
Abstract
The response of peripheral T lymphocytes (T cell) is controlled by multiple checkpoints to avoid unwanted activation against self-tissues. Two opposing costimulatory receptors, CD28 and CTLA-4, on T cells bind to the same ligands (CD80 and CD86) on antigen-presenting cells (APCs), and provide positive and negative feedback for T-cell activation, respectively. Early studies suggested that CTLA-4 is induced on activated T cells and binds to CD80/CD86 with much stronger affinity than CD28, providing a competitive inhibition. Subsequent studies by many researchers revealed the more complex mode of T-cell inhibition by CTLA-4. After T-cell activation, CTLA-4 is stored in the intracellular vesicles, and recruited to the immunological synapse formed between T cells and APCs, and inhibits further activation of T cells by blocking signals initiated by T-cell receptors and CD28. CTLA-4-positive cells can also provide cell-extrinsic regulation on other autoreactive T cells, and are considered to provide an essential regulatory mechanism for FoxP3+ regulatory T cells. Genetic deficiency of CTLA-4 leads to CD28-mediated severe autoimmunity in mice and humans, suggesting its function as a fundamental brake that restrains the expansion and activation of self-reactive T cells. In cancer, therapeutic approaches targeting CTLA-4 by humanized blocking antibodies has been demonstrated to be an effective immunotherapy by reversing T-cell tolerance against tumors. This chapter introduces CTLA-4 biology, including its discovery and mechanism of action, and discusses questions related to CTLA-4.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

1 Introduction: CD28 and CTLA-4 as Checkpoints
The activation and function of the peripheral T lymphocyte (T cell) reaction depends primarily on recognition of the antigen presented on the major histocompatibility complex (MHC) by the T-cell antigen receptor (TCR). This recognition, at the time of priming (the priming phase), causes the clonal expansion of T cells and the differentiation into effector T cells in secondary lymphoid organs. Recognition at the site of inflammation (the effector phase) results in an attack against infected tissue. The activation of self-reactive T cells can lead to an attack on self-tissue, leading to autoimmunity. To prevent this, there are many systems that have developed to keep self-reactive T cells in check.
The first signal, provided by TCR during recognition, does not cause activation of the T cells on its own. Full T-cell activation requires a second set of signals, called “costimulation,” which is mainly provided by activated antigen-presenting cells (APCs). The best-characterized costimulatory system is the CD28 receptor on T cells, triggered by its ligands, CD80 or CD86 (previously called B7-1 and B7-2, respectively) on activated professional APCs (dendritic cells, macrophages, and B cells). CD80 and CD86 are upregulated on activated APCs by microbial “danger signals” so APCs presenting microbial antigens can efficiently stimulate T-cell activation (Banchereau and Steinman 1998; Akira et al. 2001). Successful engagement of CD28 leads to IL-2 production (June et al. 1987), induction of anti-apoptotic protein Bcl-xL (Boise et al. 1995), and stimulates glucose uptake by inducing glucose transporter and glycolysis (Frauwirth et al. 2002), as well as the cell-cycle progression necessary for the massive clonal expansion of antigen-specific T cells. In contrast, resting APCs do not express a high level of CD80/CD86. T cells that are stimulated in the absence of the CD28 signal fall into an unresponsive state called “clonal anergy” and become refractory to further stimulation by the same antigen (Schwartz 2003). CTLA-4, another receptor that is structurally similar to CD28, is induced on activated T cells and binds to CD80 and CD86 with greater avidity than CD28. All CTLA-4 KO mice showed massive lymphocyte proliferation in the lymph nodes and spleen, followed by an autoimmune attack against virtually all tissues by leucocytes, and premature death (Tivol et al. 1995; Waterhouse et al. 1995; Chambers et al. 1997b). The lethal phenotype is prevented by treating young CTLA-4 KO mice with antibody-depleting CD4+, but not CD8+ T cells (Chambers et al. 1997b). The conditional deletion of CTLA-4 only on CD4+ T-cells phenocopied germline CTLA-4 KO (Klocke et al. 2016), suggesting that the disease is CD4+ “helper T cell”-dependent. A recombinant soluble CTLA-4 (CTLA-4 Ig) blocking CD28 engagement by CD80/86 (Chambers et al. 1997b), CD28/CTLA-4 double knockout mice (Mandelbrot et al. 2001), or breeding CTLA-4 KO into CD80/CD86 double knockout mice (Triple KO; Mandelbrot et al. 1999) completely prevented lymphoproliferation and autoimmunity. Also, CTLA-4 KO remains healthy in mice genetically lacking autoreactive T cells (Waterhouse et al. 1997; Chambers et al. 1999; Bachmann et al. 2001; Greenwald et al. 2001; Gajewski et al. 2001). Conversely, by limiting the complexity of the T-cell repertoire by overexpressing TCRβ chain as a transgene, it was shown that CTLA-4 knockout disease is totally self-antigen-dependent (Ise et al. 2009). These data clearly suggest that CTLA-4 provides negative feedback on the CD28-mediated activation of polyclonal, self-reactive CD4 helper T cells.
Thus, T-cell activation against self is fundamentally prevented by two “checkpoints”: 1. T cells in the absence of the CD28 signal (in the absence of CD80 and CD86 on APCs) become unresponsive. 2. Upon activation, CTLA-4 is induced, interacts with CD80/CD86, and prevents further activation of self-reactive T cells. After this discovery, several pairs of ligands and receptors were shown to have unique and functions in the immune system. It is now accepted that PD-1 and CTLA-4 blocks the T cell-mediated immune system at different levels, at the cell type and location (Chikuma 2016). CTLA-4 was also shown to be important for regulatory function. The dramatic phenotypes of CTLA-4 null mice attracted many researchers in immunology. It is now of particular importance that the artificial blockade of CTLA-4 was shown to break tolerance in the tumor setting, providing efficient immunotherapy against tumors (Leach et al. 1996). It turned out that the basis of T-cell inhibition by CTLA-4 was far more complex than previously expected. In this chapter, the history of discovery and basic research on CTLA-4 in terms of its biological roles and the mechanism of T-cell inactivation will be introduced, and the currently unsolved questions will be discussed.
2 Structure of CTLA-4
2.1 Discovery and Primary Structure of CTLA-4
CTLA-4 was originally isolated from a cDNA library derived from mouse cytotoxic T cells, hence the name cytotoxic T-cell-associated antigen-4 (Brunet et al. 1987). The CTLA-4 gene is encoded on human Ch2q33 and mouse Ch1C1. CTLA-4 consists of 4 exons and 3 introns, and encodes a protein of 223 amino acids (Ling et al. 1999). The protein is a type I transmembrane protein belonging to the immunoglobulin (Ig) superfamily, and it bears a single Ig-V (variable)-like domain on the extracellular portion. Like other receptors in the CD28 family, CTLA-4 is highly glycosylated at its extracellular domain. CTLA-4 forms a homodimer on activated T cells, which is mediated by the disulfide bond at the cysteine residue at amino acid position 120 (Linsley et al. 1995). There is a well-conserved motif (amino acid sequence: MYPPPY) on the extracellular domain that is shared by CD28, which is critical for binding its ligands CD80 and CD86 (Peach et al. 1994). CD80 and CD86 bind to CTLA-4 with stronger affinity than to CD28, which may account for CTLA-4 antagonizing to the activation signal. Dimerized CTLA-4 binds to two ligands (Linsley et al. 1995). Ostrov et al. (2000) demonstrated by solving the crystal structure of the extracellular portion that CTLA-4 shows a unique mode of dimerization with its ligand-binding domain distal to the dimerization interface. Stamper et al. (2001) and Schwartz et al. (2001) solved the crystal structures of CTLA-4 in complex with CD80 and CD86, respectively. Both groups concluded that each CTLA-4 dimer binds to two CD80 molecules, forming a lattice-like structure and providing very high avidity binding. The results suggest that CTLA-4 induction on the surface of activated T cells results in ligand sequestration of CD28. CTLA-4 also bears a short cytoplasmic tail that is almost 100% conserved among many species. The tail is reported to bind to various signaling molecules that mediate subcellular trafficking and the function of CTLA-4 (Fig. 1a, discussed later).

A. schematic structure of CTLA-4. Note that the YVKM motif binds to the clathrin adaptor complex in its unphosphorylated form whereas the same motif binds to enzymes for signal transduction when phosphorylated, B. Summary of structure-functional studies. N/D: not done
2.2 Alternatively Spliced Isoforms of CTLA-4
The splicing of messenger RNA of CTLA-4 might result in the expression of functionally important isoforms in the genetics of autoimmunity. For example, human resting blood lymphocytes express an alternatively spliced isoform of CTLA-4 lacking Exon3, encoding the entire transmembrane domain, resulting in a soluble isoform consisting solely of the extracellular domain (soluble CTLA-4: sCTLA-4; (Magistrelli et al. 1999; Oaks et al. 2000). Since the ectodomain of CTLA-4 can bind to its ligands (CD80 and CD86) with blocking activity, sCTLA-4 might be acting as a naturally occurring antagonist for CD28-mediated T-cell activation by CD80 and CD86. Indeed, Ueda et al. (2003) reported a CTLA-4 allelic variant associated with a lower mRNA level of sCTLA-4 linked with susceptibility to human autoimmune diseases. It was also reported that a naturally occurring CTLA-4 variant that is completely lacking its extracellular domain may determine autoimmune susceptibility in a particular mice strain. The “ligand-independent form” (referred to as liCTLA-4) can preferentially accumulate to the immunological synapse, and thereby provide a tonic inhibitory signal (Bour-Jordan et al. 2011 and Chikuma unpublished). Non-obese diabetes (NOD) mice that exhibit autoimmune juvenile diabetes express a reduced amount of this isoform (Ueda et al. 2003), which accounts for their genetic susceptibility (Araki et al. 2009). A transgenic overexpression of liCTLA-4 or a mutant CTLA-4 that lacks the ligand-binding domain on T cells significantly retards the death of CTLA-4 KO mice, suggesting the function of a ligand-independent CTLA-4 signal (Vijayakrishnan et al. 2004; Chikuma et al. 2005; Araki et al. 2009). Interestingly, the activation status of T cells results in dynamic change in the ratio of transcripts encoding full-length, soluble, and ligand-independent forms of CTLA-4 transcripts, suggesting the unique role of each spliced isoform in different T-cell subpopulations (Oaks et al. 2000; Vijayakrishnan et al. 2004). A super-short form of CTLA-4 that encodes only the signal peptide (Exon1) and part of the cytoplasmic tail (Exon4; out of its original translational frame) was reported to affect autoimmune mice when overexpressed (Liu et al. 2012; Ichinose et al. 2013). The expression of this super-short form may alter the mRNA of other isoforms, although the exact function of this isoform is not yet fully elucidated.
3 Function of CTLA-4
3.1 Cell-Intrinsic T-cell Inhibition by CTLA-4
It was first reported that the monoclonal antibodies (mAb) that block the binding of CTLA-4 to CD80/CD86 augment T-cell responses when added to the co-culture of T cells, APCs, and anti-CD3 antibody (which work as a TCR ligand) in a soluble form (Walunas et al. 1994). The F(ab)2 form of the same mAb, which lacks the ability to bind Fc receptor had similar activity, suggesting that genuine blocking activity of CTLA-4-CD80/CD86 binding in this culture augmented the T-cell response. In contrast to the blocking experiment, anti-CTLA-4 immobilized on the surface of plastics works to crosslink/stimulate CTLA-4 and inhibits T-cell activation, as well as IL-2 production and proliferation (Krummel and Allison 1995; Walunas et al. 1994). This inhibition was associated with inhibition of the cell-cycle progression and expression of activation markers CD69 and CD25 (Krummel and Allison 1996). A membrane-bound single-chain variable fragment (scFv) of anti-CTLA-4 antibody engineered to be expressed on a fibroblast cell line can ligate CTLA-4 in vitro and inhibits activation of co-cultured T cells, triggered by anti-CD3 and anti-CD28 on the same cell (Griffin et al. 2000). A transgenic expression of CTLA-4 scFv on B cells ameliorated autoimmune diabetes in NOD mice (Fife et al. 2006) in vivo. Importantly, this amelioration was observed in NOD mice lacking CD80/CD86, suggesting inhibition of the activation of islet-reactive diabetogenic T cells by B cells expressing the artificial CTLA-4 ligand directly, but not through inhibition of CD28 ligation. The strain also shows almost no FoxP3+ Tregs due to the absence of the CD28 signal necessary for Treg development/survival (Salomon et al. 2000; Tang et al. 2003; Tai et al. 2005). Thus, the CTLA-4 ligand-mediated inhibition of autoimmune diabetes in Tg mice is, at least in part independent of Tregs (Fife et al. 2006). An ectopic expression of CTLA-4-deficient primary mouse T cells or cell lines with CTLA-4 or its mutants reconstitutes ligand-mediated T-cell inactivation by CTLA-4 in many systems (Nakaseko et al. 1999; Chikuma et al. 2005; Cinek et al. 2000; Baroja et al. 2000; Vijayakrishnan et al. 2004). On the basis of these experiments, CTLA-4 was suggested to be a negative regulator for T-cell activation. Overexpression of CTLA-4 or its mutants to cell lines lacking endogenous CTLA-4 can be seen to mimic CTLA-4-mediated T-cell inhibition in vitro in many reports (Fig. 1b). Two groups reported that Jurkat cells (human-derived) expressing stable mouse CTLA-4 can be inhibited (in terms of IL-2 secretion) by immobilized anti-mouse CTLA-4 antibody. Subsequent mutational analysis showed that mutation of one or both tyrosine motifs to phenylalanine did not affect CTLA-4’s ability to inhibit IL-2 production in the same system. Similar results were observed in a study, where CTLA-4-negative T cell clone was reconstituted with CTLA-4 by retroviral vectors (Nakaseko et al. 1999). Restimulated cells with wild-type CTLA-4 inhibited the activation, and the tyrosine mutants did not show any defects in the inhibition. In Nakaseko’s study, CTLA-4 lacking almost all cytoplasmic regions but the membrane proximal to seven amino acids (KMLKKRS) showed suppressive activity. Although ligation of CTLA-4 alone does not cause changes in gene expression, it inhibits abundant gene expression by TCR/CD28 stimulation (Riley et al. 2002). This suppression is associated with inhibition of TCR-CD28-mediated biochemical events. It was reported that CTLA-4 ligation inhibits the phosphorylation of AKT (Parry et al. 2005), TCRζ (Lee et al. 1998), Erk (Calvo et al. 1997; Baroja et al. 2000; Chikuma et al. 2005), Jnk (Calvo et al. 1997), and Iκb (Pioli et al. 1999), all of which are triggered by TCR-CD28. Interestingly, some reports suggested that CTLA-4 can inhibit T-cell activation even without ligand engagement. In mice, the transgenic expression of CTLA-4 mutants that lack either the entire extracellular domain (Vijayakrishnan et al. 2004; Araki et al. 2009) or an essential ligand-binding motif (MYPPPY; Chikuma et al. 2005) inhibited T-cell proliferation and rescued the CTLA-4 null phenotype significantly, but not fully. The study clearly indicated the T-cell-intrinsic inhibition by CTLA-4. CTLA-4 also mediates the induction of T-cell anergy, a form of tolerance. Greenwald et al. (Greenwald et al. 2001) used a genuine genetic model in which CTLA-4 was lacking on T cells bearing single TCR with known specificity to show this. The CTLA-4 KO rag2 KO TCR Tg (classII MHC-restricted; CD4+) showed resistance to anergy induction in the transferred host when challenged by the cognate antigen, whereas CTLA-4 WT rag2 KO TCR Tg were tolerized. In contrast, CTLA-4 KO CD8+ T cells in the analogous system showed no defects in anergy induction. In a similar system, CTLA-4 on CD8+ T cells did not regulate their anergy induction (Frauwirth et al. 2000, 2001). In contrast, PD-1 deficiency in the same system had effects on CD8+ T cells which led to the augmentation of CD8+ T-cell activation and resistance to anergy (Chikuma et al. 2009), These results suggest that CTLA-4 and PD-1 sets the threshold for CD4+ and CD8+ T cells, respectively.
3.2 Cell-Extrinsic T-Cell Inhibition by CTLA-4
In contrast to the direct inhibition of T cells, CTLA-4 (or T-cell-expressing CTLA-4) can indirectly inhibit the activation of other T cells. This non-cell-autonomous tolerance mechanism was observed in lethally irradiated wild-type mice receiving mixed bone marrow from CTLA-4 wild-type and KO bone marrow when they did not develop the lymphoproliferative disease observed in CTLA-4 KO mice or recipients of CTLA-4 KO cells only (Bachmann et al. 1999). Similarly, Tivol and Gorski (2002) showed that the transfer of a mixture of Thy1+ splenocytes WT and CTLA-4 KO into Rag KO mice prevented inflammatory disease caused by the transfer of CTLA-4 KO cells only. In this setting, KO cells were eliminated in vivo, in the presence of WT T cells, suggesting that CTLA-4 on WT T cells can induce the deletion of KO T cells when the population coexist in vivo.
This non-cell-autonomous manner of CTLA-4-mediated inhibition is thought to be a major mechanism of tolerance by FoxP3+ Tregs. FoxP3+ Tregs are found to express high levels of CTLA-4 on the cell surface and intracellularly (Read et al. 2000; Takahashi et al. 2000; Salomon et al. 2000). FoxP3+ Tregs show a unique demethylation pattern on the CTLA-4 locus (Ohkura et al.), and FoxP3 can bind to CTLA-4 promoter (Wu et al. 2006) and drives its expression (Hori et al. 2003). FoxP3-deficient (germline KO or natural mutation; the so-called “scurfy” mutation) and CTLA-4 KO mice show a very similar phenotype with premature death by autoimmunity (Brunkow et al. 2001; Khattri et al. 2003; Fontenot et al. 2003). It has been, however difficult to determine if CTLA-4 expression on FoxP3+ Treg is critical for the regulatory function. For instance, FoxP3+ Tregs from CTLA-4 KO mice show suppression as efficiently as FoxP3+ Tregs from WT mice in vitro (Tang et al. 2004; Kataoka et al. 2005). To explore this question in vivo, Chikuma and Bluestone (2007) reconstituted RAG-1 KO recipients with bone marrow (BM) from CTLA-4KO (bred to a CD80/80 null background to avoid autoimmunity) mice and/or Foxp3-deficient mice. Control recipients, receiving either CTLA-4KO or Scurfy BM alone, rapidly developed interstitial pneumonitis, colitis, wasting, and dermatitis and died, which suggests that fatal autoimmunity occurred in the donor strain (Chikuma and Bluestone 2007). Next, the experiment was repeated using a 50:50 mixture of CTLA-4KO and Scurfy BM cells were tested to determine whether the addition of the CTLA-4+ FoxP3neg could overcome the pathology of and CTLA-4 or FoxP3-deficient cells in lymphocyte-null Rag1-KO recipients. Intriguingly, mixed BM chimeric recipients developed less severe autoimmunity and lived longer compared to CTLA-4KO or Scurfy BM alone, suggesting that the mixture of CTLA-4KO and Scurfy BM afforded some level of protection. The data suggest that CTLA-4 has a Treg-independent extrinsic function in the control of T-cell tolerance and homeostasis. Strikingly, the chimeric mice died with later kinetics, suggesting that CTLA-4 and Foxp3 must be expressed in cis on the same cell to fully prevent lethal autoimmunity and lymphoproliferation (Chikuma and Bluestone 2007). Wing et al. (2008) generated a mouse carrying a floxed CTLA-4 allele that allowed conditional knockout mice lacking CTLA-4 specifically on FoxP3+ Tregs when bred to mice expressing CRE under FoxP3 promotor. The resulting mice developed lethal autoimmunity, suggesting that the Treg-specific CTLA-4 expression is important for absolute maintenance of T-cell homeostasis. Again, mice showed much milder autoimmunity that affected limited organs and resulted in later death in comparison to the germline CTLA-4 KO (Wing et al. 2008). Conversely, Jain et al. (2010) showed that mice with CTLA-4 deleted only on activated conventional T cells (CTLA-4 on FoxP3+ Tregs is intact) show significant, but not complete, protection from the lethal disease phenotype. Collectively, these results (Chikuma and Bluestone 2007; Wing et al. 2008; Jain et al. 2010) suggest that CTLA-4 on both activated conventional T cells and FoxP3+ Tregs is important for complete protection from death. As for the mechanism of cell-extrinsic regulation by CTLA-4, the wild-type T cells protecting autoimmunity appeared not to require inhibitory cytokines, such as TGFβ or IL-10 (Friedline et al. 2009). Instead, the CTLA-4 regulation of APC activation and function was proposed. Using two-photon microscopy, Tang et al. (2006) showed that FoxP3+ Tregs do not directly interact with conventional autoreactive cells in an inflammatory setting. Instead, Tregs interact with APC to down-modulate their ability to activate autoreactive conventional T cells. Using an in vitro co-culture system of conventional T cells, Tregs, and APCs, Wing et al. (2008) showed that the Tregs outcompeted with conventional T cells for binding to APCs. APCs cultured with FoxP3+ Tregs are attenuated in the expression of activation markers, CD80 and CD86, and lost the ability to stimulate conventional T cells (Wing et al. 2008). Qureshi et al. (2011) reported that the downregulation of CD80/86 by CTLA-4-expressing cells is through the CTLA-4-mediated capture of CD80/86 on APC and trans-endocytosis into CTLA-4+ T cells. The trans-endocytosed CD80/86 is degraded within CTLA-4+ cells, resulting in a reduction of molecules on the APC (Qureshi et al. 2011). This trans-endocytosis model is attractive to explain cell-extrinsic regulation by CTLA-4, especially because the model works in a FoxP3 null setting in an antigen-specific system (Wang et al. 2012). On the other hand, Tai et al. (2012) showed that the internalization-defective CTLA-4 transgene on CTLA-4 KO Tregs was functional in vivo, suggesting that trans-endocytosis may not be important. CTLA-4 binding to CD80/86 is also suggested in the transmission of “reverse signaling” that induces the production of an enzyme indolamine 2,3-dioxygenase (IDO) on APCs. IDO catalyzes the degradation of the amino acid tryptophan into the immune-inhibitory metabolite kynurenine, which is important in immuno-suppression in various immunologic settings, such as pregnancy, cancer, chronic infection, autoimmunity, and allergy (reviewed in Fallarino et al. 2003; Puccetti and Grohmann 2007). These hypotheses largely resulted from in vitro experiments using cells overexpressing CTLA-4 and/or CD80/CD86 so proof in the physiological system is less clear. CTLA-4-mediated cell-extrinsic inhibition is crucial in the maintenance of self-tolerance.
4 Mechanisms of CTLA-4-Mediated Immune Regulation
4.1 The Biochemical Partners of CTLA-4
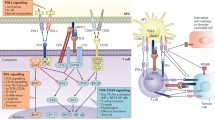
The 33 amino-acid intracellular domain of CTLA-4 is 100% conserved among species, suggesting its importance in protection against lethal autoimmunity (Fig. 1a). The domain is composed of a lysine-rich membrane-proximal motif (KMLKKRS) followed by two motifs containing tyrosine (YVKM and YFIP), with a proline-rich motif (PPTEP). The tail lacks a signaling module with enzymatic activity. Instead, the CTLA-4 tail interacts with many intracellular molecules, which control trafficking and signaling by CTLA-4.
4.1.1 SHP-2
Src-homology-containing tyrosine phosphatase-2 (SHP-2, also called Sh-PTP2 or Syp) is a cytosolic tyrosine phosphatase with a Src-homology 2 (SH2) domain. SHP-2 is involved in activation of the RAS-MAPK pathway, and transmits signals from cytokine receptor and co-inhibitory receptors (Lorenz 2009). SHP-2 interacts with CTLA-4 based on mutagenesis and immune coprecipitation studies. This association was suggested to be mediated by the SH2 of SHP-2 and the phosphotyrosine sequence YVKM within the CTLA-4 cytoplasmic tail when the motif is phosphorylated. This motif was not, however, a typical ITIM motif. The association of SHP-2 to CTLA-4 might be indirect or it may require other molecule(s) as adapter molecules (Schneider and Rudd 2000). The CTLA-4-SHP-2 interaction was proposed to cause tyrosine dephosphorylation of key signaling component required for T-\cell activation (Marengere et al. 1996). For instance, SHP-2 phosphatase activity can dephosphorylate the TCRζ chain(Lee et al. 1998) and the RAS regulator p52SHC(Marengere et al. 1996), and it has been suggested that the delivery of this enzyme by CTLA-4 to the specific region of the immunological synapse inhibits the TCR-CD3 complex-mediated signal transduction, leading to direct inhibition of T-cell activation (Lee et al. 1998). Similarly, Gab2, which binds directly to TCRζ signaling complex, recruits SHP-2 there and blocks proximal TCR signaling (Yamasaki et al. 2001).
4.1.2 PP2A
PP2A is a heterotrimeric serine-threonine phosphatase that dephosphorylates a wide variety of protein substrates involved in cellular activity. PP2A composes a very major fraction of cellular protein, and dephosphorylates many substrates (Lorenz 2009). In a yeast two-hybrid screen, Chuang et al. (2000) found that the catalytic subunit of PP2A associates with the cytoplasmic tail of CD28 and CTLA-4. Independently, Baroja et al. (2002) reported that the regulatory subunit (PP2AA) also interacted with CTLA-4. The catalytic and regulatory subunit of PP2A interacted with the lysine-rich KMLKKRS and YVKM motif within CTLA-4, respectively, suggesting tri-molecular interaction. Through mutagenesis of the KMLKKRS motif, Baroja et al. suggested that the PP2A interaction inhibits CTLA-4 suppressive activity. Parry et al. (Parry et al. 2005) showed that PP2A antagonizes phosphorylation-dependent AKT activation mediated by TCR and CD28, which was sensitive to ocadaic acid, an inhibitor of PP2A.
4.1.3 PKC-η(Eta)
Kong et al. (2014) reported that CTLA-4 associates with the η isoform of PKC. FoxP3+ Tregs contain a significant amount of phosphorylated PKC-η, which interacts with CTLA-4 at the immunological synapse. The mutation or truncation of the KMLKKRS motif greatly reduced PKC-η binding, suggesting the importance of this motif together with PP2A in CTLA-4 function. Germline PKC-η deficient mice demonstrated defects in conventional T-cell activations (Fu et al. 2011), but did not show any defects in the development of FoxP3+ Treg (Kong et al. 2014). They eventually exhibited lymphadenopathy, however, and their FoxP3+ Tregs were shown to have multiple defects in regulatory functions in vitro and in vivo. It appears that CTLA-4-PKC-η interaction is required for firm interaction of Tregs to APCs, by phosphorylation of PAK2, GIT2, two components of the focal adhesion complex, as well as a guanine nucleotide exchange factor, αPIX, at the immunological synapse. Mice with the Treg-specific deletion of PKC-η may result in Treg-specific defects in adhesion due to defective phosphorylation of the GIT2-aPIX-PAK complex.
4.1.4 Clathrin Adaptor Complex
Several groups reported that CTLA-4 interacts with AP1, and that AP2 subunits of the clathrin adaptor complex are primary components in the cell biology that induce the clathrin-mediated internalization of associated molecules (Chuang et al. 1997; Shiratori et al. 1997; Zhang and Allison 1997; Bradshaw et al. 1997; Schneider et al. 1999). AP1 and AP2 interact with the unphosphorylated form of Y201VKM, which may account for the preferential intracellular trafficking of CTLA-4.
4.1.5 Tyrosine Kinases
CTLA-4 phosphorylation and relocation to the immunological synapse are TCR-dependent. TCR-proximal kinase Lck and Fyn were shown to phosphorylate the Y201VKM motif (Chuang et al. 1999; Miyatake et al. 1998). Other tyrosine kinases, such as JAK2 (Chikuma et al. 2000) and Rlk (Schneider et al. 1998), were reported to directly bind to CTLA-4, and can phosphorylate Y201, suggesting that the broad extracellular signals that activate tyrosine kinases can stimulate CTLA-4 phosphorylation. Importantly, Lck and Fyn are membrane-associated src kinases, and their activity is TCR-dependent. The preferential recruitment of CTLA-4 to the IS may occur as a consequence of the trimolecular complex of LCK, CTLA-4, and TCR. The tyrosine motif is dispensable for CTLA-4-mediated inhibition in vitro. CTLA-4 KO mice overexpressing point-mutated CTLA-4 on tyrosine (YVKM → VKMs) develop late lymphoproliferative disease, suggesting the importance of this residue in CTLA-4 activation and function (Yi et al. 2004).
4.2 Induction and Dynamic Localization of CTLA-4 to the Immunological Synapse
CTLA-4 expression is restricted to T cells, but it shows a unique expression pattern, which is important for the inhibitory function. (1) The expression of CTLA-4 is activation-dependent. (2) CTLA-4 preferentially localizes to the intracellular vesicle compartment, (3) recycles to the T cell surface upon engagement of TCR, where it participates at the contact site of T cells and APC, called the “immunological synapse,” (4) binds to CD80 and CD86, and is internalized from the immunological synapse to intracellular compartments. This unique pattern of CTLA-4 expression is critical for its specialized function as a negative regulatory molecule. Here, we focus on such dynamics of CTLA-4 mediated inhibition.
4.2.1 Induction of CTLA-4 on T Cells
In contrast to CD28’s constitutive expression on the surface of all T cells, CTLA-4 expression is only found on activated T cells (Linsley et al. 1992). CTLA-4 on T cells is strongly induced by ConA and IL-2 (Brunet et al. 1987). TCR plus CD28 signals synergistically induce CTLA-4 expression by two mechanisms: enhanced transcription and an increase in the mRNA stability (Finn et al. 1997). A nucleotide sequence located within 335 bp upstream from the transcriptional start site of CTLA4 is sufficient for the induction (Perkins et al. 1996). FoxP3+ Tregs are poised to show demethylation on specific loci of CTLA-4, and an activated phenotype due to self-interactions during thymic development (Ohkura et al. 2012). CTLA-4 expression is sensitive to inhibition by cyclosporine, a calcineurin inhibitor. In addition, rapamycin, a small mTOR inhibitor that blocks the IL-2-mediated signaling cascade can control CTLA-4 expression. These results indicate that CTLA-4 upregulation is controlled by general T-cell activation signal. The effects of IL-2 and CD28 signaling were additive but independent, as the CD28 signal augmented CTLA4 expression in IL-2-deficient mice. In contrast, CTLA4 expression was not augmented by cytokines IL-4, IL-6, IL-7, or IL-12 (Alegre et al. 1996). Sodium butylate, an HDAC inhibitor, significantly augmented CTLA-4 expression, suggesting epigenetic silencing (Doyle et al. 2001). CD4+ CD25+ FoxP3+ Tregs showed the most abundant CTLA-4 expression in the steady-state condition, suggesting that these cells are constantly undergoing antigen recognition and activation (Takahashi et al. 2000; Read et al. 2000; Salomon et al. 2000). This concept is supported by the finding that Nur77, a proximal molecule expressed on recently activated cells, is expressed at its highest level(Zikherman et al. 2012).
4.2.2 CTLA-4 Storage
CTLA-4 is not primarily localized on the T-cell surface, but instead resides intracellularly in a region that overlaps the Golgi apparatus (Leung et al. 1995) and/or endocytic compartment(s) with perforin-containing secretory granules (Linsley et al. 1996; Iida et al. 2000). The transfer of 11 cytoplasmic residues, TTGVYVKMPPT, from the CTLA-4 cytoplasmic tail to CD28 conferred intracellular localization (Leung et al. 1995). Importantly, CTLA-4 expressed on the surface was also internalized, which explains its low levels of expression on the cell surface (Alegre et al. 1996). Consequently, on naïve, uninfected mice, CTLA-4 is hardly detected on the T-cell surface by highly sensitive multi-step fluorescent labeling and flow-cytometry detection, even in CD4+ CD25+ FoxP3+ Tregs, which have the most abundant CTLA-4 expression (Chikuma unpublished). CTLA-4 is likely transcribed and stored in the intracellular vesicle in the case of high-affinity antigen recognition.
4.2.3 CTLA-4 Localization and Inhibition at the Immunological Synapse
The dynamic movement of CTLA-4 to the contact site of APC and T cells immediately after transient calcium influx by TCR stimulation was first reported by Linsley et al. (1996). The immunological synapse has a structure composed of a central super-molecular cluster (c-SMAC) that includes TCR and signaling molecules, surrounded by peripheral SMAC containing adhesion molecules. Egen and Allison (2002) followed CTLA-4 recruitment to the immunological synapse by time-lapse microscopy. CTLA-4 is located at the uropod (the opposite site of T-cell movement), but rapidly relocated to the T-cell-APC contact site a few minutes after stimulation. Iida et al. (Iida et al. 2000) suggested that CTLA-4-containing secretory granules, also comprising perforin, rapidly relocates to the synapse upon TCR ligation. Chikuma et al. (2003) showed that CTLA-4, phosphorylated Lck, and TCR-ζ-chain form a trimolecular complex within the glycosphingolipid-enriched microdomain (also called the lipid raft) that is known to be enriched at the immunological synapse. The export of CTLA-4 to IS was dependent on TCR affinity, suggesting that CTLA-4 can preferentially inhibit CD4+ T cells that have higher affinity to antigens(Egen and Allison 2002). The CTLA-4 enrichment to the IS was dependent on CD80/CD86-binding (Pentcheva-Hoang et al. 2004), suggesting that the ligation of CTLA-4 by CD80/86 occurs before the formation of a mature immunological synapse. By observing the T-cell-APC contact surface using an artificial lipid bilayer as APC, Yokosuka et al. (2010) showed that CTLA-4 participated in the immunological synapse at a relatively later time point in mature synapse formation and sequestered CD28 out of the immunological synapse by competing for CD80/CD86 signaling. By collecting the lipid raft fraction by biochemical methods, Chikuma et al. (2003) showed that the TCR molecules within the raft decreased after the co-ligation of TCR and CTLA-4, suggesting that CTLA-4 signal sequesters TCR from the lipid raft, enriched with Lck or other signaling molecules. The recruitment of phosphatases SHP-2 (Lee et al. 1998) and PP2A (Parry et al. 2005) to CTLA-4 causes dephosphorylation or the inhibition of phosphorylation of the TCR-ζ chain (Lee et al. 1998) and/or AKT (Parry et al. 2005), respectively; however, these phosphatases are not found to form a cluster with CTLA-4 to the immunological synapse (Yokosuka et al.). The association of phosphatases is likely very transient or happens at an earlier time point, when the CTLA-4 cluster is very small.
4.2.4 Internalization and Trans-Endocytosis
CTLA-4 binds to CD80/CD86 and brings them into the intracellular compartments for degradation (Qureshi et al. 2011), specifically at late time points of the T-APC interactions (Yokosuka et al. 2010). One key question is whether the CTLA-4 molecule trans-endocytosis and recycling is designed to move CTLA-4 into and out of the immunological synapse to regulate T-cell activation.
4.3 Structure and Functional Relationship
Given the importance of motifs used for molecular interaction (i.e., MYPPPY, KMLKKRS, YVKM) within CTLA-4, to date, many efforts have been made to understand their functional importance. Figure 2 summarizes many of the results. In vitro, CTLA-4-negative cells, reconstituted with mutants that are stably expressed on the plasma membrane show inhibitory activity when ligated by anti-CTLA-4 mAb or CD80/CD86. The critical point that makes it difficult to interpret the data is that CTLA-4 has two modes of inhibiting T-cell activation, namely, ligand competition and signaling. For example, deletion of the YVKM motif may abrogate phosphatase binding, but it augments cell surface expression and the inhibitory function of CTLA-4. Since the mutant CTLA-4 molecule lacking almost the entire cytoplasmic tail retains membrane-proximal KMLKKRS that is functionally able to protect the lethal phenotype of CTLA-4 KO mice. This motif binds to PP2A and PKC-η, which may be a minimal requirement of CTLA-4 function. Transgenic overexpression of this form of CTLA-4 completely (Takahashi et al. 2005) rescued the CTLA-4 KO phenotype whereas random amino acid substitution of the entire CTLA-4 tail only partially rescued it (Masteller et al. 2000) (see Fig. 2). Trans-endocytosis and degradation of CD80/86 by CTLA-4 is suggested to account for CTLA-4-mediated regulation (Qureshi et al.). Tailless CTLA-4 used in rescue experiments, which lacks the binding site for clathrin adaptors (YVKM), is functional when overexpressed on the T-cell compartment (Takahashi et al. 2005; Masteller et al. 2000) or the Treg compartment (Tai et al.), suggesting that endocytosis is not an absolute requirement for CTLA-4-mediated inhibition.

A proposed model of spatiotemporal T-cell inhibition by CTLA-4
5 Biological Roles of CTLA-4
5.1 CTLA-4 Genetics in Autoimmunity: Mice to Humans
Germline CTLA-4 knockout mice show lymphoproliferation, and die from multi-organ failure and a cytokine storm, suggesting the indispensable role of CTLA-4 in regulating self-tolerance (Tivol et al. 1995; Waterhouse et al. 1995; Chambers et al. 1997a). In humans, single nucleotide polymorphisms in the regulatory region, Exon1, 3’untranslated region, have been suggested to be associated with various autoimmune diseases (Gough et al. 2005; Scalapino and Daikh 2008), including celiac disease (Djilali-Saiah et al. 1998), rheumatoid arthritis (Yanagawa et al. 2000), multiple sclerosis (Ligers et al. 1999), type I diabetes (Todd 1997), Graves’ disease (Yanagawa et al. 1995), Hashimoto’s thyroiditis, autoimmune Addison’s disease (Donner et al. 1997), systemic lupus erythematosus (Hudson et al. 2002) to name a few. This clearly suggests that CTLA-4 insufficiency is a genetic factor in human autoimmunity. The alternative splicing of CTLA-4 mRNA is also suggested to determine autoimmune susceptibility. Ueda et al. (2003), through a comprehensive genetic association study, showed that the alternatively spliced form of the CTLA-4 locus that determines the relative amount of soluble CTLA-4 (sCTLA-4) ligand-independent liCTLA-4 versus ligand-binding CTLA-4 (lacking the entire part of the extracellular domain) determines autoimmune susceptibility in mice and humans.
There were no case reports of nonsense mutation of CTLA-4 resulting in human autoimmunity until recently. Two groups (Schubert et al. 2014; Kuehn et al. 2014) reported familial CTLA-4 deficiency from five families that show a common variable immunodeficiency (CVID) syndrome in which patients exhibit symptoms including recurrent infection, hypogammaglobulinemia, autoimmune cytopenia, cerebral infiltration, autoimmune heamolytic anemia, autoimmune enteropathy, and granulomatous lung disease. A heterozygous nonsense mutation of CTLA-4 in exon 1 is reported in autosomal-dominant immune disorders. The penetrance of identified individuals with the mutation was not 100%, but they show low expression of CTLA-4 (especially on Tregs) and defects in trans-endocytosis of CD80 and CD86. The study suggested that even haploinsufficiency (heterozygous loss) of CTLA-4 causes severe disease in humans. Another group reported the most extensive form of CTLA-4 deficiency in humans(Lo et al. 2015). The reported autoimmunity was not due to a mutation of the CTLA-4 protein itself, but was linked to a mutation in the LRBA gene (encoding the lipopolysaccharide-responsive and beige-like anchor protein), and caused a juvenile autoimmune manifestation similar to CTLA-4 deficiency (such as humoral immune deficiency and autoimmunity associated with lymphoproliferation) (Lo et al. 2015). LRBA co-localizes with CTLA-4 in the endosomal vesicles. Mutation/deficiency alters this functionality resulting in accelerated turnover of CTLA-4 by lysosomal degradation, leading to reduced CTLA-4 protein. Inhibiting the degradation by the lysosomal inhibitor chloroquine reduced CTLA-4 degradation and amelioration of autoimmunity. The authors concluded that chemicals that inhibit lysosomes (such as chroloquine) can be used as drugs to treat autoimmunity (by stabilizing CTLA-4 protein.) As described before, in mice, CD28-CTLA-4 double KO, CTLA-4/CD80/86 triple knockout, or CTLA-4 KO treated with CD28 antagonists (CTLA-4 Ig) do not develop autoimmunity, suggesting a key role of the CD28 signal. Patients with LRBA mutation who were treated with abatacept that blocks the CD28 signal show dramatic improvement in autoimmune symptoms (Lo et al. 2015), suggesting that excess CD28 signaling is the cause of the disease. Therefore, it would be beneficial to examine if individuals showing autoimmunity have mutation/polymorphism on the CTLA-4 locus, which should predict the effectiveness of abatacept therapy(Boussiotis 2014; Schubert et al. 2014).
5.2 CTLA-4 in Infection
Some reports suggest that CTLA-4 is critical for optimal T-cell response in antigen-specific immunity. CD4+ cells from CTLA-4 KO show spontaneous Th2-type skewing, even in the genetic absence of STAT6 (Bour-Jordan et al. 2003). Accordingly, CTLA-4 KO mice (partially rescued from lymphoproliferation by the tailless CTLA-4 transgene) show defects in controlling leishmania infection (Masteller et al. 2000). Since CTLA-4 knockout mice experience early death by lymphoproliferation, it was difficult to experimentally address how CTLA-4 KO mice mount antigen-specific immunity to infection. To clarify this, CTLA-4 in infection was addressed using mice reconstituted by bone marrow from CTLA-4 KO and WT mice to protect them from autoimmunity-associated death (Bachmann et al. 1999; Homann et al. 2006). Bachmann et al. showed that although half of the T cells in these animals do not express CTLA-4 genetically, both CTLA-4 KO T cells and WT T cells responded to leishmania, lymphocytic choriomeningitis virus (LCMV), and mouse mammary tumor virus (MMTV) normally and contracted them equally. In addition, Homann et al. showed that both CTLA-4 KO T cells and WT T cells respond normally to invading viruses and decrease in number rapidly after elimination of the pathogen (Homann et al. 2006). These investigators proposed that CTLA-4 works mainly to inhibit autoreactive CD4+ helper T cells rather than by modulating/terminating ongoing immune responses against exogenous pathogens.
5.3 CTLA-4 in Cancer
The goal for cancer immune therapy is to enhance patients’ immune systems in order to reject cancer. This concept was proposed by pioneer studies that showed that tumor cells mutated from healthy cells are recognized as “non-self” by T lymphocytes (De Plaen et al. 1988; Lurquin et al. 1989). The problem is that mutations in cancer cells may not always create a strong agonistic peptides epitope for T cells, and may instead stimulate tolerance rather than protective T cell responses. Therefore, enhancement of tumor recognition and activation by T cells through manipulation of the known pathway, such as costimulation, is beneficial. It was first shown that transfecting CD80, a CD28/CTLA-4 ligand, on a poorly immunogenic cancer cell line stimulated the mouse immune system for rejection upon tumor transplantation (Chen et al. 1992; Yang et al. 1995). The data demonstrated that recognition of antigens on a tumor can be augmented by additional signals mediated by CD28 and/or CTLA-4, leading to efficient T-cell activation and attack. Allison and colleagues demonstrated that a systemic administration with blocking anti-CTLA-4 mAb in mice boosted the anti-tumor response, resulting in the rejection of a transplanted tumors (Leach et al. 1996; Kwon et al. 1997). These reports established a milestone that the blockade of negative costimulatory molecules to their physiological ligand promotes tumor immunity. This and subsequent results led to the concept of immune-checkpoint blockade in cancer treatment. Subsequently, the CTLA-4 blockade was shown to be effective in combination with tumor vaccination in mice (van Elsas et al. 1999). Shrikant et al. (1999) used an antigen-specific tumor elimination mouse model to elucidate the mechanism of augmented tumor immunity by CTLA-4 blockade. They showed that tumor-specific CD8+ cells are generally anergic, but exhibit tumor attack upon administration of anti-CTLA-4 antibody in vivo. This re-activation of CD8+ T cells was dependent on CD4+ helper T cells and IL-2 produced by this population, suggesting that re-activation of the helper response indirectly boosts the killer-mediated anticancer responses (Shrikant et al. 1999). The result also supported the notion that CTLA-4 blockade not only directly augments effector CD8+ T cells, but also indirectly boosts immune responses by acting on helper T cells.
A fully humanized chimeric antibody named ipilimumab has been shown to be effective in melanoma in monotherapy (Hodi et al. 2010), in combination with chemotherapy (Robert et al. 2011) or with anti PD-1 antibody (Wolchok et al. 2013). Although manipulation of this pathway is attractive in cancer therapies, the immune-related adverse effects are often problematic. It was initially reported that 50–60% of patients receiving anti-CTLA-4 therapy showed adverse events (Hodi et al. 2010; Robert et al. 2011) affecting various organs (reviewed inMichot et al. 2016). Analogous to mouse studies, these data suggest that there are many autoreactive T cells in the periphery that need to be under continuous control by CTLA-4. Currently, patients with immune-related adverse events higher than grade 3 are primarily treated by steroids (reviewed in Michot et al. 2016). In the future, adequate management if these side effects are likely and it is expected that there will be an abrogation of adverse events without avoiding anticancer response. However, in case of subacute, life-threatening autoimmunity, given the example of human CTLA-4 deficiency, abatacept targeting the CD28 pathway might be important.
Conclusions and perspectives
CTLA-4 is no doubt important in the maintenance of T-cell homeostasis. The longstanding basic question of why CTLA-4 deficiency results in a lethal autoimmune phenotype in mice has not, however, been solved. An interesting observation was made by two groups. An induced deletion of CTLA-4 at adulthood resulted in milder autoimmunity compared to the original germline KO (Klocke et al. 2016). Another group developed a similar system deleting CTLA-4 in adult mice and did not cause autoimmunity (Paterson et al. 2015). Although there is a technical argument regarding the efficiency of the deletion in both models, the mice did not die, unlike germline KO mice. Similarly, it was suggested that successful anti-CTLA-4 treatment on adult cancer patients, at least in part rely on Fc-receptor-mediated depletion of CTLA-4+ T cells (Simpson et al. 2013), however, this treatment does not result in overt lymphoproliferation. An assumption based on these data is that CTLA-4 expression is particularly important during the neonatal period when polyclonal T cells undergo lymphopenia-driven expansion to seed the body (Fig. 3a) (Min et al. 2003). This form of T-cell proliferation does not require high-affinity interaction of TCR with the MHC-self-antigen complex, but is driven by the CD28 signal (Hagen et al. 2004). The neonatal period is also a time for development of self-reactive FoxP3+ Tregs that prevents activation of other T cells (Itoh et al. 1999) (Yang et al. 2015). Therefore, the loss of CTLA-4 from birth causes a CD28-dependent massive expansion of nearly the entire repertoire of low-affinity self-reactive T cells without proper regulation by FoxP3+ Tregs, resulting in lymphoproliferation and death, due to a cytokine storm (Fig. 3a). After the initial wave of lymphocyte expansion ceases, CTLA-4-mediated inhibition is less important for maintaining the homeostatic condition of T cells (Klocke et al. 2016; Paterson et al. 2015).

A scheme representing the difference of CTLA-4-mediated inhibition between neonatal mice and naïve (uninfected) adult mice. A possible “immune monitoring” of cancer patients under checkpoint therapy to predict treatment outcome. The idea of Ki67-mediated detection of early-responding T-cell population is from (Huang et al. 2017) and (Kamphorst et al. 2017)
In the cancer setting, high-affinity T cells receive the advantages of CTLA-4 blockade and regain their ability to fight, because these T cells recruit more CTLA-4 to the immunological synapse and are preferentially inhibited by CTLA-4 (Egen and Allison 2002). The problem is that the autoreactive memory T cells present in patients are also activated by this treatment. The balance and affinity of existing autoreactive/anticancer memory T cells will differ from person to person, which may determine the outcome of the treatment. Monitoring individuals for these clones will thus provide more efficient anticancer immunity with fewer treatment side effects (Fig. 3b). These approaches are currently in development, combining Ki67 (a proliferation marker) to monitor T cell re-activation before and after immune checkpoint therapy using patient peripheral blood samples (Huang et al. 2017; Kamphorst et al. 2017). It will hopefully become possible to detect and follow the activation of cancer/self-specific T-cell clones in the periphery during anti-CTLA-4 therapy. This will provide useful information when considering future cancer immune therapy (Fig. 3b).
References
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2(8):675–680
Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, Thompson CB (1996) Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol 157(11):4762–4770
Araki M, Chung D, Liu S, Rainbow DB, Chamberlain G, Garner V, Hunter KM, Vijayakrishnan L, Peterson LB, Oukka M, Sharpe AH, Sobel R, Kuchroo VK, Wicker LS (2009) Genetic evidence that the differential expression of the ligand-independent isoform of CTLA-4 is the molecular basis of the Idd5.1 type 1 diabetes region in nonobese diabetic mice. J Immunol 183(8):5146–5157
Bachmann MF, Gallimore A, Jones E, Ecabert B, Acha-Orbea H, Kopf M (2001) Normal pathogen-specific immune responses mounted by CTLA-4-deficient T cells: a paradigm reconsidered. Eur J Immunol 31(2):450–458
Bachmann MF, Kohler G, Ecabert B, Mak TW, Kopf M (1999) Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol 163(3):1128–1131
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392(6673):245–252
Baroja ML, Luxenberg D, Chau T, Ling V, Strathdee CA, Carreno BM, Madrenas J (2000) The inhibitory function of CTLA-4 does not require its tyrosine phosphorylation. J Immunol 164(1):49–55
Baroja ML, Vijayakrishnan L, Bettelli E, Darlington PJ, Chau TA, Ling V, Collins M, Carreno BM, Madrenas J, Kuchroo VK (2002) Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J Immunol 168(10):5070–5078
Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB (1995) CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3(1):87–98
Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA (2011) Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol Rev 241(1):180–205
Bour-Jordan H, Grogan JL, Tang Q, Auger JA, Locksley RM, Bluestone JA (2003) CTLA-4 regulates the requirement for cytokine-induced signals in T(H)2 lineage commitment. Nat Immunol 4(2):182–188
Boussiotis VA (2014) Somatic mutations and immunotherapy outcome with CTLA-4 blockade in melanoma. N Engl J Med 371(23):2230–2232
Bradshaw JD, Lu P, Leytze G, Rodgers J, Schieven GL, Bennett KL, Linsley PS, Kurtz SE (1997) Interaction of the cytoplasmic tail of CTLA-4 (CD152) with a clathrin-associated protein is negatively regulated by tyrosine phosphorylation. Biochemistry 36(50):15975–15982
Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, Golstein P (1987) A new member of the immunoglobulin superfamily–CTLA-4. Nature 328(6127):267–270
Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F (2001) Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27(1):68–73
Calvo CR, Amsen D, Kruisbeek AM (1997) Cytotoxic T lymphocyte antigen 4 (CTLA-4) interferes with extracellular signal-regulated kinase (ERK) and Jun NH2-terminal kinase (JNK) activation, but does not affect phosphorylation of T cell receptor zeta and ZAP70. J Exp Med 186(10):1645–1653
Chambers CA, Cado D, Truong T, Allison JP (1997a) Thymocyte development is normal in CTLA-4-deficient mice. Proc Natl Acad Sci USA 94(17):9296–9301
Chambers CA, Kuhns MS, Allison JP (1999) Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc Natl Acad Sci USA 96(15):8603–8608
Chambers CA, Sullivan TJ, Allison JP (1997b) Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity 7(6):885–895
Chen L, Ashe S, Brady WA, Hellstrom I, Hellstrom KE, Ledbetter JA, McGowan P, Linsley PS (1992) Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell 71(7):1093–1102
Chikuma S (2016) Basics of PD-1 in self-tolerance, infection, and cancer immunity. International journal of clinical oncology 21(3):448–455
Chikuma S, Abbas AK, Bluestone JA (2005) B7-independent inhibition of T cells by CTLA-4. J Immunol 175(1):177–181
Chikuma S, Bluestone JA (2007) Expression of CTLA-4 and FOXP3 in cis protects from lethal lymphoproliferative disease. Eur J Immunol 37(5):1285–1289
Chikuma S, Imboden JB, Bluestone JA (2003) Negative regulation of T cell receptor-lipid raft interaction by cytotoxic T lymphocyte-associated antigen 4. J Exp Med 197(1):129–135
Chikuma S, Murakami M, Tanaka K, Uede T (2000) Janus kinase 2 is associated with a box 1-like motif and phosphorylates a critical tyrosine residue in the cytoplasmic region of cytotoxic T lymphocyte associated molecule-4. J Cell Biochem 78(2):241–250
Chikuma S, Terawaki S, Hayashi T, Nabeshima R, Yoshida T, Shibayama S, Okazaki T, Honjo T (2009) PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J Immunol 182(11):6682–6689
Chuang E, Alegre ML, Duckett CS, Noel PJ, Vander Heiden MG, Thompson CB (1997) Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J Immunol 159(1):144–151
Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, Gardner JP, Hambor JE, Neveu MJ, Thompson CB (2000) The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 13(3):313–322
Chuang E, Lee KM, Robbins MD, Duerr JM, Alegre ML, Hambor JE, Neveu MJ, Bluestone JA, Thompson CB (1999) Regulation of cytotoxic T lymphocyte-associated molecule-4 by Src kinases. J Immunol 162(3):1270–1277
Cinek T, Sadra A, Imboden JB (2000) Cutting edge: tyrosine-independent transmission of inhibitory signals by CTLA-4. J Immunol 164(1):5–8
De Plaen E, Lurquin C, Van Pel A, Mariame B, Szikora JP, Wolfel T, Sibille C, Chomez P, Boon T (1988) Immunogenic (tum-) variants of mouse tumor P815: cloning of the gene of tum- antigen P91A and identification of the tum- mutation. Proc Natl Acad Sci USA 85(7):2274–2278
Djilali-Saiah I, Schmitz J, Harfouch-Hammoud E, Mougenot JF, Bach JF, Caillat-Zucman S (1998) CTLA-4 gene polymorphism is associated with predisposition to coeliac disease. Gut 43(2):187–189
Donner H, Braun J, Seidl C, Rau H, Finke R, Ventz M, Walfish PG, Usadel KH, Badenhoop K (1997) Codon 17 polymorphism of the cytotoxic T lymphocyte antigen 4 gene in Hashimoto’s thyroiditis and Addison’s disease. J Clin Endocrinol Metab 82(12):4130–4132
Doyle AM, Mullen AC, Villarino AV, Hutchins AS, High FA, Lee HW, Thompson CB, Reiner SL (2001) Induction of cytotoxic T lymphocyte antigen 4 (CTLA-4) restricts clonal expansion of helper T cells. J Exp Med 194(7):893–902
Egen JG, Allison JP (2002) Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 16(1):23–35
Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P (2003) Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol 4(12):1206–1212
Fife BT, Griffin MD, Abbas AK, Locksley RM, Bluestone JA (2006) Inhibition of T cell activation and autoimmune diabetes using a B cell surface-linked CTLA-4 agonist. J Clin Invest 116(8):2252–2261
Finn PW, He H, Wang Y, Wang Z, Guan G, Listman J, Perkins DL (1997) Synergistic induction of CTLA-4 expression by costimulation with TCR plus CD28 signals mediated by increased transcription and messenger ribonucleic acid stability. J Immunol 158(9):4074–4081
Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat Immunol 4(4):330–336
Frauwirth KA, Alegre ML, Thompson CB (2000) Induction of T cell anergy in the absence of CTLA-4/B7 interaction. J Immunol 164(6):2987–2993
Frauwirth KA, Alegre ML, Thompson CB (2001) CTLA-4 is not required for induction of CD8(+) T cell anergy in vivo. J Immunol 167(9):4936–4941
Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB (2002) The CD28 signaling pathway regulates glucose metabolism. Immunity 16(6):769–777
Friedline RH, Brown DS, Nguyen H, Kornfeld H, Lee J, Zhang Y, Appleby M, Der SD, Kang J, Chambers CA (2009) CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J Exp Med 206(2):421–434
Fu G, Hu J, Niederberger-Magnenat N, Rybakin V, Casas J, Yachi PP, Feldstein S, Ma B, Hoerter JA, Ampudia J, Rigaud S, Lambolez F, Gavin AL, Sauer K, Cheroutre H, Gascoigne NR (2011) Protein kinase C eta is required for T cell activation and homeostatic proliferation. Sci Signal 4 (202):ra84
Gajewski TF, Fallarino F, Fields PE, Rivas F, Alegre ML (2001) Absence of CTLA-4 lowers the activation threshold of primed CD8+ TCR-transgenic T cells: lack of correlation with Src homology domain 2-containing protein tyrosine phosphatase. J Immunol 166(6):3900–3907
Gough SC, Walker LS, Sansom DM (2005) CTLA4 gene polymorphism and autoimmunity. Immunol Rev 204:102–115
Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH (2001) CTLA-4 regulates induction of anergy in vivo. Immunity 14(2):145–155
Griffin MD, Hong DK, Holman PO, Lee KM, Whitters MJ, O’Herrin SM, Fallarino F, Collins M, Segal DM, Gajewski TF, Kranz DM, Bluestone JA (2000) Blockade of T cell activation using a surface-linked single-chain antibody to CTLA-4 (CD152). J Immunol 164(9):4433–4442
Hagen KA, Moses CT, Drasler EF, Podetz-Pedersen KM, Jameson SC, Khoruts A (2004) A role for CD28 in lymphopenia-induced proliferation of CD4 T cells. J Immunol 173(6):3909–3915
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723
Homann D, Dummer W, Wolfe T, Rodrigo E, Theofilopoulos AN, Oldstone MB, von Herrath MG (2006) Lack of intrinsic CTLA-4 expression has minimal effect on regulation of antiviral T-cell immunity. J Virol 80(1):270–280
Hori S, Nomura T, Sakaguchi S (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299(5609):1057–1061
Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, Adamow M, Kuk D, Panageas KS, Carrera C, Wong P, Quagliarello F, Wubbenhorst B, D’Andrea K, Pauken KE, Herati RS, Staupe RP, Schenkel JM, McGettigan S, Kothari S, George SM, Vonderheide RH, Amaravadi RK, Karakousis GC, Schuchter LM, Xu X, Nathanson KL, Wolchok JD, Gangadhar TC, Wherry EJ (2017) T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545(7652):60–65
Hudson LL, Rocca K, Song YW, Pandey JP (2002) CTLA-4 gene polymorphisms in systemic lupus erythematosus: a highly significant association with a determinant in the promoter region. Hum Genet 111(4–5):452–455
Ichinose K, Zhang Z, Koga T, Juang YT, Kis-Toth K, Sharpe AH, Kuchroo V, Crispin JC, Tsokos GC (2013) Brief report: increased expression of a short splice variant of CTLA-4 exacerbates lupus in MRL/lpr mice. Arthritis Rheum 65(3):764–769
Iida T, Ohno H, Nakaseko C, Sakuma M, Takeda-Ezaki M, Arase H, Kominami E, Fujisawa T, Saito T (2000) Regulation of cell surface expression of CTLA-4 by secretion of CTLA-4-containing lysosomes upon activation of CD4+ T cells. J Immunol 165(9):5062–5068
Ise W, Kohyama M, Nutsch KM, Lee HM, Suri A, Unanue ER, Murphy TL, Murphy KM (2009) CTLA-4 suppresses the pathogenicity of self antigen-specific T cells by cell-intrinsic and cell-extrinsic mechanisms. Nat Immunol 11(2):129–135
Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, Sakaguchi S (1999) Thymus and autoimmunity: production of CD25+ CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol 162(9):5317–5326
Jain N, Nguyen H, Chambers C, Kang J (2010) Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci USA 107(4):1524–1528
June CH, Ledbetter JA, Gillespie MM, Lindsten T, Thompson CB (1987) T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol 7(12):4472–4481
Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, Behera M, Wu H, McCausland M, Chen Z, Zhang C, Khuri FR, Owonikoko TK, Ahmed R, Ramalingam SS (2017) Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci USA
Kataoka H, Takahashi S, Takase K, Yamasaki S, Yokosuka T, Koike T, Saito T (2005) CD25(+)CD4(+) regulatory T cells exert in vitro suppressive activity independent of CTLA-4. Int Immunol 17(4):421–427
Khattri R, Cox T, Yasayko SA, Ramsdell F (2003) An essential role for Scurfin in CD4+ CD25+ T regulatory cells. Nat Immunol 4(4):337–342
Klocke K, Sakaguchi S, Holmdahl R, Wing K (2016) Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci USA 113(17):E2383–E2392
Kong KF, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, Becart S, Kim G, Yates JR 3rd, Kronenberg M, Saito T, Gascoigne NR, Altman A (2014) Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat Immunol 15(5):465–472
Krummel MF, Allison JP (1995) CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182(2):459–465
Krummel MF, Allison JP (1996) CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med 183(6):2533–2540
Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, Frucht DM, Dumitriu B, Scheinberg P, Folio LR, Frein CA, Price S, Koh C, Heller T, Seroogy CM, Huttenlocher A, Rao VK, Su HC, Kleiner D, Notarangelo LD, Rampertaap Y, Olivier KN, McElwee J, Hughes J, Pittaluga S, Oliveira JB, Meffre E, Fleisher TA, Holland SM, Lenardo MJ, Tangye SG, Uzel G (2014) Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345(6204):1623–1627
Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, Burg MB, Allison JP (1997) Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci USA 94(15):8099–8103
Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271(5256):1734–1736
Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, Straus D, Samelson LE, Thompson CB, Bluestone JA (1998) Molecular basis of T cell inactivation by CTLA-4. Science 282(5397):2263–2266
Leung HT, Bradshaw J, Cleaveland JS, Linsley PS (1995) Cytotoxic T lymphocyte-associated molecule-4, a high-avidity receptor for CD80 and CD86, contains an intracellular localization motif in its cytoplasmic tail. J Biol Chem 270(42):25107–25114
Ligers A, Xu C, Saarinen S, Hillert J, Olerup O (1999) The CTLA-4 gene is associated with multiple sclerosis. J Neuroimmunol 97(1–2):182–190
Ling V, Wu PW, Finnerty HF, Sharpe AH, Gray GS, Collins M (1999) Complete sequence determination of the mouse and human CTLA4 gene loci: cross-species DNA sequence similarity beyond exon borders. Genomics 60(3):341–355
Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS (1996) Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 4(6):535–543
Linsley PS, Greene JL, Tan P, Bradshaw J, Ledbetter JA, Anasetti C, Damle NK (1992) Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med 176(6):1595–1604
Linsley PS, Nadler SG, Bajorath J, Peach R, Leung HT, Rogers J, Bradshaw J, Stebbins M, Leytze G, Brady W et al (1995) Binding stoichiometry of the cytotoxic T lymphocyte-associated molecule-4 (CTLA-4). A disulfide-linked homodimer binds two CD86 molecules. J Biol Chem 270(25):15417–15424
Liu SM, Sutherland AP, Zhang Z, Rainbow DB, Quintana FJ, Paterson AM, Sharpe AH, Oukka M, Wicker LS, Kuchroo VK (2012) Overexpression of the Ctla-4 isoform lacking exons 2 and 3 causes autoimmunity. J Immunol 188(1):155–162
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, Zhang Y, Liu Z, Fritz JM, Marsh R, Husami A, Kissell D, Nortman S, Chaturvedi V, Haines H, Young LR, Mo J, Filipovich AH, Bleesing JJ, Mustillo P, Stephens M, Rueda CM, Chougnet CA, Hoebe K, McElwee J, Hughes JD, Karakoc-Aydiner E, Matthews HF, Price S, Su HC, Rao VK, Lenardo MJ, Jordan MB (2015) AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349(6246):436–440
Lorenz U (2009) SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev 228(1):342–359
Lurquin C, Van Pel A, Mariame B, De Plaen E, Szikora JP, Janssens C, Reddehase MJ, Lejeune J, Boon T (1989) Structure of the gene of tum- transplantation antigen P91A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell 58(2):293–303
Magistrelli G, Jeannin P, Herbault N, Benoit De Coignac A, Gauchat JF, Bonnefoy JY, Delneste Y (1999) A soluble form of CTLA-4 generated by alternative splicing is expressed by nonstimulated human T cells. Eur J Immunol 29(11):3596–3602
Mandelbrot DA, McAdam AJ, Sharpe AH (1999) B7-1 or B7-2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). J Exp Med 189(2):435–440
Mandelbrot DA, Oosterwegel MA, Shimizu K, Yamada A, Freeman GJ, Mitchell RN, Sayegh MH, Sharpe AH (2001) B7-dependent T-cell costimulation in mice lacking CD28 and CTLA4. J Clin Invest 107(7):881–887
Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW (1996) Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 272(5265):1170–1173
Masteller EL, Chuang E, Mullen AC, Reiner SL, Thompson CB (2000) Structural analysis of CTLA-4 function in vivo. J Immunol 164(10):5319–5327
Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, Bahleda R, Hollebecque A, Massard C, Fuerea A, Ribrag V, Gazzah A, Armand JP, Amellal N, Angevin E, Noel N, Boutros C, Mateus C, Robert C, Soria JC, Marabelle A, Lambotte O (2016) Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 54:139–148
Min B, McHugh R, Sempowski GD, Mackall C, Foucras G, Paul WE (2003) Neonates support lymphopenia-induced proliferation. Immunity 18(1):131–140
Miyatake S, Nakaseko C, Umemori H, Yamamoto T, Saito T (1998) Src family tyrosine kinases associate with and phosphorylate CTLA-4 (CD152). Biochem Biophys Res Commun 249(2):444–448
Nakaseko C, Miyatake S, Iida T, Hara S, Abe R, Ohno H, Saito Y, Saito T (1999) Cytotoxic T lymphocyte antigen 4 (CTLA-4) engagement delivers an inhibitory signal through the membrane-proximal region in the absence of the tyrosine motif in the cytoplasmic tail. J Exp Med 190(6):765–774
Oaks MK, Hallett KM, Penwell RT, Stauber EC, Warren SJ, Tector AJ (2000) A native soluble form of CTLA-4. Cell Immunol 201(2):144–153
Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, Osaki M, Tanaka Y, Yamashita R, Nakano N, Huehn J, Fehling HJ, Sparwasser T, Nakai K, Sakaguchi S (2012) T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 37(5):785–799
Ostrov DA, Shi W, Schwartz JC, Almo SC, Nathenson SG (2000) Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science 290(5492):816–819
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL (2005) CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25(21):9543–9553
Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, Arancibia-Carcamo CV, Sobel RA, Rudensky AY, Kuchroo VK, Freeman GJ, Sharpe AH (2015) Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med 212(10):1603–1621
Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, Linsley PS (1994) Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med 180(6):2049–2058
Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP (2004) B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 21(3):401–413
Perkins D, Wang Z, Donovan C, He H, Mark D, Guan G, Wang Y, Walunas T, Bluestone J, Listman J, Finn PW (1996) Regulation of CTLA-4 expression during T cell activation. J Immunol 156(11):4154–4159
Pioli C, Gatta L, Frasca D, Doria G (1999) Cytotoxic T lymphocyte antigen 4 (CTLA-4) inhibits CD28-induced IkappaBalpha degradation and RelA activation. Eur J Immunol 29(3):856–863
Puccetti P, Grohmann U (2007) IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol 7(10):817–823
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, Walker LS, Sansom DM (2011) Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332(6029):600–603
Read S, Malmstrom V, Powrie F (2000) Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med 192(2):295–302
Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, Gregson BP, June CH, Linsley PS (2002) Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci U S A 99(18):11790–11795
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364(26):2517–2526
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA (2000) B7/CD28 costimulation is essential for the homeostasis of the CD4+ CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12(4):431–440
Scalapino KJ, Daikh DI (2008) CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev 223:143–155
Schneider H, Martin M, Agarraberes FA, Yin L, Rapoport I, Kirchhausen T, Rudd CE (1999) Cytolytic T lymphocyte-associated antigen-4 and the TCR zeta/CD3 complex, but not CD28, interact with clathrin adaptor complexes AP-1 and AP-2. J Immunol 163(4):1868–1879
Schneider H, Rudd CE (2000) Tyrosine phosphatase SHP-2 binding to CTLA-4: absence of direct YVKM/YFIP motif recognition. Biochem Biophys Res Commun 269(1):279–283
Schneider H, Schwartzberg PL, Rudd CE (1998) Resting lymphocyte kinase (Rlk/Txk) phosphorylates the YVKM motif and regulates PI 3-kinase binding to T-cell antigen CTLA-4. Biochem Biophys Res Commun 252(1):14–19
Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schaffer AA, Gruning BA, Unger S, Frede N, Baumann U, Witte T, Schmidt RE, Dueckers G, Niehues T, Seneviratne S, Kanariou M, Speckmann C, Ehl S, Rensing-Ehl A, Warnatz K, Rakhmanov M, Thimme R, Hasselblatt P, Emmerich F, Cathomen T, Backofen R, Fisch P, Seidl M, May A, Schmitt-Graeff A, Ikemizu S, Salzer U, Franke A, Sakaguchi S, Walker LS, Sansom DM, Grimbacher B (2014) Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 20(12):1410–1416
Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC (2001) Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 410(6828):604–608
Schwartz RH (2003) T cell anergy. Annu Rev Immunol 21:305–334
Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, Saito T (1997) Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity 6(5):583–589
Shrikant P, Khoruts A, Mescher MF (1999) CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity 11(4):483–493
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA (2013) Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 210(9):1695–1710
Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, Stahl ML, Seehra J, Somers WS, Mosyak L (2001) Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature 410(6828):608–611
Tai X, Cowan M, Feigenbaum L, Singer A (2005) CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol 6(2):152–162
Tai X, Van Laethem F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, Granger L, Kruhlak M, Lindsten T, Thompson CB, Feigenbaum L, Singer A (2012) Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood 119(22):5155–5163
Takahashi S, Kataoka H, Hara S, Yokosuka T, Takase K, Yamasaki S, Kobayashi W, Saito Y, Saito T (2005) In vivo overexpression of CTLA-4 suppresses lymphoproliferative diseases and thymic negative selection. Eur J Immunol 35(2):399–407
Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S (2000) Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med 192(2):303–310
Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA (2006) Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol 7(1):83–92
Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA (2004) Distinct roles of CTLA-4 and TGF-beta in CD4+ CD25+ regulatory T cell function. Eur J Immunol 34(11):2996–3005
Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, Bluestone JA (2003) Cutting edge: CD28 controls peripheral homeostasis of CD4 + CD25 + regulatory T cells. J Immunol 171(7):3348–3352
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH (1995) Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3(5):541–547
Tivol EA, Gorski J (2002) Re-establishing peripheral tolerance in the absence of CTLA-4: complementation by wild-type T cells points to an indirect role for CTLA-4. J Immunol 169(4):1852–1858
Todd JA (1997) Genetics of type 1 diabetes. Pathol Biol (Paris) 45(3):219–227
Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RC, Howlett S, Healy B, Nutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker NM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia A, Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehto J, Bingley P, Gillespie KM, Undlien DE, Ronningen KS, Guja C, Ionescu-Tirgoviste C, Savage DA, Maxwell AP, Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SC (2003) Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423(6939):506–511
van Elsas A, Hurwitz AA, Allison JP (1999) Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 190(3):355–366
Vijayakrishnan L, Slavik JM, Illes Z, Greenwald RJ, Rainbow D, Greve B, Peterson LB, Hafler DA, Freeman GJ, Sharpe AH, Wicker LS, Kuchroo VK (2004) An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity 20(5):563–575
Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA (1994) CTLA-4 can function as a negative regulator of T cell activation. Immunity 1(5):405–413
Wang CJ, Kenefeck R, Wardzinski L, Attridge K, Manzotti C, Schmidt EM, Qureshi OS, Sansom DM, Walker LS (2012) Cutting edge: cell-extrinsic immune regulation by CTLA-4 expressed on conventional T cells. J Immunol 189(3):1118–1122
Waterhouse P, Bachmann MF, Penninger JM, Ohashi PS, Mak TW (1997) Normal thymic selection, normal viability and decreased lymphoproliferation in T cell receptor-transgenic CTLA-4-deficient mice. Eur J Immunol 27(8):1887–1892
Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW (1995) Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270(5238):985–988
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S (2008) CTLA-4 control over Foxp3 + regulatory T cell function. Science 322(5899):271–275
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M (2013) Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369(2):122–133
Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A (2006) FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126(2):375–387
Yamasaki S, Nishida K, Hibi M, Sakuma M, Shiina R, Takeuchi A, Ohnishi H, Hirano T, Saito T (2001) Docking protein Gab2 is phosphorylated by ZAP-70 and negatively regulates T cell receptor signaling by recruitment of inhibitory molecules. J Biol Chem 276(48):45175–45183
Yanagawa T, Gomi K, Nakao EI, Inada S (2000) CTLA-4 gene polymorphism in Japanese patients with rheumatoid arthritis. J Rheumatol 27(12):2740–2742
Yanagawa T, Hidaka Y, Guimaraes V, Soliman M, DeGroot LJ (1995) CTLA-4 gene polymorphism associated with Graves’ disease in a Caucasian population. J Clin Endocrinol Metab 80(1):41–45
Yang G, Hellstrom KE, Hellstrom I, Chen L (1995) Antitumor immunity elicited by tumor cells transfected with B7-2, a second ligand for CD28/CTLA-4 costimulatory molecules. J Immunol 154(6):2794–2800
Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D (2015) Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348(6234):589–594
Yi LA, Hajialiasgar S, Chuang E (2004) Tyrosine-mediated inhibitory signals contribute to CTLA-4 function in vivo. Int Immunol 16(4):539–547
Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, Yagita H, Tokunaga M, Saito T (2010) Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity 33(3):326–339
Zhang Y, Allison JP (1997) Interaction of CTLA-4 with AP50, a clathrin-coated pit adaptor protein. Proc Natl Acad Sci USA 94(17):9273–9278
Zikherman J, Parameswaran R, Weiss A (2012) Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature 489(7414):160–164
Acknowledgements
I would like to thank everyone who works/worked on CTLA-4. I especially thank Dr. Jeff Bluestone (University of California, San Francisco) for comments on the manuscript. This work was supported by Kakenhi (17H05801).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Chikuma, S. (2017). CTLA-4, an Essential Immune-Checkpoint for T-Cell Activation. In: Yoshimura, A. (eds) Emerging Concepts Targeting Immune Checkpoints in Cancer and Autoimmunity. Current Topics in Microbiology and Immunology, vol 410. Springer, Cham. https://doi.org/10.1007/82_2017_61
Download citation
DOI: https://doi.org/10.1007/82_2017_61
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-68928-9
Online ISBN: 978-3-319-68929-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)