA4 blockade is more efficient with respect to tumor therapy. Thus, Icos-/- or Icosl-/- mice have impaired T cell response against tumor with anti-CTLA4 treatment. (Fan et al. 2014). Not surprisingly, a patent (US8709417B2) has been awarded for the co-treatment of ICOS stimulation with anti-CTLA4 blockade in enhancing anti-tumor therapy.
Therapeutic monoclonal antibodies to PD1 such as Nivolumab target the interaction site between PD1/PDL1 (Zak et al. 2017). PD1 blockade reduces the levels of SHP-2 causing higher Th1 responses in tumor infiltrating lymphocytes, thereby reversing the immunosuppression in the tumor microenvironment (Li and Ferris 2014). Treatment with anti-PD1 led to lowering of tumors in patients with several types of tumors, e.g. melanoma, non–small-cell lung cancer and renal cell cancer, and the efficacy ranged from 18– 28% (Topalian et al. 2012). In addition, anti-PD1 is less toxic than other chemotherapeutic drugs (Brahmer et al. 2015). Nivolumab (anti-PD1) belongs to the IgG4 isotype which lowers complement activation and antibody dependent cell cytotoxicity (Wang et al. 2014).
The ligands of PD1 are expressed on non-hematopoeitic cells and tissues (Keir et al. 2008), leading to the notion that CTLA4 is required early during T cell activation whereas PD1 inhibition works at a later stage, i.e at the tissue level. It is important to understand that CTLA4 and PD1 act in different ways: CTLA4 inhibits AKT via the phospohatase PP2A; on the other hand, PD1 inhibits the CD28-mediated activation of AKT (Parry et al. 2005). The E3 ubiquitin ligase, Cbl-b, is known to lower T cell responses. CD28 activation lowers Cbl-b amounts whereas CTLA4 is important for its expression (Li et al. 2004). Interestingly, anti-tumor responses in Cbl-b-/- mice are more responsive to CTLA4, but not PD1, blockade (Peer et al. 2017). In addition, checkpoint inhibtors lead to the upregulation of different classes of T cells against tumors, i.e. the peripheral T cell differentiation process is altered. Anti-CTLA4 therapy leads to the upregulation of both ICOS+ Th1 like CD4+ effector T cells and exhausted CD8+ T cells whereas anti-PD1 therapy leads to the upregulation of only the latter (Wei et al. 2017). A recent study has shown that Ctla4-/- mice form Th2 skewed CD4+ T cells, T follicular helper cells, T follicular helper regulatory cells and Tregs. On the other hand Pd1-/- mice show expansion of PDL1+Sca1+IRF4+CD8+ cells (Wei et al. 2019a, b). To increase the efficacy of the drugs, combinatorial therapy was considered. A combination of anti-PD1 (Nivolumab) and anti-CTLA4 (Ipilimumab) was approved by the FDA in 2015 for treating melanoma patients. In 2018, the combination of anti-CTLA4 plus anti-PD1 was approved for the treatment of renal cell carcinoma. It is likely that the combination of anti-CTLA4 and anti-PD1 uniquely affects tumor infiltrating T cell populations, e.g. terminally differentiated effector CD8+ T cells and Th1-like effector CD4+ T cells, as opposed to monotherapy (Wei et al. 2019a, b).
8 Factors affecting the efficacy of checkpoint inhibitors
Although checkpoint inhibitor therapy has saved thousands of lives of patients afflicted with cancer, there are some challenges that need to be addressed as only 20–25% of patients respond to this form of therapy (Syn et al. 2017). Therefore, understanding the factors that affect the outcome of therapy is extremely important (Yan et al. 2018). One of the primary factors that play major roles are the patient’s age and sex. Ageing generally is associated with a less effective immune system. It has been observed that high expression of PD1 on the surface of T cells in mice suggests a dampened immune response against tumors (Mirza et al. 2010). It is also possible that patients who are unresponsive to the checkpoint inhibitors may lack sufficient numbers of anti-tumor T cells. Sex dependent effects of checkpoint therapy have also been reported with males benefitting more compared to females (Conforti et al. 2018). Also, anti-CTLA4 therapy, compared to anti-PD1 therapy, is more sex dependent benefitting male patients more than females (Wu et al. 2018).
It appears that a higher immunosuppressive environment in the tumors is inversely correlated with the lack of efficacy of checkpoint inhibitor therapy. The number of Tregs and myeloid derived suppressor cells (MDSCs) contribute to the immunosuppressive environment near the tumor making it difficult for the checkpoint therapy to work (Kalathil et al. 2013). The presence of high amounts of T cell cytokines is a good indication that they are effective in reducing tumor growth (Peng et al. 2012). In addition, it has been observed that the presence of CD8+ T cells near the tumor determines whether therapies such as anti-PD1 therapy work (Daud et al. 2016). The amount of mutational burden in a tumor has been found to correlate with efficacy of checkpoint therapy. The loss of function of DNA repair genes causes reduction in DNA repair followed by microsatellite instability. Consequently, greater the microsatellite instability the higher is the probability that the checkpoint therapy is working (Le et al. 2015). Patients who do not respond to anti-CTLA4 (Ipilimumab) have been found to contain tumors with genomic defects in the IFNγ signaling pathway (Gao et al. 2016). Patients likely to respond to anti-CTLA4 display higher ICOS+ T cells and lower neutrophil-to-lymphocyte ratio (Di Giacomo et al. 2013). Recent studies have shown important roles for T cell factor 1, a transcription factor, during checkpoint inhibitor therapy (Kurtulus et al. 2019; Siddiqui et al. 2019).
Finally, diet profiles, body mass index and gut microbiota may also be important in determining the efficacy of checkpoint therapy. There is a direct correlation of obesity to improved efficiency of anti-PD1 therapy compared to individuals having normal body mass index with respect to metastatic melanoma (McQuade et al. 2018). Our gut microbiota is of paramount importance in maintaining our health by helping in absorbing important nutrients, protecting against potential pathogens etc. The checkpoint inhibitor therapy has been found to be ineffective in mice reared in germ free environments or undergoing antibiotic therapy which will deplete the gut microflora (Vétizou et al. 2015; Routy et al. 2018). Particular bacterial species have been identified that play roles in enhancing checkpoint blockade therapy. For example, the amounts of Bifidobacterium (Sivan et al. 2015) or Akkermansia muciniphila (Routy et al. 2018) determines the efficacy of the anti-PDL1 therapy. On the other hand, treatment of melanoma with anti-CTLA4 is dependent on two specific Bacteroides species: B. fragile and B. thetaiotaomicron (Vétizou et al. 2015). Overall, it is clear that further studies are required to comprehend the roles of high fiber diet, probiotics and additional factors associated with checkpoint therapy. There is no doubt that this is an area of active investigation and is likely to unearth molecular players that may lead to potential therapies to boost the efficacy of checkpoint therapy.
9 Challenges faced with the use of checkpoint inhibitors
The two main concerns with this form of therapy are cost and immune-related adverse effects (IRAEs). The affordability of immune checkpoint therapy is a major concern, given its estimated cost to be ~$100,000 to $250,000 per patient, depending on factors such as the type of therapy, duration of treatment and route of administration. Combined therapies may cost even more; for example joint treatment with Nivolumab and Ipilimumab can cost up to $300,000 per patient, thus reducing their affordability (Andrews 2015). Given such high costs, it is imperative to consider cost effectiveness with respect to selection of patients who are likely to benefit with this form of therapy (Verma et al. 2018).
The other main problem with immune checkpoint therapy is the augmentation of autoimmune diseases known as immune related adverse effects (IRAEs), which leads to serious health complications, associated with excessive inflammatory responses. This is not surprising as once the brakes on T cells are curtailed, anti-tumor as well as autoimmune T cells are likely to be unleashed. As mentioned earlier, negative costimulatory molecules (checkpoints) are crucial for self-tolerance and their blockade can predispose patients to autoimmune conditions, resulting in IRAEs. The collateral damage of IRAEs extends to normal tissues and organs including gut, skin, hepatic, pulmonary and endocrine systems. Around 90% of patients treated with anti-CTLA4 and 70% of those treated with anti-PD1/PDL1 experience some form of IRAEs. IRAEs usually occur within a period of 3–6 months once the anti-CTLA4 or anti-PD1 treatment begins (Michot et al. 2016). The most common IRAEs affecting endocrine system for anti-PD1 are thyroid dysfunctions like hypothyroidism and thyrotoxicosis and hypophysitis for anti CTLA4 antibodies (Ferrari et al. 2019). Ipilimumab, the first cancer immunotherapy drug to get an FDA approval for treatment of melanoma can cause dermatitis, enterocolitis and hepatitis. Hypophysitis induced by anti-CTLA4 (Ipilimumab) mostly causes adenohypophysis hormone deficiency, predominantly ACTH and TSH (Min 2016). In case of anti-CTLA4 treatment, incidents of autoimmune colitis, and organ damage (mainly dermal and gastrointestinal) have been reported (Bertrand et al. 2015). Anti-CTLA4 increases Th17 cells in peripheral blood of patients with metastatic melanoma and this study may provide insights into the pathogenesis of anti-CTLA4-induced toxicities (von Euw et al. 2009). Therefore, efforts are directed towards identifying altered version of anti-CTLA4 that display high anti-tumor potential but low IRAEs (Pai et al. 2019). PD1 targeting Nivolumab has been reported to be linked with autoimmune thyroid dysfunction (Byun et al. 2017). It has been observed that curtailing Type 1 interferon responses reduces autoimmunity without affecting anti-tumor responses (Walsh et al. 2019).
Another recent issue that has come to light is “hyperprogression” or increase in tumor progression upon treatment with anti-PD1 or anti-PDL1. However the mechanisms behind this condition are currently under investigation. One group has discovered that 6 out of 155 cancer patients receiving anti-PD1/PDL1 therapy who became progressors had MDM2/MDM4 amplification. On the other hand, 2 out of 10 patients harboring EGFR alterations showed similar behavior (Kato et al. 2017). On the other hand, another group studying patients with non-small cell lung carcinoma found 39 out of 187 patients undergoing anti-PD1 treatment showed hyperprogression, and discovered that their tumor tissues were enriched with tumor associated macrophages (Russo et al. 2019). About 10% of advanced gastric cancer patients treated with anti-PD1 display hyperprogression. Interestingly, this correlates with the development of highly proliferative FoxP3+ Treg cells which may suppress the tumor reactive PD1+ effector T cells (Kamada et al. 2019). Clearly, this is a matter of concern and further studies are required to fully uncover the reasons for this behavior in a small number of patients.
Studies in the area of T cell costimulation therapy can be tricky and a cautionary tale that is worth recounting is the phase 1 clinical trial of the CD28 superagonist TGN1412 possessing a humanized IgG4 isotype. Upon injection of this potential drug, healthy human volunteers suffered from life-threatening excessive inflammatory conditions (Attarwala 2010). Further investigations to uncover the differences between pre-clinical and clinical trials revealed that, unlike humans, macaque effector and memory T cells (TEM) undergo loss of CD28 expression upon maturity (Eastwood et al. 2010). This is one the reason as to why TGN1412 did not stimulate high amounts of cytokines in monkeys during the pre-clinical studies but did so in humans during the trial. Due to the failure of the trails, the company TeGenero had to file for bankruptcy. Investigations revealed that the volunteers were dosed with high amounts of the drug. Perhaps, sequential dosing would be a better idea. Subsequent studies have shown lower doses of TGN1412 increases IL10 probably due to activation of Tregs and the cytokine burst is not observed (Brown 2018). Therefore, a better understanding of the mechanisms by which antibodies to costimulatory molecules function is important.
10 Effects of polymorphisms in costimulatory receptors
Polymorphisms in CD28, CTLA4 and PD1 could hinder T cell regulation leading to higher incidences of autoimmune disorders, tumors etc. Single nucleotide polymorphisms (SNPs) associated with CD28 are associated with breast cancer (Yan and Zhang 2017). It has been found that a group of four polymorphisms in ICOS is responsible for B cell chronic lymphocytic leukemia in Polish populations (Karabon et al. 2011). The close relationship between CD28 and ICOS may explain the reasons for the same disease due to polymorphisms in these two costimulatory receptors. As CD80 and CD86 are the main interacting partners of CD28 and CTLA4, it is obvious that polymorphisms in these may also be associated with diseases. Two polymorphisms i.e. rs6641T>G (CD80) and rs17281995G>C (CD86) are involved with MS along with CD28 (Wagner et al. 2015). In addition, it is observed that CD86 +1054G/A polymorphisms increase the risk in colorectal cancer (Pan et al. 2010).
Polymorphisms in CTLA4 are known to affect the responsiveness of T cells to stimulation (Maier et al. 2007) and a large number of variants are found in the MYPPPY motif (Siggs et al. 2019). One of the first autoimmune diseases that was found to be associated with CTLA4 polymorphism is Grave’s disease, an autoimmune disorder leading to hyperthyroidism, in which affected individuals display excessive amounts of thyroid hormone. A microsatellite marker is found to be responsible for these phenomena with a single allele of an (AT)n repeat sequence which has a higher frequency in 3’-UTR region of the gene, which may affect RNA stability (Gough et al. 2005). Interestingly, this microsatellite is not present in mouse Ctla4. Another C-T SNP in intron 1 at +1822 position is also responsible for Grave’s disease. It is possible that A-G SNP in exon-1 is a cause for Type1 diabetes in a number of family data sets as well as individual cases (Ihara et al. 2001). This same SNP is prevalent in other autoimmune diseases such as Addison’s disease, systemic lupus erythematosus (SLE), celiac disease etc. Interestingly, a splice variant of Ctla4 associated with Type 1 diabetes lacks the binding motif MYPPPY and inhibits T cell activation by dephosphylating the TCR-zeta chain (Vijayakrishnan et al. 2004).
Multiple polymorphisms in the PD1 are associated with a number of autoimmune diseases such as SLE, Rheumatoid Arthritis (RA), etc. A link between the roles of PD1 to RA is shown in the Caucasian population where the (PD1.3G/A) was significantly associated with the disease (Zou et al. 2017). With respect to cancer such as esophagogastric junction adenocarcinoma, three different polymorphisms in PD1 are associated with this disease (Tang et al. 2017). PDL1 and PD1 polymorphisms are also associated with diseases such as non-small cell cancer (Nomizo et al. 2017). In another study PDL1 rs4143615 C>G and PD1 rs2227982 C>T were associated with ovarian cancer (Tan et al. 2018).
11 Efficacy of CTLA4-Ig in the treatment of autoimmune diseases
In general, checkpoint inhibitors exacerbate autoimmune symptoms due to the aforementioned reasons. In an experimental model of autoimmune encephalomyelitis, anti-CTLA4 as well as its F(ab)2 fragments increases production of proinflammatory cytokines and T cell proliferation, resulting in accelerated disease (Karandikar et al. 1996). However, CTLA4-Ig, a soluble fusion protein consisting of the extracellular domain of human CTLA4 linked to Fc portion of modified human IgG1 is very useful in lowering immune responses especially during autoimmune diseases (Linsley et al. 1992a). Due to its higher affinity for CD80 and CD86, CTLA4-Ig inhibits the early phase of T cell activation, differentiation and survival (Salomon and Bluestone 2001). Mice expressing CTLA4-Ig display lower T cell-dependent B cell responses with impaired class switching and absence of germinal center formation in spleen and lymph nodes (Lane et al. 1994). Hyper-activation of T lymphocytes contributes to the pathogenesis of autoimmune diseases through the production of inflammatory cytokines and autoantibodies. Fully humanized CTLA4-Ig is commercially named as Abatacept (under commercial trade name Orencia) and is being used as a therapeutic molecule in autoimmunity and transplantation (Najafian and Sayegh 2000). Listed below are some diseases where studies with CTLA4-Ig have been performed and some examples are as follows:
RA is a common autoimmune disease which is characterized by the hyper-activated immune system attacking healthy body tissue. Clinical trials have demonstrated CTLA4-Ig to be efficacious in patients with RA, especially in those patients who are refractory to TNFα inhibitors or methotrexate treatment (Genovese et al. 2008). Evaluation of biomarkers in CTLA4-Ig treated RA patients confirmed an effective reduction in rheumatoid factor, TNF-α, IL-6, soluble IL-2 receptor, soluble intracellular adhesion,etc. (Weisman et al. 2006).
Psoriasis is an autoimmune disease and lesions are characterized by inflammatory infiltrates, consisting largely of T cells and APCs. In phase I clinical study, 46% of total admitted patients achieved >50% sustained improvement in clinical disease severity upon treatment with CTLA4-Ig. The improvement was associated with a quantitative reduction in the skin-infiltrating T cells and epidermal hyperplasia (Abrams et al. 1999).
Multiple sclerosis is a chronic demyelinating inflammatory disease that affects the central nervous system. In a phase I dose escalation study, in patients with relapsing & remitting multiple sclerosis received a single intravenous CTLA4-Ig and immunological assessments showed a decreased IFNγ production and reduction in myelin basic protein proliferation within two months of treatment (Viglietta et al. 2008).
SLE: In a phase II clinical trial, CTLA4-Ig treatment has shown amelioration of disease severity, improvement in health parameters and enhanced quality of life in CTLA4-Ig treated subjects (Merrill et al. 2010). Similar results were observed in lupus-prone mice treated with CTLA4-Ig in combination with anti-CD40 ligand (Wang et al. 2002).
Graft versus host rejection. Blockade with CTLA4-Ig is useful for acute graft versus host disease prevention, which may occur during unrelated-donor hematopoietic cell transplantation. Patients receiving CTLA4-Ig plus cyclosporine/methotrexate demonstrated significant inhibition of early CD4+ T cell proliferation and activation compared to the cyclosporine/methotrexate cohort group. The CTLA4-Ig cohort patients demonstrated a low rate of acute GVHD, despite robust immune reconstitution (Koura et al. 2013).
CTLA4-Ig has transitioned from being a basic immunological investigation tool to a therapeutic molecule. It is approved for treatment of RA (in adult patients) and polyarticular juvenile idiopathic arthritis (in pediatric patients) (Blair and Deeks 2017). A patient with polymorphisms in the MYPPP motif of CTLA4 demonstrating autoimmunity responded well to treatment with CTLA4-Ig (Siggs et al. 2019). Despite improving disability and enhancing the quality of patient’s life, low percentage of successful cure, reactivations of latent infection and adverse effects are still a concern. In addition, several important aspects of CTLA4-mediated immune-suppression still require further study. CTLA4-Ig is shown to be less effective at inhibiting memory T cells and CD8+ T cells than naïve CD4+ T cells (Salomon and Bluestone 2001; Blair and Deeks 2017). Predominantly T cell-mediated autoimmune diseases such as RA respond to CTLA4-Ig alone. However, other diseases such as transplantation and SLE which are regulated by both T and B cells respond partially to CTLA4-Ig therapy alone (Wang et al. 2002; Genovese et al. 2008). Prolonged CTLA4-Ig therapy may lead to the complete suppression of immune responses which is a major concern (Reddy et al. 2001). Further studies are required to optimize the treatment of various autoimmune disease and/or in disease and phase-specific manner.
12 Checkpoint inhibitors and other diseases
The recent success of immune checkpoint blockade in tumor immunotherapy suggests that targeting these checkpoints could also be effective for curing a range of infectious diseases. Currently, checkpoint blockade is being evaluated for reversing T cell exhaustion which occurs during chronic infections and cancer. Recent studies have shown that microbial infections up-regulate PD1 and CTLA4 on host immune cells which leads to the reversible immune dysfunction (Bhadra et al. 2011; Butler et al. 2012; Palmer et al. 2013; Habib et al. 2018). Therefore, targeting immune checkpoint may be a prudent approach for the treatment of infectious diseases.
Viral diseases: Checkpoint inhibitor pathways limit the functioning of pathogen-specific T cell responses during chronic infection. Upregulation of checkpoint inhibitors CTLA4 and PD1 by HIV-specific CD4+ T cells correlate with disease progression. Blockade of CTLA4 and PDL1 lowers HIV viral loads, enhances HIV-specific CD8+ T cell function and killing of infected target cells (Kaufmann and Walker 2009; Palmer et al. 2013). Similar results are observed in simian immunodeficiency virus-infected rhesus macaques upon administration of anti-PD1 antibody (Velu et al. 2009).
Bacterial disease: CTLA4 and PD1 expression are associated with bacterial load and progressive dysfunction of pathogen-specific T cell responses (Rowe et al. 2009; Day et al. 2018). Blockade of CTLA4 increases the pathogen-specific CD4+ and CD8+ T cells and confers protection against Listeria monocytogenes in an experimental murine infection model (Rowe et al. 2009). In a mouse model of burn injury, anti-PDL1 treatment improves the bacterial clearance and survival following Pseudomonas aeruginosa or Staphylococcus aureus infection (Patil et al. 2018). In a Mycobacterium tuberculosis infection model, CTLA4 blockade enhances the immune response but fails to augment the clearance of the infection (Kirman et al. 1999); however, Pd1-/- mice are highly sensitive to infection by Mycobacterium tuberculosis and display immunopathology (Lázár-Molnár et al. 2010). Interestingly, one of the side effects of anti-PD1 therapy is the reactivation of M. tuberculosis in latently infected patients (Fujita et al. 2016). Consequently, some patients undergoing anti-PD1 therapy for cancer may lead to reactivation of tuberculosis (Barber et al. 2019).
Parasite diseases: The exhaustion of T cells is one of the hallmarks of chronic parasite infections. There is strong evidence that CD8+ T cell exhaustion plays a pivotal role in the reactivation of chronic Toxoplasmosis. The blockade of PD1 during chronic Toxoplasmosis controls the recrudescence disease by rescuing dysfunctional CD8+ T cells (Bhadra et al. 2011). T cell exhaustion in Leishmania infection is associated with an increase in the expression of PD1 expression on immune T cells. Blocking PDL1 signaling in vivo restores protective type 1 responses with a significant decrease in the parasite burden (Esch et al. 2013, Habib et al. 2018). Similarly CTLA4 blockade during Trypanosoma cruzi infection in mice reduces the mortality by about 50% (Graefe et al. 2004).
CTLA4 expression reduces immune associated pathology during infection with malaria (Jacobs et al. 2002). In a mouse model of malaria infection, PD1 enhances the protective response by regulating CD8+ T cells (Horne-Debets et al. 2016). In vivo blockade of the PD1 ligand in combination with LAG-3 or OX40 signaling restores CD4+ T cell function, increases the number of follicular helper T cells and T cell mediated B cell activation and plasmablasts. It also enhances protective antibodies and rapid clearance of blood-stage malaria parasites in mice (Butler et al. 2012, Zander et al. 2015). Patients infected with Plasmodium display an unique population of PD1+CTLA4+CD4+ T cells that produces IFNγ and IL10 that suppress the activation of other T cells (Mackroth et al. 2016). Also, PD1 deficiency or blockade enhances the humoral immune response to malaria antigens and this observation may be important in developing vaccine strategies (Liu et al. 2015b).
Checkpoint inhibition therapy for infectious diseases is in its infancy and all the studies till date are restricted to the animal models of infections. In some models, checkpoint blockade results in rapid pathogen clearance, whereas in other models pathogen clearance is outweighed by the collateral tissue injuries due to the hyper-activation of immune cells. Nevertheless, these studies are encouraging and the possible use of checkpoint inhibitors in clinical studies, with respect to viral latency, etc., in the near future needs to be explored.
13 The two Nobel laureates involved in the discovery of checkpoint inhibitors
It is perhaps useful to better understand the background of the two scientists whose work on checkpoint inhibitors was awarded with the 2018 Nobel prize in physiology/medicine. Prof. James P. Allison’s foray into T cell immunology began as an Assistant Professor in the basic sciences research campus in Smithsville, University of Texas. He was the first one to identify and biochemically characterize the αβ TCR by generating a monoclonal antibody, 124–40, to a T cell lymphoma, C6XL (Allison et al. 1982). Subsequently, other groups identified the αβ TCR using serological and molecular approaches (Mak 2007). Following this discovery, he moved as Director of the Cancer Research Laboratory and Professor to the University of California, Berkeley where he spent the next twenty years (1984–2004). During this period, his laboratory contributed to better understanding in diverse areas of T cell biology: the γδ T cell receptor (Allison et al. 1991), thymic maturation (Havran and Allison 1988) and costimulation. Initially, it was thought that CTLA4 performed positive costimulatory roles similar to CD28 (Linsley et al. 1992b). Subsequent studies by Prof. Allison’s group showed that antibodies to CTLA4, under soluble conditions, increased T cell activation whereas it had to the opposite effect upon crosslinked conditions (Krummel and Allison 1995). The generation of Ctla4-/- mice which died within 3–4 weeks due to hyper-proliferation of CD4+ T cells confirmed the negative role of Ctla4 during T cell activation (Waterhouse et al. 1995; Tivol et al. 1995; Chambers et al. 1997). It was in Berkeley that his laboratory discovered that antibodies to mouse CTLA4 reduce the growth of tumors (Leach et al. 1996). Subsequently, companies were involved in generating antibodies to human CTLA4 and performing clinical trials that demonstrated its efficacy in lowering tumor growth in patients (table 1). Notably, this is a good example of basic research in mice that led to translation benefits in humans. It is possible that Prof. Allison moved his laboratory to Memorial Sloan Kettering hospital in New York (2004–2012) to monitor the clinical trials with anti-CTLA4. Prof. Allison’s somewhat iconoclastic and irreverent traits are best reflected in the following statements: In college, I studied biochemistry, which was a good way to start because it teaches you precision, quantitation, and the fundamentals. But I got fascinated by the immune system, where there wasn’t much precision. The professor that taught me immunology in undergrad wasn’t even sure that there was such a thing as T cells…………….In science, you don’t have to be right. You have a hypothesis, and you test it, and you only have to be right some of the time. I thought that being wrong a lot was more fun (Neill 2016). The state of Texas has played a huge role in shaping Prof. Allison’s life: he was born there and did all his education including graduate schooling in Texas. He started his laboratory as an Assistant Professor in Texas and he is now back in the M D Anderson hospital, Texas as head of the Immunology program. In keeping with the theme “work hard and play hard”, Prof. Allison is a big fan of the country singer, Willie Nelson, and plays the harmonica as a member of the band, Checkpoints!
Prof. Tasuku Honjo from the Kyoto University, Japan, has made some stellar contributions in Immunology: First, his group identified S regions which are responsible for class switch recombination in Ig genes (Shimizu et al. 1982; Honjo 2008). Class switch recombination ensures that the heavy chain is changed during B cell differentiation, e.g. from μ to γ, without changing the antigenic specificity of Igs. In other words, the variable part of the Igs is unaltered whereas the constant region of the Igs is changed. Double strand breaks are generated at switch regions which are upstream to the heavy chain constant region genes and the intervening sequences are deleted (Kataoka et al. 1981). Second, one of the enzymes that is involved in CSR and somatic hyper mutation is Activation Induced Deaminase, a part of the RNA editing deaminase family, which was discovered by Honjo’s group (Muramatsu et al. 1999; Honjo 2008). In fact, deficiency in Activation Induced Deaminase leads to absence of class switching, resulting in hyper IgM. On the other hand, over expression leads to higher class switching from IgM to IgA (Muramatsu et al. 2000). Third, Honjo’s group was also responsible for the characterization of the 75 kDa subunit of the IL2 receptor which combines with another 55 kDa protein (Tac antigen) to form the high affinity IL2 receptor which is important for the autocrine proliferation of T cells (Kondo et al. 1987). Fourth, his laboratory has studied the roles of the Notch signaling pathways in the immune system with an emphasis on RBP-J, a transcription factor involved in Notch signaling, and Kyo-T which interacts with RBP-J and regulates its function (Taniguchi et al. 1998). Also, the roles of Mint, an endogenous inhibitor of the Notch signaling pathway, during T cell development was shown (Tsuji et al. 2007). During the course of his multi-faceted studies, his laboratory identified Pd1 to be upregulated during apoptosis (Ishida et al. 1992). Further studies on PD1 led the identification of its ligands (Freeman et al. 2000; Latchman et al. 2001) and roles in anti-tumor responses (Iwai et al. 2002). Currently, anti-PD1 has a large market share and is effective against a wide range of tumors (Topalian et al. 2012; Andrews 2015).
14 Future goals
Some of the issues listed above have led to the search for alternate and or combinatorial strategies that may lead to higher efficacies with lower toxic side effects. Precise scheduling of different immunotherapy regimens and dosing based upon circulating tumor DNA, neutrophil-lymphocyte ratio, cytokine release, etc. are taken into consideration for achieving proper efficacy. In case of a mammary tumor model, delaying the treatment with anti-PD1 is more effective at reducing tumor growth during combination therapy with anti-OX40 (Messenheimer et al. 2017). In fact, a more prudent approach to determine the order in which antibodies will be administered may be decided upon understanding the natural rhythms of anti-tumor immune responses (Rothschilds and Wittrup 2019).
The identification of antibodies against CTLA4 and PD1 as checkpoint inhibitors led to the search for other molecules that can also play similar roles. Activating monoclonal antibodies targeting different TNF superfamily members e.g. OX40, 4–1BB, CD40, GITR are known to sustain the proliferation and survival of activated T cells. Utomilumab (anti-4–1BB antibody) has been applied for clinical trials in patients with solid tumors and Merkel cell carcinoma where partial and complete responses had been reported. However, several of the treated patients demonstrated signs of fatigue, pyrexia, rashes and decreased appetite (Segal et al. 2018). In addition, clinical trials with anti-OX40 (BMS 986178), in combination with the TLR9 agonist SD-101 and radiation therapy, are underway (https://clinicaltrials.gov/ct2/show/record/NCT03410901?view=record). Also, antagonistic monoclonal antibodies against Lymphocyte activation gene-3 (LAG-3), V-domain Ig suppressor of T cells activation (VISTA), T cell Ig and mucin domain-3 (TIM-3) etc are also in clinical trials (Dempke et al. 2017). In the coming years, the efficacy of these additional costimulatory receptors in anti-tumor and other immune responses will be unearthed.
It is likely that existing and emerging technologies may combine with checkpoint inhibitor therapy to greatly reduce cancer progression and some are mentioned below. In particular, the combination of newer technologies with immune checkpoint inhibition is likely to result in long-term clinical benefits (Sharma and Allison 2015). Genomic profiling of tumor cells in cancer patients greatly influences the selection of particular anti-cancer chemotherapeutic drugs and this mode of genomic targeted selection of drugs is a useful tool. For example, the combination of anti-PD1 with a tyrosine kinase inhibitor shows greater reduction in tumor load compared to monotherapy in mice models (Tu et al. 2019). Lately, there has been an interest in identifying small molecular inhibitors that can act similar to checkpoint inhibitors. These will have the advantage of enhanced lipophilicity to infiltrate tumor microenvironment and may be less toxic with lower production costs (Sasikumar and Ramachandra 2018). However, further studies are required to evaluate their clinical efficacy.
Another form of therapy known as the Chimeric Antigen Receptor (CAR) T cell therapy involves genetic modification of autologous T cells, isolated through leukapheresis (a procedure to separate white blood cells from blood). The extracellular domain of CAR T cells contains binding moieties towards target antigen, so that immune response is mounted against the tumor expressing the antigen of interest (Gross et al. 1989). Therapeutic strategy using CAR T cells targeted to bind CD19 has been approved by the FDA in adults with diffuse large B cell lymphoma and children and young adults with B cell acute lymphoblastic leukemia. This approach too comes with a number of clinical complications such as cytokine release syndrome, neurotoxicity and inflammatory reactions (Grupp et al. 2016). Studies are in progress to broaden and improve the efficacy of CAR therapy (Crowther et al. 2020) and lower the toxicity in large number of tumors.
15 Summary
T cell costimulation is important for the maintenance of peripheral tolerance. Aberrant activation of T cells in the absence of proper context can lead to hyper-inflammation, including autoimmunity. Therefore, understanding the molecular players in this area is an active and important area of investigation. This review gives an overview of the field of T cell costimulation, which led to the development of anti-tumor therapy. In fact, it underscores the importance of basic research that may reap translational benefits. In addition, the development of CTLA4-Ig, which aids in dampening the immune response, is approved for the treatment against RA. We now know that there are multiple players orchestrating the immune response. The identification of the two important checkpoint players, CTLA4 and PD1, and their success has led to a new area of immunotherapy. Although, we have come a long way in our fight against cancer, there are issues and challenges with this form of therapy. Better understanding of additional players together with clinical trials is likely to lead to strategies, either singly or in combination, to increase the efficacy of anti-tumor therapy in a vast majority of patients with lower toxicity and cost. In fact, the success of checkpoint immunotherapy has led to the hope that the fight to win against cancer is not too distant in the future.
Abbreviations
- APC:
-
antigen presenting cells
- CAR:
-
chimeric antigen receptor
- CDRs:
-
complementarity determining regions
- CTLA4:
-
cytotoxic T lymphocyte associated antigen 4
- DC:
-
dendritic cells
- HIV:
-
Human immunodeficiency virus
- HVEM:
-
herpes virus entry mediator
- ICOS:
-
inducible T cell costimulator
- ICOSL:
-
inducible T cell costimulatory ligand
- IFNγ:
-
interferon gamma
- Ig:
-
immunoglobulin
- IL:
-
interleukin
- IRAEs:
-
immune-related adverse effects
- ITIM:
-
immunoreceptor tyrosine-based inhibitory motif
- ITSM:
-
immunoreceptor tyrosine-based switch motif
- Lck:
-
lymphocyte-specific protein tyrosine kinase
- MHC:
-
major histocompatibility complex
- PD1:
-
programmed death 1
- PDL1:
-
programmed death ligand 1
- PDL2:
-
programmed death ligand 2
- PI3K:
-
phosphoinositide 3-kinase
- PKB:
-
protein kinase B
- RA:
-
rheumatoid arthritis
- SLE:
-
systemic lupus erythematosus
- SNP:
-
single nucleotide polymorphisms
- TCR:
-
T cell receptor
- TGFβ:
-
transforming growth factor beta
- TNFα:
-
tumor necrosis factor alpha
- Tregs:
-
regulatory T cells
References
Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, Menter A, Lowe NJ, Krueger G, Brown MJ, Weiner RS, Birkhofer MJ, Warner GL, Berry KK, Linsley PS, Krueger JG, Ochs HD, Kelley SL and Kang S 1999 CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J. Clin. Invest. 103 1243–1252
Ahmed A, Mukherjee S and Nandi D 2009 Intracellular concentrations of Ca(2+) modulate the strength of signal and alter the outcomes of cytotoxic T-lymphocyte antigen-4 (CD152)-CD80/CD86 interactions in CD4(+) T lymphocytes. Immunology 126 363–377
Ahmed A and Nandi D 2011 T cell activation and function: role of signal strength; in Mathematical Models and Immune Cell Biology Lythe G and Molina-Paris C (Eds) (Springer, Berlin)
Allison JP, McIntyre BW and Bloch D 1982 Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J. Immunol. 129 2293–300
Allison JP, Asarnow DM, Bonyhadi M, Carbone A, Havran WL, Nandi D and Noble J 1991 Gamma delta T cells in murine epithelia: origin, repertoire, and function. Adv. Exp. Med. Biol. 292 63–69
Anderson DE, Bieganowska KD, Bar-Or A, Oliveira EML, Carreno B, Collins M and Hafler DA 2000 Paradoxical inhibition of T-cell function in response to CTLA-4 blockade; heterogeneity within the human T-cell population. Nat. Med. 6 211–214
Andrews A 2015 Treating with checkpoint inhibitors—figure $1 million per patient. Am. Health Drug Benefits 8 9
Arimura Y, Kato H, Dianzani U, Okamoto T, Kamekura S, Buonfiglio D, Miyoshi-Akiyama T, Uchiyama T and Yagi J 2002 A co-stimulatory molecule on activated T cells, H4/ICOS, delivers specific signals in T(h) cells and regulates their responses. Int. Immunol. 14 555–566
Aruffo A and Seed B 1987 Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proc. Natl. Acad. Sci. USA 84 8573–8577
Attarwala H 2010 TGN1412: from discovery to disaster. J. Young Pharm. 2 332–336
Bachmann MF, Köhler G, Ecabert B, Mak TW and Kopf M 1999 Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J. Immunol. 163 1128–1131
Bajorath J, Peach RJ and Linsley PS 1994 Immunoglobulin fold characteristics of B7–1 (CD80) and B7–2 (CD86) Protein Sci. 3 2148–2150
Balkwill FR, Capasso M and Hagemann T 2012 The tumor microenvironment at a glance. J. Cell Sci. 125 5591–5596
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ and Ahmed R 2006 Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439 682–687
Barber DL, Sakai S, Kudchadkar RR, Fling SP, Day TA, Vergara JA, Ashkin D, Cheng JH, Lundgren LM, Raabe VN, Kraft CS, Nieva JJ, Cheever MA, Nghiem PT, Sharon E 2019 Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci. Transl. Med. 11 eaat2702
Barnes MJ, Griseri T, Johnson AM, Young W, Powrie F and Izcue A 2013 CTLA-4 promotes Foxp3 induction and regulatory T cell accumulation in the intestinal lamina propria. Mucosal Immunol. 6 324–334
Bernard D, Riteau B, Hansen JD, Phillips RB, Michel F, Boudinot P and Benmansour A 2006 Costimulatory receptors in a teleost fish: typical CD28, elusive CTLA4. J. Immunol. 176 4191–4200
Bartkowiak T and Curran MA 2015 4–1BB Agonists: multi-potent potentiators of tumor immunity. Front. Oncol. 5 117
Bertrand A, Kostine M, Barnetche T, Truchetet ME and Schaeverbeke T 2015 Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med. 13 211
Beyersdorf N, Kerkau T and Hünig T 2015 CD28 co-stimulation in T-cell homeostasis: a recent perspective. Immunotargets Ther. 4 111–122
Bhadra R, Gigley JP, Weiss LM and Khan IA 2011 Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1–PDL-1 blockade. Proc. Natl. Acad. Sci. 108 9196–9201
Blair HA and Deeks ED 2017 Abatacept: a review in rheumatoid arthritis. Drugs 77 1221–1233
Blank C and Mackensen A 2007 Contribution of the PD-L1/PD-1 pathway to T-cellexhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 56 739–745
Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T and Thompson CB 1995 CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3 87–98
Boomer JS and Green JM 2010 An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2 a002436
Boomer JS, Deppong CM, Shah DD, Bricker TL and Green JM 2014 A double-mutant knockin of the CD28 YMNM and PYAP motifs reveals a critical role for the YMNM motif in regulation of T cell proliferation and Bcl-xL expression. J. Immunol. 192 3465–3469
Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, Strom TB, Simpson EM, Freeman GJ, Sharpe AH 1997 B7–1 and B7–2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity 6 303–313
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, et al. 2015 Nivolumab versus Docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373 123–35
Bretscher P and Cohn M 1970 A theory of self-nonself discrimination. Science 169 1042–9
Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA and Freeman GJ 2003 Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J. Immunol. 170 1257–66
Brown KE 2018 Revisiting CD28 Superagonist TGN1412 as potential therapeutic for pediatric B cell leukemia: a review. Diseases 6 41
Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG and Golstein P 1987 A new member of the immunoglobulin superfamily–CTLA-4. Nature 328 267–270
Buckley RH 2000 Primary immunodeficiency diseases due to defects in lymphocytes. N. Engl. J. Med. 343 1313–1324
Burnett DL, Parish IA, Masle-Farquhar E, Brink R, Goodnow CC 2017 Murine LRBA deficiency causes CTLA-4 deficiency in Tregs without progression to immune dysregulation. Immunol. Cell Biol. 95 775–788
Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, Crompton PD and Harty JT 2012 Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol. 13 188–195
Byun DJ, Wolchok JD, Rosenberg LM and Girotra M 2017 Cancer immunotherapy—immune checkpoint blockade and associated endocrinopathies. Nat. Rev. Endocrinol. 13 195–207
Cai G and Freeman GJ 2009 The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol. Rev. 229 244–258
Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ and Carreno BM 2002 PD-1:PD-L inhibitory pathway affects both CD4(+) andCD8(+) T cells and is overcome by IL-2. Eur. J. Immunol. 32 634–643
Céfaï D, Schneider H, Matangkasombut O, Kang H, Brody J and Rudd CE 1998 CD28 receptor endocytosis is targeted by mutations that disrupt phosphatidylinositol 3-kinase binding and costimulation. J. Immunol. 160 2223–2230
Chambers CA, Sullivan TJ and Allison JP 1997 Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity 7 885–895
Chattopadhyay K, Lazar-Molnar E, Yan Q, Rubinstein R, Zhan C, Vigdorovich V, Ramagopal UA, Bonanno J, Nathenson SG and Almo SC 2009 Sequence, structure, function, immunity: structural genomics of costimulation. Immunol. Rev. 229 356–386
Chou PY and Fasman GD 1974 Conformational parameters for amino acids in helical, β-sheet, and random coil regions calculated from proteins. Biochemistry 13 211–222
Chuang E, Lee KM, Robbins MD, Duerr JM, Alegre ML, Hambor JE, Neveu MJ, Bluestone JA and Thompson CB 1999 Regulation of cytotoxic T lymphocyte-associated molecule-4 by Src kinases. J. Immunol. 162 1270–1277
Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA and Davis SJ 2002 The interaction properties of costimulatory molecules revisited. Immunity 17 201–210
Collins M, Ling V and Carreno BM 2005 The B7 family of immune-regulatory ligands. Genome Biol. 6 223
Conforti F, Pala L, Bagnardi V, De Pas T, Martinetti M, Viale G, Gelber RD and Goldhirsch A 2018 Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. Lancet Oncol. 19 737–746
Crowther MD, Dolton G, Legut M, Caillaud ME, Lloyd A, Attaf M, Galloway SAE, Rius C, et al. 2020 Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 21 178–185
Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, TsaiK, Nosrati A, Nardo L, Alvarado MD, Algazi AP, Pampaloni MH, Lobach IV, Hwang J, Pierce RH, Gratz IK, Krummel MF and Rosenblum MD 2016 Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Invest. 126 3447–52
Day CL, Abrahams DA, Bunjun R, Stone L, de Kock M, Walzl G, Wilkinson RJ, Burgers WA and Hanekom WA 2018 PD-1 Expression on Mycobacterium tuberculosis—specific CD4 T cells is associated with bacterial load in human tuberculosis. Front. Immunol. 9 1995
Dempke WCM, Fenchel K, Uciechowski P and Dale SP 2017 Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur. J Cancer 74 55–72
Dennehy KM, Elias F, Zeder-Lutz G, Ding X, Altschuh D, Lühder F and Hünig T 2006 Cutting edge: monovalency of CD28 maintains the antigen dependence of T cell costimulatory responses J. Immunol. 176 5725–5729
Dodson LF, Boomer JS, Deppong CM, Shah DD, Sim J, Bricker TL, Russell JH and Green JM 2009 Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol. Cell Biol. 29 3710–37121
Dong H, Zhu G, Tamada K and Chen L 1999 B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 5 1365–1369
Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM and Chen L 2004 B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity 2004 20 327–336
Di Giacomo AM, Calabrò L, Danielli R, Fonsatti E, Bertocci E, Pesce I, Fazio C, Cutaia O, Giannarelli D, Miracco C, Biagioli M, Altomonte M and Maio M 2013 Long-term survival and immunological parameters in metastatic melanoma patients who responded to ipilimumab 10 mg/kg within an expanded access programme. Cancer Immunol. Immunother. 62 1021–1028
Eastwood D, Findlay L, Poole S, Bird C, Wadhwa M, Moore M, Burns C, Thorpe R and Stebbings R 2010 Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T‐cells. Br. J. Pharmacol. 161 512–526
Egen JG and Allison JP 2002 Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 16 23–35
Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, et al. 2016 Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N. Engl. J. Med. 375 1845–1855
Esensten JH, Helou YA, Chopra G, Weiss A and Bluestone JA 2016 CD28 costimulation: from mechanism to therapy. Immunity 44 973–988
Evans EJ, Esnouf RM, Manso-Sancho R, Gilbert RJ, James JR, Yu C, Fennelly JA, Vowles C, Hanke T, Walse B, Hünig T, Sørensen P, Stuart DI and Davis SJ 2005 Crystal structure of a soluble CD28-Fab complex. Nat. Immunol. 6 271–279
Esch KJ, Juelsgaard R, Martinez PA, Jones DE and Petersen CA 2013 Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J. Immunol. 191 5542–5550
Fan X, Quezada SA, Sepulveda MA, Sharma P and Allison JP 2014 Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 211 715–725
Ferguson SE, Han S, Kelsoe G and Thompson CB 1996 CD28 is required for germinal center formation. J. Immunol. 156 4576–4581
Ferrari SM, Fallahi P, Elia G, Ragusa F, Ruffilli I, Patrizio A, Galdiero MR, Baldini E, Ulisse S, Marone G and Antonelli A 2019 Autoimmune endocrine dysfunctions associated with cancer immunotherapies. Int. J. Mol. Sci. 20(10) 2560
Fournier S, Rathmell JC, Goodnow CC and Allison JP 1997 T cell-mediated elimination of B7.2 transgenic B cells. Immunity 6 327–339
Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH and Thompson CB 2002 The CD28 signaling pathway regulates glucose metabolism. Immunity 16 769–777
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, et al. 2000 Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192 1027–1034
Fu T, He Q and Sharma P 2011 The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 71 5445–5454
Fujita K, Terashima T and Mio T 2016 Anti-PD1 antibody treatment and the development of acute pulmonary tuberculosis. J. Thorac. Oncol. 11 2238–2240
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, Chen PL, Hwu P, Allison JP, Futreal A, Wargo JA and Sharma P 2016 Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167 397–404
Genovese MC, Schiff M, Luggen M, Becker JC, Aranda R, Teng J, Li T, Schmidely N, Le Bars M and Dougados M 2008 Efficacy and safety of the selective co-stimulation modulator abatacept following 2 years of treatment in patients with rheumatoid arthritis and an inadequate response to anti-tumour necrosis factor therapy. Ann. Rheum. Dis. 67 547–554
Gibbs K, Purcell L.D., Liu C and Files D 2018 PD-1 inhibition promotes Th1 inflammation and alters lymphocyte activation in mice after LPS-lung injury. Am. J. Resp. Crit. Care Med. 197 A7539
Gmünder H and Lesslauer W 1984 A 45-kDa human T-cell membrane glycoprotein functions in the regulation of cell proliferative responses. Eur. J Biochem. 142 153–160
Gough SC, Walker LS and Sansom DM 2005 CTLA4 gene polymorphism and autoimmunity. Immunol. Rev. 204 102–15
Graefe SE, Jacobs T, Wächter U, Bröker BM and Fleischer B 2004 CTLA-4 regulates the murine immune response to Trypanosoma cruzi infection. Parasite Immunol. 26 19–28
Green JM, Noel PJ, Sperling AI, Walunas TL, Gray GS, Bluestone JA and Thompson CB 1994 Absence of B7-dependent responses in CD28-deficient mice. Immunity 1 501–508
Gross G, Waks T and Eshhar Z 1989 Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 86 10024–10028
Gross JA, Callas E and Allison JP 1992 Identification and distribution of the costimulatory receptor CD28 in the mouse. J. Immunol. 149 380–388
Grupp SA, Laetsch TW, Buechner J, Bittencourt H, Maude SL, Verneris MR, Myers GD, Boyer MW, et al. 2016 Analysis of a global registration trial of the efficacy and safety of CTL019 in pediatric and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL). Blood 128 221
Habib S, El Andaloussi A, Elmasry K, Handoussa A, Azab M, Elsawey A, Al-Hendy A and Ismail N 2018 PDL-1 blockade prevents T cell exhaustion, inhibits autophagy, and promotes clearance of Leishmania donovani. Infect. Immun. 86 e00019-18
Hansen JA, Martin PJ and Nowinski RC 1980 Monoclonal antibodies identifying a novel T-Cell antigen and Ia antigens of human lymphocytes. Immunogenetics 10 247–260
Hansen JD, Du Pasquier L, Lefranc MP, Lopez V, Benmansour A and Boudinot P 2009 The B7 family of immunoregulatory receptors: a comparative and evolutionary perspective. Mol. Immunol. 46 457–472
Harding FA, McArthur JG, Gross JA, Raulet DH and Allison JP 1992 CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 356 607–609
Havran WL and Allison JP 1988 Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature 335 443–445
Honjo T 2008 A memoir of AID, which engraves antibody memory on DNA. Nat. Immunol. 9 335–337
Holsti MA, McArthur J, Allison JP and Raulet DH 1994 Role of IL-6, IL-1, and CD28 signaling in responses of mouse CD4+ T cells to immobilized anti-TCR monoclonal antibody. J. Immunol. 152 1618–1628
Horne-Debets JM, Karunarathne DS, Faleiro RJ, Poh CM, Renia L and Wykesa MN 2016 Mice lacking programmed cell death-1 show a role for CD8+ T cells in long-term immunity against blood-stage malaria. Sci. Rep. 6 26210
Ikemizu S, Gilbert RJ, Fennelly JA, Collins AV, Harlos K, Jones EY, Stuart DI and Davis SJ 2000 Structure and dimerization of a soluble form of B7–1. Immunity 12 51–60
Ingram JR, Blomberg OS, Rashidian M, Ali L, Garforth S, Fedorov E, Fedorov AA, Bonanno JB, Le Gall C, Crowley S, Espinosa C, Biary T, Keliher EJ, Weissleder R, Almo SC, Dougan SK, Ploegh HL, Dougan M 2018 Anti-CTLA-4 therapy requires an Fc domain for efficacy. Proc. Natl. Acad. Sci. USA 115 3912–3917
Ishida Y, Agata Y, Shibahara K and Honjo T 1992 Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11 3887–3895
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T and Minato N 2002 Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 99 12293–12297
Ihara K, Ahmed S, Nakao F, Kinukawa N, Kuromaru R, Matsuura N, Iwata I, Nagafuchi S, Kohno H, Miyako K and Hara T 2001 Association studies of CTLA-4, CD28, and ICOS gene polymorphisms with type 1 diabetes in the Japanese population. Immunogenetics 53 447–454
Jacobs T, Graefe SE, Niknafs S, Gaworski I, Fleischer B 2002 Murine malaria is exacerbated by CTLA-4 blockade. J. Immunol. 169 2323–2329
Jenkins MK and Schwartz RH 1987 Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 165 302–319
Jenkins MK, Taylor PS, Norton SD and Urdahl KB 1991 CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J. Immunol. 147 2461–2466
Jones RG, Elford AR, Parsons MJ, Wu L, Krawczyk CM, Yeh WC, Hakem R, Rottapel R, Woodgett JR and Ohashi PS 2002 CD28-dependent activation of protein kinase B/Akt blocks Fas-mediated apoptosis by preventing death-inducing signaling complex assembly. J. Exp. Med. 196 335–348
June CH, Ledbetter JA, Gillespie MM, Lindsten T and Thompson CB 1987 T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol. Cell Biol. 7 4472–4481
Kalathil S, Lugade AA, Miller A, Iyer R and Thanavala Y 2013 Higher frequencies of GARP(+) CTLA-4(+) Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 73 2435–2444
Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, Morikawa H, Kawazoe A, Kinoshita T, Shitara K, Sakaguchi S and Nishikawa H 2019 PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 116 9999–10008
Karabon L, Jedynak A, Tomkiewicz A, Wolowiec D, Kielbinski M, Woszczyk D, Kuliczkowski K and Frydecka I 2011 ICOS gene polymorphisms in B-cell chronic lymphocyticleukemia in the Polish population. Folia Histochem. Cytobiol. 49 49–54
Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD and Bluestone JA 1996 CTLA-4: a negative regulator of autoimmune disease. J. Exp. Med. 184 783–788
Kassu A, Marcus RA, D’Souza MB, Kelly-McKnight EA, Golden-Mason L, Akkina R,Fontenot AP, Wilson CC and Palmer BE 2010 Regulation of virus-specific CD4+ T cell function by multiple costimulatory receptors during chronic HIV infection. J. Immunol. 185 3007–3018
Kataoka T, Miyata T and Honjo T 1981 Repetitive sequences in class-switch recombination regions of immunoglobulin heavy chain genes. Cell 23 357–368
Kato T and Nariuchi H 2000 Polarization of naive CD4+ T cells toward the Th1 subset by CTLA-4 costimulation. J. Immunol. 164 3554–3562
Kato S, Goodman A,Walavalkar V, Barkauskas DA, Sharabi A and Kurzrock R 2017 Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin. Cancer Res. 23 4242–4250
Kavanagh B, O’Brien S, Lee D, Hou Y, Weinberg V, Rini B, Allison JP, Small EJ and Fong L 2008 CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood 112 1175–1183
Kaufmann DE and Walker BD 2009 PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J. Immunol. 182 5891–5897
Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M and Escors D 2011 PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 3 581–592
Keir ME, Butte MJ, Freeman GJ and Sharpe AH 2008 PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26 677–704
Kirman J, McCoy K, Hook S, Prout M, Delahunt B, Orme I, Frank A and Le Gros G 1999 CTLA-4 blockade enhances the immune response induced by mycobacterial infection but does not lead to increased protection. Infect. Immun. 67 3786–3792
Khattri R, Auger JA, Griffin MD, Sharpe AH and Bluestone JA 1999 Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J. Immunol. 162 5784–5791
Kondo S, Kinoshita M, Shimizu A, Saito Y, Konishi M, Sabe H and Honjo T 1987 Expression and functional characterization of artificial mutants of interleukin-2 receptor. Nature 327 64–67
Kong KF, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, Becart S, Kim G, Yates JR 3rd, Kronenberg M, Saito T, Gascoigne NR and Altman A 2014 Protein kinase C-η controls CTLA-4-mediated regulatory T cell function. Nat. Immunol. 15 465–472
Koura DT, Horan JT and Langston AA et al. 2013 In vivo T cell costimulation blockade with abatacept for acute graft-versus-host disease prevention: a first-in-disease trial. Biol. Blood Marrow Transplant. 19 1638–1649
Krummel MF and Allison JP 1995 CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182 429–465
Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, et al. 2014 Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345 1623–1627
Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, Kuchroo VK, Regev A and Anderson AC 2019 Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1-CD8+ Tumor-Infiltrating T Cells. Immunity 50 181–194
Lafage-Pochitaloff M, Costello R, Couez D, Simonetti J, Mannoni P, Mawas C and Olive D 1990 Human CD28 and CTLA-4 Ig superfamily genes are located on chromosome 2 at bands q33-q34. Immunogenetics 31 198–201
Lafferty KJ and Cunningham AJ 1975 A new analysis of allogeneic interactions. Aust. J. Exp. Biol. Med. Sci. 53 27–42
Laman JD, Claassen E and Noelle RJ 2017 Functions of CD40 and its ligand, gp39 (CD40L). Crit. Rev. Immunol. 37 371–420
Lane P, Burdet C, Hubele S, Scheidegger D, Müller U, McConnell F and Kosco-Vilbois M 1994 B cell function in mice transgenic for mCTLA4-H gamma 1: lack of germinal centers correlated with poor affinity maturation and class switching despite normal priming of CD4+ T cells. J. Exp. Med. 179 819–830
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, et al. 2001 PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2 261–268
Lathrop SK, Huddleston CA, Dullforce PA, Montfort MJ, Weinberg AD and Parker DC 2004 A signal through OX40 (CD134) allows anergic, autoreactive T cells to acquire effector cell functions. J. Immunol. 172 6735–6743
Lázár-Molnár E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, Porcelli SA, Almo SC, Nathenson SG and Jacobs WR Jr 2010 Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc. Natl. Acad. Sci. USA 107 13402–13407
Lázár-Molnár E, Scandiuzzi L, Basu I, Quinn T, Sylvestre E, Palmieri E, Ramagopal UA, Nathenson SG, Guha C and Almo SC 2017 Structure-guided development of a high-affinity human Programmed Cell Death-1: Implications for tumor immunotherapy. EBioMedicine 17 30–44
Leach DR, Krummel MF and Allison JP 1996 Enhancement of antitumor immunity by CTLA-4 blockade. Science 271 1734–6
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, et al. 2015 PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372 2509–2520
Lee HW, Park SJ, Choi BK, Kim HH, Nam KO and Kwon BS 2002 4–1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J. Immunol. 169 4882–4888
Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, Straus D, Samelson LE, Thompson CB and Bluestone JA 1998 Molecular basis of T cell inactivation by CTLA-4. Science 282 2263–2266
Lee GR 2018 The balance of Th17 versus Treg cells in autoimmunity. Int. J. Mol. Sci. 19 730
Li D, Gál I, Vermes C, Alegre ML, Chong AS, Chen L, Shao Q, Adarichev V, Xu X, Koreny T, Mikecz K, Finnegan A, Glant TT and Zhang J 2004 Cbl-b: one of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J. Immunol. 173 7135–7139
Li J and Ferris RL 2014 PD-1/SHP-2 negatively regulate Tc1/Th1 phenotypic responses and activation of T cells in the tumor microenvironment. J. Immunother. Cancer 2 P221
Liakou C, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, Logothetis C and Sharma P 2008 CTLA-4 blockade increases IFN gamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA 105 14987–14992
Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK and Ledbetter JA 1991a Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp. Med. 173 721–730
Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK and Ledbetter JA 1991b CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174 561–569
Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, Singh C and Tepper MA 1992a Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science 257 792–795
Linsley PS, Greene JL, Tan P, Bradshaw J, Ledbetter JA, Anasetti C and Damle NK 1992b Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 176 1595–1604
Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL and Mittler RS 1996 Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 4 535–543
Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, Luo D, Qi H 2015a T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature 517 214–218
Liu T, Lu X, Zhao C, Fu X, Zhao T, Xu W 2015b PD-1 deficiency enhances humoral immunity of malaria infection treatment vaccine. Infect. Immun. 83 2011–2017
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, Zhang Y, Liu Z, et al. 2015 Autoimmune disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349 436–440
Lonberg N 2005 Human antibodies from transgenic animals. Nat. Biotechnol. 23 1117–1125
Lucas PJ, Negishi I, Nakayama K, Fields LE, Loh DY 1995 Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J. Immunol. 154 5757–5768
Mackroth MS, Abel A, Steeg C,zur Wiesch JS and Jacobs T 2016 Acute malaria induces PD1+CTLA4+ effector T cells with cell-extrinsic suppressor function. PLoS Pathog. 12 e1005909
Maier LM, Anderson DE, De Jager PL,Wicker LS and Hafler DA 2007 Allelic variant in CTLA4 alters T cell phosphorylation patterns. Proc. Natl. Acad. Sci. USA 104 18607–18612
McQuade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, Park JJ, Haydu LE, et al. 2018 Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol. 19 310–322
Mak TW 2007 The T cell antigen receptor: “The Hunting of the Snark”. Eur. J. Immunol. 37(Suppl 1) S83–S93
Majumdar S, Pathak S and Nandi D 2018 Thymus: the site for development of cellular immunity. Resonance 2 197–217
Mandelbrot DA, McAdam AJ and Sharpe AH 1999 B7–1 or B7–2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). J. Exp. Med. 189 435–440
Masteller EL, Chuang E, Mullen AC, Reiner SL and Thompson CB 2000 Structural analysis of CTLA-4 function in vivo. J. Immunol. 164 5319–5327
Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D’Cruz D, Wallace DJ, Bae SC, Sigal L, Becker JC, Kelly S, Raghupathi K, Li T, Peng Y, Kinaszczuk M and Nash P 2010 The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 62 3077–8730
Michel F, Attal-Bonnefoy G, Mangino G, Mise-Omata S and Acuto O 2001 CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity 15 935–945
Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, et al. 2016 Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur. J. Cancer 54 139–148
Min L 2016 Immune-related endocrine disorders in novel immune checkpoint inhibition therapy. Genes Dis. 3 252–256
Mirza N, Duque MA, Dominguez AL, Schrum AG, Dong H and Lustgarten J 2010 B7-H1expression on old CD8+ T cells negatively regulates the activation of immune responses in aged animals. J. Immunol. 184 5466–5474
Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki I-m and Okazaki T 2019 PD-1 Primarily targets TCR signal in the inhibition of functional T cell activation. Front. Immunol. https://doi.org/10.3389/fimmu.2019.00630
Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO and Honjo T 1999 Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274 18470–18476
Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai S and Honjo T 2000 Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102 553–563
Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, Gough MJ, Urba WJ and Fox BA 2017 Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. Clin. Cancer Res. 23 6165–6177
Mukherjee S, Maiti PK and Nandi D 2002 Role of CD80, CD86, and CTLA4 on mouse CD4(+) T lymphocytes in enhancing cell-cycle progression and survival after activation with PMA and ionomycin. J. Leukoc. Biol. 72 921–931
Najafian N and Sayegh MH 2000 CTLA4-Ig: a novel immunosuppressive agent. Exp. Opin. Invest. Drugs 9 2147–2157
Neill US 2016 A conversation with James Allison. J. Clin. Invest. 126 3–4
Nomizo T, Ozasa H, Tsuji T, Funazo T, Yasuda Y, Yoshida H, Yagi Y, Sakamori Y,Nagai H, Hirai T and Kim YH 2017 Clinical impact of single nucleotide polymorphism in PD-L1 on response to nivolumab for advanced non-small-cell lung cancer patients. Sci. Rep. 7 45124
Nishimura H, Minato N, Nakano T and Honj T 1998 Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 10 1563–1572
Nishimura H, Nose M, Hiai H, Minato N and Honjo T 1999 Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying Immunoreceptor. Immunity 11 141–151
Oaks MK and Hallett KM 2000 A soluble form of CTLA-4 in patients with autoimmune thyroid disease. J. Immunol. 164 5015–5018
Oosterwegel MA, Mandelbrot DA, Boyd SD, Lorsbach RB, Jarrett DY, Abbas AK and Sharpe AH 1999 The role of CTLA-4 in regulating Th2 differentiation. J. Immunol. 163 2634–2639
Oestreich KJ, Yoon H, Ahmed R and Boss JM 2008 NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol. 181 4832–4839
Ogawa S, Watanabe M, Sakurai Y, Inutake Y, Watanabe S, Tai X and Abe R 2013 CD28 signaling in primary CD4(+) T cells: identification of both tyrosine phosphorylation-dependent and phosphorylation-independent pathways. Int. Immunol. 25 671–681
Okazaki T, Otaka Y, Wang J, Hiai H, Takai T, Ravetch JV and Honjo T 2005 Hydronephrosis associated with antiurothelial and antinuclear autoantibodies in BALB/c-Fcgr2b-/-Pdcd1-/- mice. J. Exp. Med. 202 1643–1648
Ostrov DA, Shi W, Schwartz JC, Almo SC and Nathenson SG 2000 Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science 290 816–819
Pai CS, Simons DM, Lu X, Evans M, Wei J, Wang YH, Chen M, Huang J, et al. 2019 Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J. Clin. Invest. 129 349–363
Pan XM, Gao LB, Liang WB, Liu Y, Zhu Y, Tang M, Li YB and Zhang L 2010 CD86 +1057 G/Apolymorphism and the risk of colorectal cancer DNA. Cell Biol. 29 381–386
Pandiyan P, Gärtner D, Soezeri O, Radbruch A, Schulze-Osthoff K and Brunner-Weinzierl MC 2004 CD152 (CTLA-4) determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3 kinase function. J. Exp. Med. 199 831–842
Palmer BE, Neff CP, Lecureux J, Ehler A, Dsouza M, Remling-Mulder L, Korman AJ, Fontenot AP and Akkina R 2013 In vivo blockade of the PD-1 receptor suppresses HIV-1 viral loads and improves CD4+ T cell levels in humanized mice. J. Immunol. 190 211–219
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB and Riley JL 2005 CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 25 9543–9553
Patil NK, Luan L, Bohannon JK, Hernandez A, Guo Y and Sherwood ER 2018 Anti-PD-L1 protects against infection with common bacterial pathogens after burn injury. J. Leukoc. Biol. 103 23–33
Peach RJ, Bajorath J, Naemura J, Leytze G, Greene J, Aruffo A and Linsley PS 1995 Both extracellular immunoglobin-like domains of CD80 contain residues critical for binding T cell surface receptors CTLA-4 and CD28. J. Biol. Chem. 270 21181–21187
Peer S, Baier G and Gruber T 2017 Cblb-deficient T cells are less susceptible to PD-L1-mediated inhibition. Oncotarget 8 41841–41853
Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, Yagita H, Overwijk WW, Lizée G, Radvanyi L and Hwu P 2012 PD-1 blockade enhances T-cell migration to tumors by elevating IFN-γ inducible chemokines. Cancer Res. 72 5209–5218.
Podder S, Rakshit S, Ponnusamy M and Nandi D 2016 Efficacy of bacteria in cancer immunotherapy: special emphasis on the potential of mycobacterial species. Clin. Cancer Drugs 3 100–108
Porciello N, Grazioli P, Campese AF, Kunkl M, Caristi S, Mastrogiovanni M, Muscolini M, Spadaro F, Favre C, Nunès JA, Borroto A, Alarcon B, Screpanti I, Tuosto L 2018 A non-conserved amino acid variant regulates differential signalling between human and mouse CD28. Nat. Commun. 9 1080
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, Walker LS and Sansom DM 2011 Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332 600–603
Ramagopal UA, Liu W, Garrett-Thomson SC, Bonanno JB, Yan Q, Srinivasan M, Wong SC, Bell A, Mankikar S, Rangan VS, Deshpande S, Korman AJ and Almo SC 2017 Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab Proc. Natl. Acad. Sci. USA 114 E4223–E4232
Rakshit S, Ponnusamy M, Papanna S, Saha B, Ahmed A and Nandi D 2012 Immunotherapeutic efficacy of Mycobacterium indicus pranii in eliciting anti-tumor T cell responses: critical roles of IFNγ. Int. J. Cancer 130 865–875
Reddy B, Gupta S, Chuzhin Y, Kalergis AM, Budhai L, Zhang M, Droguett G, Horwitz MS, Chowdhury JR, Nathenson SG and Davidson A 2001 The effect of CD28/B7 blockade on alloreactive T and B cells after liver cell transplantation. Transplantation 71 801–811
Redmond WL, Ruby CE and Weinberg AD 2009 The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit. Rev. Immunol. 29 187–201
Routy B, Le Chatelier, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, et al. 2018 Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359 91–97
Rowe JH, Johanns TM, Ertelt JM, Lai JC and Way SS 2009 Cytotoxic T-lymphocyte antigen 4 blockade augments the T-cell response primed by attenuated Listeria monocytogenes resulting in more rapid clearance of virulent bacterial challenge. Immunology 128 e471–e478
Rowshanravan B, Halliday N and Sansom DM 2018 CTLA-4: a moving target in immunotherapy. Blood 131 58–67
Rothschilds AM and Wittrup KD 2019 What, why, where, and when: bringing timing to immuno-oncology. Trends Immunol. 40 12–21
Ribas A, Comin-Anduix B, Economou JS, Donahue TR, de la Rocha P, Morris LF, Jalil J, Dissette VB, Shintaku IP, Glaspy JA, Gomez-Navarro J and Cochran AJ 2009 Intratumoral immune cell infiltrates, FoxP3, and indoleamine 2,3-dioxygenase in patients with melanoma undergoing CTLA4 blockade. Clin. Cancer 15 390–399
Riley JL, Blair PJ, Musser JT, Abe R, Tezuka K, Tsuji T and June CH 2001 ICOS costimulation requires IL-2 and can be prevented by CTLA-4 engagement. J. Immunol. 166 4943–4948
Rudd CE, Taylor A and Schneider H 2009 CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 229 12–26
Russo, G. L., Moro, M., Sommariva, M., Cancila, V., Boeri, M., Centonze, G. and Milione M 2019 Antibody–Fc/FcR interaction on macrophages as a mechanism for hyperprogressive disease in non–small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin. Cancer Res. 25 989–999
Salomon B and Bluestone JA 2001 Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Ann. Rev. Immunol. 19 225–252
Sansom DM, Manzotti CN and Zheng Y 2003 What’s the difference between CD80 and CD86? Trends Immunol. 24 314–319
Sasikumar PG and Ramachandra M 2018 Small-molecule immune checkpoint inhibitors targeting PD-1/PD-L1 and other emerging checkpoint pathways. BioDrugs 32 481–497
Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, Cesari R, Chen Y, Davis CB, Huang B, Thall AD and Gopal AK 2018 Phase I study of single-agent Utomilumab (PF-05082566), a 4–1BB/CD137 agonist, in patients with advanced cancer. Clin. Cancer Res. 24 1816–1823
Schwartz JC, Zhang X, Fedorov AA, Nathenson SG and Almo SC 2001 Structural basis for co-stimulation by the human CTLA-4/B7–2 complex. Nature 410 604–608
Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, et al. 2014 Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 20 1410–1416
Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M and Korman AJ 2013 Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 1 32–42
Sharma P and Allison JP 2015 Immune checkpoint targeting in cancer therapy: towards combination strategies with curative potential. Cell 161 205–214
Shi J, Hou S, Fang Q, Liu X, Liu X and Qi H 2018 PD-1 controls follicular T helper cell positioning and function. Immunity 49 264–274
Shimizu A, Takahashi N, Yaoita Y and Honjo T 1982 Organization of the constant-region gene family of the mouse immunoglobulin heavy chain. Cell 28 499–506
Shinohara T, Taniwaki M, Ishida Y, Kawaichi M and Honjo T 1994 Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics 23 704–706
Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS and Saito T 1997 Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity 6 583–589
Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE and Held W 2019 Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50 195–211
Siggs OM, Russell A, Singh-Grewal D, Wong M, Chan P, Craig ME, O’Loughlin T, Stormon M and Goodnow CC 2019 Preponderance of CTLA4 Variation Associated With Autosomal Dominant Immune Dysregulation in the MYPPPY Motif. Front. Immunol. 10 1544
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB and Gajewski TF 2015 Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350 1084–1089
Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, Stahl ML, Seehra J, Somers WS and Mosyak L 2001 Crystal structure of the B7–1/CTLA-4 complex that inhibits human immune responses. Nature 410 608–611
Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, Cui G, Li MO and Kaech SM 2014 The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity 41 802–814
Suvas S, Singh V, Sahdev S, Vohra H and Agrewala JN 2002 Distinct role of CD80 and CD86 in the regulation of the activation of B cell and B cell lymphoma. J. Biol. Chem. 277 7766–7775
Syn NL, Teng MWL, Mok TSK and Soo RA 2017 De-novo and acquired resistance to immune checkpoint targeting Lancet Oncol. 18 e731–e741
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ and Mering CV 2019 STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019 47 D607–D613
Tai X, Van Laethem F, Sharpe AH and Singer A 2007 Induction of autoimmune disease in Ctla4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc. Natl. Acad. Sci. USA 104 13756–13761
Tamura K, Peterson D, Peterson N, Stecher G, Nei M and Kumar S 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28 2731–2739
Tan D, Sheng L and Yi QH 2018 Correlation of PD-1/PD-L1 polymorphisms and expressions with clinicopathologic features and prognosis of ovarian cancer. Cancer Biomark. 21 287–297
Tang AL, Teijaro JR, Njau MN, Chandran SS, Azimzadeh A, Nadler SG, Rothstein DM and Farber DL 2008 CTLA4 expression is an indicator and regulator of steady-state CD4+ FoxP3+ T cell homeostasis. J. Immunol. 181 1806–1813
Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M and Bluestone JA 2004 Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34 2996–3005
Tang W, Chen S, Chen Y, Lin J, Lin J, Wang Y, Liu C and Kang M 2017 Programmed death-1 polymorphisms is associated with risk of esophagogastric junction adenocarcinoma in the Chinese Han population: a case-control study involving 2,740 subjects. Oncotarget 8 39198–39208
Taniguchi Y, Furukawa T, Tun T, Han H and Honjo T 1998 LIM protein KyoT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol. Cell Biol. 18 644–654
Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T and Honjo T 2011 IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated Immunity J. Immunol. 186 2772–2779
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA and Sharpe AH 1995 Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3 541–547
Tivol EA, Boyd SD, McKeon S, Borriello F, Nickerson P, Strom TB and Sharpe AH 1997 CTLA4Ig prevents lymphoproliferation and fatal multiorgan tissue destruction in CTLA-4-deficient mice. J. Immunol. 158 5091–5094
Townsend SE and Allison JP 1993 Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science 259 368–370
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, et al. 2012 Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366 2443–2454
Tsuji M, Shinkura R, Kuroda K, Yabe D and Honjo T 2007 Msx2-interacting nuclear target protein (Mint) deficiency reveals negative regulation of early thymocyte differentiation by Notch/RBP-J signaling. Proc. Natl. Acad. Sci. USA 104 1610–1615
Tu MM, Lee FYF, Jones RT, Kimball AK, Saravia E, Graziano RF, Coleman B, Menard K, et al. 2019 Targeting DDR2 enhances tumor response to anti-PD-1 immunotherapy. Sci. Adv. 5 2437
Thompson CB, Lindsten T, Ledbetter JA, Kunkel SL, Young HA, Emerson SG, Leiden JM and June CH 1989 CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc. Natl. Acad. Sci. USA 86 1333–1337
Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM and Tsuchiya H 2001 B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193 839–846
Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G and Ferrari C 2006 PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol. 80 11398–11403
UniProt Consortium 2019 UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47 D506–D515
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, et al. 2015 Tissue-based map of the human proteome. Science 347 1260419
Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R and Amara RR 2009 Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458 206–210
Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, et al. 2015 Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350 1079–1084
Verma V, Sprave T, Haque W, Simone CB, Chang JY, Welsh JW and Thomas CR 2018 A systematic review of the cost and cost-effectiveness studies of immune checkpoint inhibitors. J. Immunother. Cancer 6 128
Viglietta V, Bourcier K, Buckle GJ, Healy B, Weiner HL, Hafler DA, Egorova S, Guttmann CR, Rusche JR and Khoury SJ 2008 CTLA4Ig treatment in patients with multiple sclerosis: an open-label, phase 1 clinical trial. Neurology 71 917–924
Viisanen T, Ihantola EL, Näntö-Salonen K, Hyöty H, Nurminen N, Selvenius J,Juutilainen A, Moilanen L, Pihlajamäki J, Veijola R, Toppari J, Knip M, Ilonen J and Kinnunen T 2017 Circulating CXCR5+PD-1+ICOS+ Follicular T helper cells are increased close to the diagnosis of type 1 diabetes in children with multiple autoantibodies. Diabetes 66 437–447
Vijayakrishnan L, Slavik JM, Illés Z, Greenwald RJ, Rainbow D, Greve B, Peterson LB, Hafler DA, Freeman GJ, Sharpe AH, Wicker LS and Kuchroo VK 2004 An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity 20 563–575
Von Euw E, Chodon T, Attar N, Jalil J, Koya RC, Comin-Anduix B and Ribas A 2009 CTLA4 blockade increases Th17 cells in patients with metastatic melanoma. J. Transl. Med. 7 35
Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, Garcia C, Wu Y, Kuhne M, Srinivasan M, Singh S, Wong S, Garner N, Leblanc H, Bunch RT, Blanset D, Selby MJ and Korman AJ 2014 In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol. Res. 2 846–856
Wang CJ, Heuts F, Ovcinnikovs V, Wardzinski L, Bowers C, Schmidt EM, Kogimtzis A, Kenefeck R, Sansom DM and Walkera LSK 2015 CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc. Natl. Acad. Sci. USA 112 524–529
Wang S, Bajorath J, Flies DB, Dong H, Honjo T and Chen L 2003 Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J. Exp. Med. 197 1083–1091
Wang X, Teng F, Kong L and Yu J 2016 PD-L1 expression in human cancers and its association with clinical outcomes. Oncotargets Ther. 9 5023–5039
Wang X, Huang W, Mihara M, Sinha J and Davidson A 2002 Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J. Immunol. 168 2046–2053
Wagner M, Sobczyński M, Karabon L, Bilińska M, Pokryszko-Dragan A, Pawlak-Adamska E, Cyrul M, Kuśnierczyk P and Jasek M 2015 Polymorphisms in CD28, CTLA-4, CD80 and CD86 genes may influence the risk of multiple sclerosis and its age of onset. J. Neuroimmunol. 288 79–86
Walker LS and Sansom DM 2011 The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 11 852–863
Walsh SR, Bastin D, Chen L, Nguyen A, Storbeck CJ, Lefebvre C, Stojdl D, Bramson JL, Bell JC and Wan Y 2019 Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J. Clin. Invest. 129 518–530
Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB and Bluestone JA 1994 CTLA-4 can function as a negative regulator of T cell activation. Immunity 1 405–413
Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H and Mak TK 1995 Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270 985–988
Weaver CT, Elson CO, Fouser LA, Kolls JK 2013 The Th17 pathway and inflammatory diseases of the intestines, lungs and skin. Annu. Rev Pathol. 8 477–512. https://doi.org/10.1146/annurev-pathol-011110-130318. Epub 2012 Nov 15. Review.
Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, WangJ, Wargo JA, Pe’er D and Allison JP 2017 Distinct cellular mechanisms underlie anti-CTLA4 and anti-PD1 checkpoint blockade. Cell 170 1120–1133
Wei SC, Sharma R, Anang NAS, Levine JH, Zhao Y, Mancuso JJ, Setty M, Sharma P, Wang J, Pe’er D and Allison JP 2019a Negative co-stimulation constrains T cell differentiation by imposing boundaries on possible cell states. Immunity 50 1084–1098
Wei SC, Anang N-A A S, Sharma R, Andrews MC, Reuben A, Levine JH, Cogdill AP, Mancuso JJ, Wargo JA, Pe’er D and Allison JP 2019b Combination anti-CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies Proc. Natl. Acad. Sci. USA 116 22699–22709
Weisman MH, Durez P, Hallegua D, Aranda R, Becker JC, Nuamah I, Vratsanos G, Zhou Y and Moreland LW 2006 Reduction of inflammatory biomarker response by abatacept in treatment of rheumatoid arthritis. J. Rheumatol. 33 2162–2166
Wherry EJ 2011 T cell exhaustion. Nat. Immunol. 12 492–499
Wikenheiser DJ and Stumhofer JS 2016 ICOS co-stimulation: friend or foe? Front. Immunol. 7 304
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T and Sakaguchi S 2008 CTLA-4 control over Foxp3+ regulatory T cell function. Science 322 271–275
Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, O’Day SJ, Hoos A, Humphrey R, Berman DM, Lonberg N and Korman AJ 2013 Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. NY Acad. Sci. 1291 1–13
Wu Y, Ju Q, Jia K, Yu J, Shi H, Wu H and Jiang M 2018 Correlation between sex and efficacy of immune checkpoint inhibitors (PD-1 and CTLA-4 inhibitors). Int. J. Cancer 143 45–51
Wykes MN and Lewin SR 2018 Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 18 91–104
Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H,Pardoll DM, Okumura K, Azuma M and Yagita H 2002 Expression of programmed death 1ligands by murine T cells and APC. J. Immunol. 169 5538–5545
Yan Y and Zhang X 2017 The association between CD28 gene rs3116496 polymorphism andbreast cancer risk in Chinese women. BioSci. Rep. 37 BSR20170884
Yan X, Zhang S, Deng Y, Wang P, Hou Q and Xu H 2018 Prognostic factors for checkpoint inhibitor based immunotherapy: an update with new evidences. Front. Pharmacol. 9 1050
Yashiro Y, Tai XG, Toyo-oka K, Park CS, Abe R, Hamaoka T, Kobayashi M, Neben S and Fujiwara H 1998 A fundamental difference in the capacity to induce proliferation of naive T cells between CD28 and other co-stimulatory molecules. Eur. J. Immunol. 28 926–935
Ying H, Yang L, Qiao G, Li Z, Zhang L, Yin F, Xie D and Zhang J 2010 Cutting edge: CTLA-4–B7 interaction suppresses Th17 cell differentiation. J. Immunol. 185 1375–1378
Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM and Ahmed R 2011 Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity 35 400–412
Zak KM, Grudnik P, Magiera K, Dömling A, Dubin G and Holak TA 2017 Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2. Structure 25 1163–1174
Zander RA, Obeng-Adjei N, Guthmiller JJ, Kulu DI, Li J, Ongoiba A, Traore B, Crompton PD and Butler NS 2015 PD-1 Co-inhibitory and OX40 co-stimulatory crosstalk regulates helper T cell differentiation and anti-plasmodium humoral immunity. Cell Host Microbe 17 628–641
Zhang Y and Allison JP 1997 Interaction of CTLA-4 with AP50, a clathrin-coated pit adaptor protein. Proc. Natl. Acad. Sci. USA 94 9273–8
Zhang X, Schwartz JC, Almo SC and Nathenson SG 2003 Crystal structure of the receptor-binding domain of human B7-2: insights into organization and signaling Proc. Natl. Acad. Sci. USA 100 2586–2591
Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, Cammer M, Chen L, Zhang ZY, Edidin MA, Nathenson SG and Almo SC 2004 Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20 337–347
Zhang Y, Chung Y, Bishop C, Daugherty B, Chute H, Holst P, Kurahara C, Lott F, Sun N, Welcher AA and Dong C 2006 Regulation of T cell activation and tolerance by PDL2. Proc. Natl. Acad. Sci. USA 103 11695–11700
Zheng J, Chan PL, Liu Y, Qin G, Xiang Z, Lam KT, Lewis DB, Lau YL and Tu W 2013 ICOS regulates the generation and function of human CD4+ Treg in a CTLA-4 dependent manner. PLoS One 8 e82203
Zou Y, Zhang Z, Liu Y, Liu D and Xu W 2017 Are programmed cell death 1 gene polymorphisms correlated with susceptibility to rheumatoid arthritis? A meta-analysis. Medicine 96 e7805
Acknowledgements
This review has been written to honor mentors who influenced the early scientific growth of DpN: Ranjan Bhat (University of Bombay), VV Modi (MS University Baroda), Howard Bern, Satyabrata Nandi, Jim Allison (all three from the University of California, Berkeley), Frank Talamantes (University of California, Santacruz) and John J Monaco (University of Cincinnati, Ohio). The encouragement of all members of the DpN lab (1997 – present) is greatly appreciated. However, a special mention needs to be made of students from the DpN laboratory who have contributed to the area of T cell biology: Prasanta Maiti, Sambudhho Mukherjee, Asma Ahmed, Srabanti Rakshit, Mukta Deobagkar-Lele and Shamik Majumdar. We greatly appreciate the comments and suggestions on this manuscript by Udipi Ramagopal, Ramray Bhat, Mohit Jolly and Vrinda Nandi. Finally, we are thankful to the numerous questions asked by curious students during talks on this topic. Indeed, these questions have shaped this manuscript and helped to clear our own doubts. Consequently, we hope that this review will be useful to the community at large. The funding from multiple agencies, i.e. DBT, SERB/DST, CSIR and the infrastructural support from the DBT-IISc partnership program, is greatly appreciated as it has allowed our laboratory to explore our scientific curiosity.
Funding
This study was supported by Science and Engineering Research Board (Grant No. EMR/2015/002486).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Rajiv K Saxena.
Corresponding editor: Rajiv K Saxena
Rights and permissions
About this article

Cite this article
Nandi, D., Pathak, S., Verma, T. et al. T cell costimulation, checkpoint inhibitors and anti-tumor therapy. J Biosci 45, 50 (2020). https://doi.org/10.1007/s12038-020-0020-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-020-0020-2