
Abstract
An abnormal increase in the expression of nuclear receptor subfamily 6 group A member 1 (NR6A1) in the hippocampus has been reported to result in depressive-like behavior in mice. However, the role of NR6A1 in the progression of neuronal death induced by ischemic stroke remains unknown. In this study, we observed an increase in NR6A1 in neurons in both in vivo and in vitro cerebral ischemic models. We found that knocking down NR6A1 in HT-22 neuronal cells subjected to oxygen–glucose deprivation/reoxygenation (OGD/R) attenuated mitochondrial dysfunction and endoplasmic reticulum (ER) stress. Conversely, NR6A1 overexpression exacerbated neuronal damage following OGD/R. NR6A1 hindered the transcription of mitonfusin 2 (MFN2), leading to a decrease in its expression. In contrast, MFN2 conferred the protective effect of NR6A1 silencing against both mitochondrial dysfunction and ER stress. In addition, NR6A1 silencing also attenuated brain infarction, ER stress, neuronal apoptosis, and loss of MFN2 in mice subjected to middle cerebral artery occlusion/reperfusion. These findings indicate that NR6A1 is a promising target for the treatment of neuronal death following cerebral ischemia. Furthermore, these results confirm the involvement of MFN2 in the effects of NR6A1 silencing. Therefore, targeting NR6A1 has potential as a viable strategy for the treatment of ischemic stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is a major contributor to mortality and disability worldwide [1]. While vascular recanalization therapy has become a major treatment for ischemic stroke, improving patient outcomes, its effectiveness is limited by a restricted therapeutic timeframe [2]. Moreover, even in cases where recanalization is successful, patients are at risk of suffering injuries caused by ischemia/reperfusion [3]. Therefore, developing strategies to salvage neurons within the ischemic penumbra is highly important for intervention in cerebral ischemia.
The pathophysiological underpinnings of neuronal injury following ischemic stroke are multifaceted, and involve intricate damage across a spectrum of cellular organelles. Central to this destructive cascade, mitochondrial dysfunction and endoplasmic reticulum (ER) stress are pivotal events that exacerbate ischemia–reperfusion injury [4]. Cerebral ischemia leads to the production of many free radicals in neuronal mitochondria and induces neuronal damage [5]. Ischemia also results in the activation of ER stress pathways and induces neuronal death [6]. Therefore, finding therapeutic targets that mitigate ER and mitochondrial impairments is crucial for diminishing the detrimental effects of cerebral ischemia.
Mitofusin (MFN), a protein localized to the outer membrane of mitochondria, orchestrates the critical process of mitochondrial fusion. Comprising two isoforms, MFN1 and MFN2, it is MFN2 that exhibits a dual localization, not only to the mitochondrial periphery but also to the ER membrane [7]. As a pivotal tethering molecule, MFN2 facilitates structural and functional coupling between mitochondria and the ER, thereby playing an indispensable role in sustaining the integrity and physiological operations of these organelles [8]. MFN2 deficiency results in the uncoupling of mitochondria from the ER, which in turn precipitates neuronal death [9, 10]. In the context of cerebral ischemia, the loss of MFN2 exacerbates neuronal loss [11]. Our previous investigations revealed a notable decrease in MFN2 levels in neurons post-cerebral ischemia, an occurrence coupled with mitochondrial impairment and ER stress [12]. Thus, enhancing the expression of MFN2 has emerged as a potentially effective therapeutic intervention for cerebral ischemia.
Nuclear receptor subfamily 6 group A member 1 (NR6A1) belongs to the nuclear receptor superfamily and is classified as an orphan nuclear receptor, given the absence of a known ligand [13]. Initial research concerning NR6A1 predominantly focused on its role in development [14]. However, more recent studies have revealed its involvement in diverse biological processes, including lipid metabolism [15], carcinogenesis [16], and atherosclerosis [17]. Intriguingly, an increase in the hippocampal expression of NR6A1 induced depressive-like behaviors in mice [18]. However, the role of NR6A1 in the pathology of cerebral ischemia remains unknown and requires further investigation.
In this study, we sought to determine whether NR6A1 is involved in neuronal death after cerebral ischemia. This study helps clarify the role of NR6A1 in cerebral ischemia and provides a potential target for the treatment of ischemic stroke.
Methods and Materials
Reagents
MFN2 antibody (#9482), caspase 3 antibody (#9662), eukaryotic translation initiation factor 2α antibody (eIF2α, #5324), phosphorylated eIF2α antibody (p-eIF2α, Ser51, #3597), c-Jun N-terminal kinase antibody (JNK, #9252), phosphorylated JNK antibody (p-JNK, Thr183/Tyr185, #9251), and inositol-requiring enzyme-1α antibody (IRE1α, #3294) were purchased from Cell Signaling Technology (MA, USA). Phosphorylated IRE1α antibody (Ser724, #ab48187), NeuN antibody (#ab177487), and Mounting Medium with DAPI (#ab104139) were purchased from Abcam (Cambridge, UK). Sh-NR6A1, si-NR6A1, the pGL4.10-MFN2 promoter plasmid, the pRL-CMV plasmid, and the NR6A1 plasmid were obtained from OBiO Technology (Shanghai, China). A Pierce™ Rapid Gold BCA Protein Assay Kit (#A53225), Opti-MEM™ Reduced Serum Medium (#31985070), fetal bovine serum (FBS, #A3161001C), Live Cell Imaging Solution (#A14291DJ), glucose-free DMEM (#11966025), CellROX™ Deep Red Reagent (#C10422), Lipofectamine 3000 Reagent (#L3000008), tetramethylrhodamine ethyl ester perchlorate (TMRE, #T669), and Mito-tracker (#M7512) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). β-tubulin antibody (#FD0064), Cell Counting Kit-8 (CCK-8, #FD3788), and RIPA buffer (#FD009) were purchased from FDBIO SCIENCE (Hangzhou, Zhejiang, China). Goat anti-mouse IgG Dylight 549 (#A23310) and Goat anti-rabbit IgG Dylight 488 (#A23220) were obtained from Abbkine Scientific (Wuhan, Hubei, China). An RNA Easy Fast Tissue/Cell Kit (#DP451), FastKing gDNA Dispelling RT SuperMix (#KR118), and FastReal qPCR PreMix (SYBR Green, #FP217) were purchased from TIANGEN Biotech (Beijing, China). NR6A1 antibody (for western blot, #12712–1-AP), GAPDH antibody (#60004–1-Ig), HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H + L) (#SA00001-2), and HRP-conjugated Affinipure Goat Anti-Mouse IgG (H + L) (#SA00001-1) were obtained from Proteintech (Wuhan, Hubei, China). NR6A1 antibody (for immunofluorescence, #sc-271733) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). 2,3,5-triphenyltetrazolium chloride (TTC, #T8877) was obtained from Merck Corp. (Darmstadt, Germany). A Dual-Luciferase® Reporter Assay System (#E1910) was purchased from Promega (Madison, WI, USA). An ATP detection kit (#S0026) was purchased from Beyotime (Shanghai, China).
Animals
Gender differences in neuronal injury within experimental stroke models have been well studied [19]. Research indicates that due to the neuroprotective effects of estrogen, there are differences in susceptibility and response to ischemic stroke between male and female rodents [20,21,22]. Therefore, this study used C57BL/6 J male mice to eliminate potential neuroprotective effects influenced by estrogen. Mice (20–25 g) were obtained from Guangdong Zhiyuan Biomedical Technology Corporation. The mice were housed in specific pathogen-free environment (22 ℃ ± 2 ℃, 12-h light/dark cycle) with free access to food and water. The experiments were approved by the Laboratory Animal Ethics Committee of Southern Medical University. Animal studies were conducted in compliance with the NIH Guide for the Care and Use of Laboratory Animals (NIH, revised 1996) and ARRIVE guidelines [23]. Power calculations were performed to determine the lowest number of mice. Using the two-tailed test, a sample of 6 mice per group was needed to detect a difference in infarct volume between the NC and sh-NR6A1 groups by TTC staining with 95% confidence and 80% power. The experiments were performed in a blinded manner: animals were randomly assigned to each group via random numbers generated via Excel software; the experimenters who conducted the surgery were blinded to the animal treatment conditions; and the observers with neurological deficit scores were blinded to the animal group assignments and treatment conditions.
Middle Cerebral Artery Occlusion/Reperfusion (MCAO/R) Model
The MCAO/R model establishment was established according to previously described methods [12]. Mice were anesthetized with isoflurane. After an incision was made, the internal carotid artery (ICA) was carefully exposed, and a 0.20-mm silicone-coated filament was inserted through the ICA to block the middle cerebral artery. During surgery, the body temperature of the mice was maintained at 37 ± 0.5 ℃ via a heated blanket. After 1 h of MCAO, the filament was removed for reperfusion. The mice were excluded from the experiments if they met the following conditions: neurological deficit score = 0, hemorrhage, or death.
Western Blot
Brain tissues and cell samples were lysed in RIPA buffer. Protein samples were boiled in 1 × reducing loading buffer and separated using SDS-PAGE. The proteins were transferred to 0.22 µm PVDF membranes. The membranes were blocked with 5% skim milk in TBST at room temperature (RT) for 2 h, incubated with primary antibodies at 4 ℃ overnight, and washed with TBST and incubated with HRP-conjugated antibodies at RT for 1 h. Finally, the membranes were washed with TBST and exposed to ECL, and immunoreactivity was detected via a chemiluminescence imager (5200, Tanon, Shanghai, China).
Immunofluorescence
Mouse brains were harvested via perfusion with 0.9% saline and 4% PFA. The brains were continuously fixed with 4% PFA overnight and then transferred to 30% sucrose until they sank to the bottom. The brains were subsequently cut into 25-µm sections via a freezing microtome (Leica, Germany). The slices were blocked with 5% BSA and 0.5% Triton X-100 in PBS at RT for 2 h. After being washed with PBS 5 times, the slices were incubated with antibodies against NR6A1 and NeuN at 4 ℃ overnight. Then, the slices were incubated with secondary antibodies at RT for 2 h, washed with PBS 5 times, and mounted using with Mounting Medium With DAPI. The infarct ipsilateral cortex penumbra was defined as the cortex near the NeuN+ cell loss area and was visualized via a confocal microscope (ECLIPSE Ti, Nikon). The NR6A1 fluorescence inside NeuN+ cell was qualified using Image J software.
Culture of HT-22 Neuronal Cells
HT-22 neuron cell line is commonly used for the study of central nervous system (CNS) disorders, including cerebral ischemia [24]. HT-22 cells were cultured as previously described [12]. Briefly, HT-22 cells were cultured in DMEM containing 10% FBS in a humidified atmosphere (5% CO2 + 95% air, 37 ℃). HT-22 cells were passaged every 2 days, and the passages within 15 were used for the experiments.
Knockdown of NR6A1 and MFN2 in HT-22 Cells
In vitro silencing of NR6A1 or MFN2 in HT-22 cells was achieved via the transfection of siRNAs specific for NR6A1 (si-NR6A1) or MFN2 (si-MFN2). The target sequence for NR6A1 mRNA was as follows: CACCUAUGUUGAUUGAAGAUG. The target sequence for MFN2 mRNA was as follows: GCGCCAGCUUCCUUGAAGA. Briefly, negative control (NC) or si-NR6A1 was incubated with P3000 reagent for 5 min and mixed with Lipofectamine 3000 reagent. After 20 min of incubation, the mixture was added to HT-22 cells, and the cells were cultured for 24 h. The knockdown efficacy was verified by western blot.
Overexpression of NR6A1 in HT-22 Cells
In vitro overexpression of NR6A1 in HT-22 cells was achieved via transfection with the NR6A1 plasmid. Briefly, the vector or NR6A1 plasmid was incubated with P3000 reagent for 5 min, and then mixed with Lipofectamine 3000 reagent. After 20 min of incubation, this mixture was added to HT-22 cells, and the cells were cultured for 24 h. The overexpression efficacy was determined by western blot.
Oxygen and Glucose Deprivation/Reperfusion (OGD/R) Model
The OGD/R cell model was established as previously described [25]. HT-22 cells were washed with PBS, and the medium was changed to glucose-free DMEM. Then, the cells were transferred to a hypoxia chamber (Billups-Rothenberg, Inc., San Diego, CA, USA) filled with 95% N2 and 5% CO2, and the chamber was maintained at 37 ℃ for 6 h. After OGD insult, the medium was changed to normal DMEM, and the cells were maintained in a normoxic environment for reoxygenation.
Detection of Cell Viability
The variance of cell viability was detected by using the CCK-8 assay as previously reported [25]. Briefly, HT-22 cells were exposed to OGD/R insult. At 24 h after reoxygenation, the cells were incubated with CCK-8 reagent for 4 h. Finally, the OD value (450 nm) was detected with a microplate reader (Synergy HT; Biotech, Winooski, VT, USA).
Calcein/PI Staining
Calcein/PI staining was used to visualize cell death after OGD/R insult. Briefly, at 24 h after OGD/R insult, HT-22 cells were incubated with Caclein-AM and PI dyes for 10 min at RT. The medium was subsequently changed to Live Cell Imaging Solution. Finally, the cell death was visualized via a fluorescence microscope and calculated using Image J software.
TMRE Staining for Mitochondrial Membrane Potential (MMP) Detection
The variance in the MMP was determined by TMRE staining according to our previous report [26]. Briefly, after OGD/R insult, HT-22 cells were incubated with 100 nM TMRE dye for 10 min, and the medium was then changed to Live Cell Imaging Solution. Finally, the cells were visualized via a confocal microscope.
CellROX Staining for Analysis of Reactive Oxygen Species (ROS)
Oxidative stress inside HT-22 cells was assessed using CellROX staining as previously described [12]. Briefly, after 6 h of OGD insult, HT-22 cells were reoxygenated for 1 h. Then, the cells were incubated with CellROX reagent for 30 min. The medium was changed to Live Cell Imaging Solution. Finally, the cells were visualized via a confocal microscope.
Mito-Tracker Staining
HT-22 cells were plated on confocal dishes at a density of 50–60%. Following OGD/R, the cells were stained with Mito-tracker dye at 37 ℃ for 15 min to visualize the mitochondria in live cells. After incubation, cells were washed with PBS and examined under a confocal microscope.
Determination of the Intracellular ATP Level
The intracellular ATP level was determined as previously reported [27]. Briefly, after OGD/R treatment, lysis buffer for ATP analysis was quickly added to the cells, and the cells were lysed with shaking on ice. Then, the lysate was centrifuged at the speed of 12,000 rpm at 4 ℃ for 5 min. The supernatant was collected for ATP assays via an ATP detection kit. The ATP detection reagent was prepared by diluting the stock solution with the provided diluent at a 1:9 ratio. In each well, 100 µL of the ATP detection work solution was added, followed by the addition of 20 µL of the supernatant. The relative light units (RLU) were measured using a luminometer. ATP concentrations were determined by comparing RLU values to a standard curve generated using the ATP standard solution. The protein level of supernatant was quantified via a BCA assay. Finally, the ATP level was defined as the ATP concentration/protein concentration.
qPCR Assay
Total RNA was extracted from HT-22 cells via an RNA Easy Fast Tissue/Cell Kit. The concentration of RNA was measured via a Nano Drop 2000 system (Thermo Fisher Scientific, USA). Reverse transcription was performed to obtain cDNA via FastKing gDNA Dispelling RT SuperMix. qPCR was performed on LightCycler480 (Roche, Rotkreuz, Switzerland) using FastReal qPCR PreMix (SYBR Green). Relative mRNA expression was calculated using the 2−ΔΔCT method. The sequences of MFN2 and GAPDH primers were showed as follows: MFN2: “AGCAAGTTGACATCACCCGA” and “TGTCCAACCAGCCAGCTTTA”; GAPDH: “TGTGAACGGATTTGGCCGTA” and “ACTGTGCCGTTGAATTTGCC.”
Luciferase Activity Analysis
HT-22 cells were seeded in 12-well plates at 70–80% confluence. For the overexpression of NR6A1, the cells were co-transfected with 1000 ng/well vector or NR6A1 plasmid and 500 ng/well pGL4.10-MFN2 promoter plasmid and 500 ng/well pRL-CMV plasmid overnight. The medium then was changed to DMEM containing 1% FBS. For NR6A1 knockdown, cells were transfected with 500 ng/well pGL4.10-MFN2 promoter plasmid and 500 ng/well pRL-CMV plasmid overnight. Then, the cells were transfected with NC or siNR6A1 for 6 h, and the medium was changed to DMEM containing 1% FBS. After 24 h of culture, the cells were harvested for luciferase activity detection via the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA). The relative luciferase activity was defined as the relative value of firefly luciferase activity to renilla luciferase activity.
Silencing NR6A1 Expression in C57BL/6 J Mice
The sequence of short hairpin RNA (shRNA) targeting NR6A1 (sh-NR6A1) was “CCCAGAUGAUCGAGCUGAA.” Sh-NR6A1 or NC was and inserted into a vector (pAAV-U6-shRNA-CMV-EGFP-WPRE), and then packed into AAV2/9. Two weeks before the MCAO procedure, AAV encoding NC or sh-NR6A1 was injected into the lateral ventricle. After being anesthetized with isoflurane, the mice were fixed on a stereotaxic apparatus (Stoelting, Chicago, IL, USA). Then, AAV was injected intracerebroventricularly with a microsyringe pump (KD Scientific Inc., Holliston, MA, USA). The stereotaxic coordinate was − 0.5 mm anteroposterior, 1 mm lateral, and − 2.5 mm dorsoventral relative to the bregma. The microsyringe was held on for 10 min in case of leakage. The knockdown efficacy was verified via western blot.
Neurological Deficit Scores Measurement
Neurological deficit scores were measured at 24 h after MCAO/R insult as previously described [27]. The observers were blinded to the assignments of the experimental groups. The neurological deficit scores were as follows: “0” no neurological deficit was recorded; “1” when the mice were lifted, their paws failed to extend the contralateral forepaw freely; “2” turned to the contralateral side when the mice walked; “3” slumped to the contralateral side at rest and walking; and “4” the mice had no spontaneous walking.
Infarct Volume Detection
The infarct volume was determined via TTC staining as previously described [28]. Briefly, the brains were quickly isolated at 24 h after MCAO and frozen at − 20 ℃ for 10 min. Then, the brains were sliced coronally into 1.5-mm thick sections using the brain matrix, and incubated in 1% TTC solution for 30 min, followed by fixation in 4% PFA. Finally, the brain sections were scanned with a scanner. The infarct volume was analyzed using Image J software. For elimination of the influence of cerebral edema on the infarction calculation, the following correction formula was used: infarct volume (%) = (contralateral volume − noninfarct ipsilateral volume)/contralateral volume × 100%.
Statistical Analysis
All the data are presented as the mean ± standard deviation (SD). Statistical analysis was performed via GraphPad Prism 8.3 (GraphPad Prism Software, San Diego, CA, USA). Student’s t test was used for comparisons between two groups, and one-way analysis of variance followed by Tukey’s post hoc test was used for comparisons among more than two groups. n represents the number of independent cultures or the number of mice. P < 0.05 was defined as statistically significant. Animals were assigned to each group via the random number program in SPSS software.
Results
NR6A1 Silencing Attenuates Cerebral Ischemia–Induced Neuronal Injury
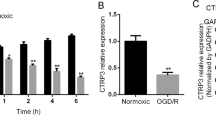
First, the expression of NR6A1 in the ischemic penumbra was determined by western blot at 3, 12, and 24 h after MCAO surgery. As shown in Fig. 1a and b, the level of NR6A1 was significantly increased in the ischemic penumbra at 3, 12, and 24 h after MCAO (P < 0.01). The expression of NR6A1 at 24 h of reperfusion in ischemic penumbra neurons was measured in the sham or MCAO/R-treated mice by immunostaining. As shown in Fig. 1c and d, the NR6A1 signal was lower in the neurons of the sham mice, whereas it was substantially greater in neurons of the MCAO/R-treated mice (P < 0.01). Next, we identified changes in NR6A1 in HT-22 neuronal cells after OGD/R. As shown in Fig. 1e and f, the expression of NR6A1 increased after OGD/R insult (P < 0.01). Then, si-NR6A1 was used to silence NR6A1 expression in HT-22 cells. The knockdown efficacy was determined by western blot (Fig. 1g and h, P < 0.01). We conducted a CCK-8 assay to identify differences in the variance of cell viability after OGD/R insult. As shown in Fig. 1i, OGD/R significantly decreased cell viability to 46.92 ± 6.96% (P < 0.01), whereas NR6A1 knockdown increased cell viability to 75.27 ± 4.57% (P < 0.01). Moreover, Calcein/PI staining was used to confirm our results. As shown in Fig. 1j and k, OGD/R caused substantial cell death, whereas NR6A1 knockdown attenuated the death of HT-22 cells (P < 0.01). Taken together, these data suggest that NR6A1 knockdown attenuates OGD/R-induced neuronal injury.

Silencing of NR6A1 ameliorates ischemia-induced neuronal injury. C57 mice was suffered from MCAO for 1 h and reperfusion for 24 h, then the mice was sacrificed. (a and b) After 3, 12, and 24 h of reperfusion, the change of NR6A1 was determined by western blot (n = 4 mice each group). (c and d) The brain was fixed with 4% PFA, and the variance of NR6A1 in neuron was visualized by double staining with NeuN and NR6A1. The intensity of NR6A1 in NeuN+ cells was qualified by Image J (n = 3 mice each group). (e and f) HT-22 cells were suffered from OGD/R insult, the variance of NR6A1 was detected by western blot, and the relative expression of NR6A1/GAPDH was calculated by densitometry of the blot (n = 3). (g and h) HT-22 cells were transfected with NC or si-NR6A1 for 24 h, then knockdown efficacy was detected by western blot, and the relative expression of NR6A1/GAPDH was measured by densitometry of the blot (n = 3). HT-22 cells pre-transfected with NC or si-NR6A1 were suffered from OGD/R insult. At 24 h after reoxygenation, (i) the cell viability was detected by CCK-8 assay (n = 4), and (j and k) the cell death percentage was visualized by Calcein/PI staining (n = 3). *P < 0.05, **P < 0.01, versus the indicated group
NR6A1 Silencing Ameliorates Mitochondrial Dysfunction and ER Stress in HT-22 Cells Following OGD/R Insult
Mitochondrial dysfunction is the major cause of neuronal death during cerebral ischemia [29]. Thus, we investigated the role of NR6A1 in mitochondrial deficit. TMRE staining was applied to determine the change in the MMP. Treatment with OGD/R significantly reduced the MMP, and NR6A1 knocked down the MMP (Fig. 2a and b, P < 0.01). Mito-tracker staining was applied to visualize the morphological changes in the mitochondria. As shown in Fig. 2c and d, OGD/R induced mitochondrial fragmentation and loss, and NR6A1 silencing attenuated the fragmentation and loss of mitochondria (P < 0.01). We also detected the variance in the intracellular ATP level. OGD/R insult caused a decrease in the ATP level to 56.59 ± 6.20% (Fig. 2e, P < 0.01), whereas NR6A1 silencing increased ATP level to 82.76 ± 16.30% (P < 0.05). As mitochondria are the major source of ROS, we detected the variance in intracellular ROS levels via CellROX staining. As shown in Fig. 2f and g, OGD/R caused an increase in the ROS signal (P < 0.01), whereas NR6A1 silencing decreased the ROS level (P < 0.01). Overaccumulation of ROS contributes to ER stress, which causes neuronal death [30]. Thus, we further studied the effect of NR6A1 silencing in OGD/R-induced ER stress. We found that OGD/R induced ER stress, as evidenced by increased levels of p-IRE1α (Fig. 2h and i, P < 0.01), p-JNK (Fig. 2h and j, P < 0.01), and p-eIF2α (Fig. 2h and k, P < 0.01), whereas NR6A1 silencing resulted in decreased levels of p-IRE1α (P < 0.01), p-JNK (P < 0.01), and p-eIF2α (P < 0.01). These results indicate that NR6A1 silencing attenuated OGD/R-induced neuronal mitochondrial dysfunction and ER stress.

Silencing of NR6A1 attenuates ER stress and mitochondrial dysfunction following OGD/R challenge. HT-22 cells pre-transfected with NC or si-NR6A1 were suffered from OGD/R insult. At 1 h after oxygenation, (a and b) the change of MMP was detected by TMRE staining (n = 3); (c and d) the morphological change was visualized by Mito-tracker staining (n = 3); (e) the change of intracellular ATP level was detected (n = 4); (f and g) the change of intracellular ROS was determined by CellROX staining (n = 3); (h–k) the variance of p-IRE1α, IRE1α, p-JNK, JNK, p-eIF2α, and eIF2α was detected by western blot, and the relative expression of p-IRE1α/IRE1α (n = 3), p-JNK/JNK (n = 4), and p-eIF2α/eIF2α (n = 4) was calculated by densitometry of the blot. *P < 0.05, **P < 0.01, versus the indicated group
Overexpression of NR6A1 Exacerbates Neuronal Injury After OGD/R Insult
Above results confirmed the role of NR6A1 silencing in neuronal injury after OGD/R insult. Next, we investigated the effect of NR6A1 overexpression in HT-22 cells. First, the overexpression efficacy was confirmed by western blot (Fig. 3a and b, P < 0.01). We found that NR6A1 overexpression had no effect on cell viability, and exacerbated the decrease in cell viability from 53.64 ± 7.29 to 31.46 ± 5.41% after OGD/R insult (Fig. 3c, P < 0.01). CellROX staining was used to determine intracellular ROS levels. We found that NR6A1 overexpression exacerbated the accumulation of intracellular ROS after OGD/R insult (Fig. 3d and e, P < 0.05). Finally, we showed that NR6A1 overexpression exacerbated ER stress, as evidenced by increased levels of p-IRE1α (Fig. 3f and g, P < 0.05) and p-eIF2α (Fig. 3f and h, P < 0.05). Collectively, these results indicate that the overexpression of NR6A1 exacerbates OGD/R-induced neuronal injury.

NR6A1 overexpression aggravates OGD/R-induced neuronal injury. (a and b) HT-22 cells were transfected with vector or NR6A1 plasmid. At 24 h after transfection, the expression of NR6A1 was verified by western blot, and the relative expression of NR6A1/GAPDH was calculated by densitometry of the blot (n = 3). (c) HT-22 cells were transfected with vector or NR6A1 plasmid, then cells were suffered from OGD/R insult. At 24 h after reoxygenation, cell viability was detected by CCK8 assay (n = 3). (d and e) HT-22 cells were transfected with vector or NR6A1 plasmid, then cells were suffered from OGD/R insult. At 1 h after reoxygenation, intracellular ROS level was measured by CellROX staining and quantified by Image J (n = 4). (f and g) HT-22 cells were transfected with vector or NR6A1 plasmid, then cells were suffered from OGD/R insult. The variance of p-IRE1α, IRE1α, p-eIF2α, and eIF2α was detected by western blot, and the relative expression of p-IRE1α/IRE1α (n = 3) and p-eIF2α/eIF2α (n = 3) was calculated by densitometry of the blot. *P < 0.05, **P < 0.01, versus the indicated group
NR6A1 Reduces MFN2 Expression by Inhibiting Its Transcription
MFN2 is important for maintaining ER and mitochondrial function [8]. Our previous study revealed that OGD/R caused a reduction in MFN2 mRNA expression [12]. And NR6A1 has been identified as a transcriptional repressor [31]. Thus, we investigated whether NR6A1 regulates MFN2 expression after OGD/R insult. As shown in Fig. 4a and b, NR6A1 knockdown rescued MFN2 loss caused by OGD/R (P < 0.01). We found that overexpression of NR6A1 further induced MFN2 loss (Fig. 4c and d, P < 0.05). Next, we determined MFN2 mRNA expression by qPCR. As shown in Fig. 4e and f, knockdown of NR6A1 increased MFN2 mRNA expression (P < 0.01), whereas NR6A1 overexpression reduced MFN2 mRNA expression (P < 0.01). Finally, a luciferase assay was used to detect the changes in MFN2 promoter activity after NR6A1 was silenced or overexpressed. We showed that NR6A1 knockdown increased MFN2 promoter activity (Fig. 4g, P < 0.05) and that NR6A1 overexpression decreased MFN2 promoter activity (Fig. 4h, P < 0.05). Hence, our results indicate that NR6A1 reduces MFN2 expression by inhibiting its transcription.

NR6A1 downregulates MFN2 expression. (a and b) HT-22 cells pre-transfected with NC or si-NR6A1 were suffered from OGD/R insult. The variance of NR6A1 was detected by western blot, and the relative expression of NR6A1/GAPDH was calculated by densitometry of the blot (n = 4). (c and d) HT-22 cells pre-transfected with vector or NR6A1 plasmid were suffered from OGD/R insult. The variance of NR6A1 was detected by western blot, and the relative expression of NR6A1/β-tubulin was calculated by densitometry of the blot (n = 3). (e) HT-22 cells were transfected with siNR6A1 or NC, then the change of MFN2 mRNA was detected by qPCR (n = 3). (f) HT-22 cells were transfected with vector or NR6A1 plasmid, then the change of MFN2 mRNA was detected by qPCR (n = 4). (g) HT-22 cells were transfected with siNR6A1 or NC, then the variance in MFN2 promoter activity was detected by luciferase assay (n = 4). (h) HT-22 cells were transfected with vector or NR6A1 plasmid, then the variance in MFN2 promoter activity was detected by luciferase assay (n = 3). *P < 0.05, **P < 0.01, versus the indicated group
MFN2 Mediates the Protective Effects of NR6A1 Silencing Against Mitochondrial Dysfunction and ER Stress
We further investigated whether MFN2 mediates the protective effect of NR6A1 knockdown. First, western blot was used to investigate the variance of MFN2 after OGD/R. As shown in Fig. 5a and b, NR6A1 knockdown rescued the loss of MFN2 (P < 0.01), whereas si-MFN2 blocked the rescue effect of NR6A1 silencing (P < 0.01). A CCK-8 assay was subsequently used to detect the change of cell viability. As shown in Fig. 5c, si-MFN2 blocked the ability of NR6A1 silencing to increase cell viability (P < 0.01). TMRE and CellROX staining were used to detect changes in the MMP and ROS. Knockdown of MFN2 abolished the protective effect of si-NR6A1 on MMP loss (Fig. 5d and e, P < 0.05) and intracellular ROS accumulation (Fig. 5f and g, P < 0.01) after OGD/R. Moreover, si-MFN2 abolished the protective effect of NR6A1 silencing against ER stress, as demonstrated by the increased levels of p-IRE1α (Fig. 5h and i, P < 0.01), p-JNK (Fig. 5j and k, P < 0.01), and p-eIF2α (Fig. 5l and m, P < 0.01). These data indicate that MFN2 is crucial for the protective effect of NR6A1 silencing on OGD/R-induced neuronal damage.

MFN2 mediates the protective effect of NR6A1 silencing against OGD/R-induced ER stress and mitochondrial dysfunction. HT-22 cells pre-transfected with NC or si-MFN2 or si-NR6A1 were suffered from OGD/R insult. (a and b) The variance of MFN2 was detected by western blot, and the relative expression of MFN2/β-tubulin was calculated by densitometry of the blot (n = 3). (c) At 24 h after reoxygenation, the change of cell viability was detected by CCK-8 assay (n = 3). (d and e) At 1 h after reoxygenation, the variance of MMP was detected by TMRE staining (n = 3). (f and g) At 1 h after reoxygenation, the variance of intracellular ROS was detected by CellROX staining (n = 3). (h–m) The variance of p-IRE1α, IRE1α, p-JNK, JNK, p-eIF2α, and eIF2α was detected by western blot, and the relative expression of p-IRE1α/IRE1α (n = 3), p-JNK/JNK (n = 4), and p-eIF2α/eIF2α (n = 3) was calculated by densitometry of the blot. *P < 0.05, **P < 0.01, versus the indicated group
NR6A1 Knockdown Protects C57BL/6 J Mice from Cerebral Ischemia
We showed that the knockdown of NR6A1 protected against ischemia/reperfusion-induced injury in vitro. Next, we studied the protective effect of NR6A1 knockdown in vivo. We knocked down NR6A1 expression by injecting AAV-sh-NR6A1 intracerebroventricularly (i.c.v.). We confirmed that this type of AAV can be used to transfect neurons and that intracerebroventricular injection of AAV can be used to transfect the cortex, stratum, and hippocampus [12]. The experimental schedule is illustrated in Fig. 6a. Western blot was used to verify the efficacy of NR6A1 silencing in the cortex (Fig. 6b and c). We established an ischemia/reperfusion model by induction of MCAO/R in C57 mice. At 24 h after reperfusion, the neurological deficit scores of the mice were measured. As shown in Fig. 6d, MCAO/R induced neurological disorders in the mice, whereas NR6A1 knockdown reversed these disorders. After evaluating mouse neurological function, we detected infarction in C57 mice using TTC staining. MCAO insult increased the infarct volume to 46.47 ± 10.26% (Fig. 6e and f, P < 0.05), whereas NR6A1 knockdown reduced the infarct volume to 20.02 ± 7.13% (P < 0.05). We further verified the effect of NR6A1 knockdown on MFN2 expression in vivo. Consistent with the in vitro results, NR6A1 knockdown rescued the loss of MFN2 induced by MCAO/R (Fig. 6g and h, P < 0.05). We also found that NR6A1 knockdown attenuated MCAO/R-induced ER stress and apoptosis, as evidenced by the reduced levels of p-eIF2α (Fig. 6i and j, P < 0.05) and cleaved caspase 3 (Fig. 6k and l, P < 0.05). In general, these data indicate that NR6A1 knockdown attenuates MCAO/R-induced neuronal damage in C57 mice.

Silencing of NR6A1 protectes against MCAO/R-induced C57 mice. (a) The diagram of the in vivo experiment. (b and c) C57 mice were injected with NC or sh-NR6A1, then the knockdown efficacy in cortex was verified by western blot (n = 3 per group). C57 mice was suffered from MCAO for 1 h and reperfusion for 24 h; (d) the neurological deficit scores were measured (n = 7 per group); (e and f) then the mice was sacrificed; the TTC staining was used to detect infarct volume (n = 7 per group). (g–l) C57 mice was suffered from MCAO for 1 h and reperfusion for 24 h; the variance of MFN2, p-eIF2α, and caspase 3 was detected by western blot, and the relative expression of MFN2/GAPDH (n = 3 per group), p-eIF2α/eIF2α (n = 3 per group), cleaved caspase 3/caspase 3 (n = 3 per group) was calculated by densitometry of the blot. *P < 0.05, **P < 0.01, versus the indicated group
Discussion
In this study, our objective was to explore the contribution of NR6A1 to neuronal death after ischemia. We observed the upregulation of NR6A1 in neurons in both in vitro and in vivo experimental stroke models. Knockdown of NR6A1 mitigated mitochondrial dysfunction and ER stress in HT-22 cells subjected to OGD/R, whereas NR6A1 overexpression exacerbated neuronal damage. Notably, our results demonstrate that NR6A1 impedes the transcription of MFN2 and reduces its expression. MFN2 conferred the protective effect of NR6A1 silencing against neuronal injury. Importantly, NR6A1 silencing attenuated brain infarction, ER stress, neuronal apoptosis, and loss of MFN2 in mice subjected to MCAO/R. A summary of our findings is shown in Fig. 7. Overall, our findings underscore the essential role of MFN2 in the protective effect of NR6A1 silencing against neuronal injury after cerebral ischemia. To the best of our knowledge, this research represents the first exploration into the function of NR6A1 in the context of cerebral ischemia, as well as the regulatory interplay between NR6A1 and MFN2.

Protective effect of NR6A1 silencing against ischemia-induced neuronal injury. The role of NR6A1 in the progression of neuronal death induced by ischemic stroke remains unknown. In this study, we found that NR6A1 hinders the transcription of MFN2, while MFN2 conferred the protective effect of NR6A1 silencing against both mitochondrial dysfunction and ER stress. These findings indicate that targeting NR6A1 holds potential as a viable strategy for the treatment of ischemic stroke
A recent study revealed that an increase in NR6A1 expression within the hippocampus can induce depressive-like behavior in mice [18]. This finding suggests the potential involvement of NR6A1 in the neuronal dysfunction characteristic of depressive states. However, the role of NR6A1 in cerebral ischemia remains unknown. Research conducted by Cai et al. revealed that NR6A1 contributes to the apoptosis of vascular smooth muscle cells [17]. Inspired by these pivotal studies, we propose that NR6A1 could play a role in neuronal death following cerebral ischemia. Herein, we showed that the NR6A1 knockdown ameliorated neuronal injury induced by cerebral ischemia. Conversely, the overexpression of NR6A1 intensified the neuronal damage caused by OGD/R. Our findings reveal for the first time that NR6A1 is involved in neuronal damage following cerebral ischemia. This finding innovatively reveals the influence of NR6A1 on the determination of neuronal survival and positions NR6A1 as a potential therapeutic target for ameliorating neuronal death. Inhibiting NR6A1 might be a beneficial strategy for the treatment of cerebral ischemia. However, to date, NR6A1 inhibitors are lacking. Moreover, the scope of this effect, whether it is exclusive to cerebral ischemia–induced neuronal death, warrants further investigation.
Post-stroke depression (PSD) is a prevalent mood disorder that impacts the patient’s recovery and quality of life following stroke, although its precise etiology remains elusive [32]. Previously, a report has shown that NR6A1 aggregated the pathological of depression [18]. As our study demonstrates, NR6A1 contributes to the neuronal injury of cerebral ischemia and MFN2 is essential for the protective effect of NR6A1 silencing. Additionally, MFN2 in neurons has been reported to act as a suppressor of depressive-like behavior in mice [33]. Thus, we propose that NR6A1 might be the contributor of PSD and that MFN2 might confer to the protective effect of NR6A1 silencing against PSD. While our study indicates that targeting NR6A1 holds therapeutic potential in stroke treatment, we recognize the necessity to consider potential adverse effects carefully. NR6A1 is implicated in various physiological processes, such as stem cell pluripotency regulation [14], lipid metabolism regulation [15], and cancer cell proliferation and migration [16]. Dysregulation of NR6A1 might lead to unintended consequences. Further studies are warranted to evaluate the long-term effects of NR6A1 modulation on other physiological systems.
Mitochondrial dysfunction and ER stress are two crucial pathological changes induced by cerebral ischemia [4]. Our research revealed that NR6A1 knockdown diminishes the adverse effects of cerebral ischemia on mitochondrial integrity and ER stress. Given that the dynamic interactions between mitochondria and the ER dictate cellular outcomes [34], NR6A1 may play a role in the cross-talk between these two organelles. Consequently, the potential of NR6A1 to influence ER-mitochondrial tethering during cerebral ischemia warrants additional inquiry. Furthermore, our data suggest that NR6A1 suppresses the expression of MFN2, a protein that is integral to maintaining ER-mitochondria contact [7]. Our results further reveal that MFN2 confers the protective effect of NR6A1 silencing against cerebral ischemia. This finding suggests that MFN2 is a key factor in how NR6A1 modulates the interaction between the ER and mitochondria, a topic that requires further investigation. It is currently unclear whether the impact of NR6A1 on ER stress and mitochondrial dysfunction is specific to cerebral ischemia or if it extends to other CNS disorders. Therefore, exploring NR6A1 as a possible therapeutic target for the treatment of neuronal mitochondrial dysfunction and ER stress across a broader spectrum of CNS diseases is important.
In our study, we observed that the knockdown of NR6A1 alleviates IRE1α/JNK and eIF2α pathways activation under conditions of OGD/R. Notably, the activation of IRE1 and the subsequent processing of XBP1α mRNA are recognized as protective mechanisms in the context of cerebral ischemia, as indicated in previous studies [35, 36]. Consequently, it is essential for future research to delve into the impact of NR6A1 on the IRE1α/XBP1 axis and to elucidate the underlying mechanisms. This will contribute to a more comprehensive understanding of the ER stress pathways. The increase in pIRE1α levels following si-MFN2 treatment, despite the silencing of NR6A1, suggests a complex interplay between mitochondrial dynamics and ER stress responses. Furthermore, our findings indicate that in the context of MFN2 deficiency, this IRE1α/JNK pathway might play a role in negating the protective effects of NR6A1 knockdown against ER stress. Further exploration is necessary to fully grasp these intricate mechanisms and their potential implications for the pathophysiology of stroke.
Understanding the influence of NR6A1 on MFN2 expression is essential, especially given that MFN2 is key to the neuroprotective effects of NR6A1 silencing in cerebral ischemia. Our study revealed that NR6A1 knockdown led to increased MFN2 transcription. Conversely, overexpression of NR6A1 had an inhibitory effect on MFN2 transcription. These findings suggest that the silencing of NR6A1 promotes MFN2 expression through enhancing transcription after cerebral ischemia, thereby mitigating neuronal damage. Our results innovatively reveal that NR6A1 inhibits MFN2 transcription. However, the exact mechanisms by which NR6A1 regulates MFN2 transcription remain unknown. Functionally, NR6A1 acts as a transcriptional repressor, attaching to the DR0 response element in the promoters of target genes to repress their transcription [37]. NR6A1 was also reported to inhibit the transcriptional activity of CREB [38]. Since CREB is implicated in the upregulation of PGC1α [39], which can promote MFN2 transcription [40], it remains unknown whether NR6A1 affects MFN2 transcription directly or via this CREB/PGC1α/MFN2 axis. Thus, further investigation is needed to ascertain whether the modulation of MFN2 by NR6A1 is direct or indirect.
In conclusion, our findings contribute to the current understanding of stroke pathogenesis by identifying NR6A1 as a novel molecular target potentially involved in neuronal injury post-stroke. The study indicates that targeting NR6A1 could represent a promising therapeutic strategy for treating neuronal death following cerebral ischemia. By modulating NR6A1 levels, it may be feasible to influence the expression of MFN2, and thereby alleviating the effects of mitochondrial dysfunction and ER stress. This strategy could provide a novel avenue for the treatment of stroke.
Data Availability
The data which support this study are available from the corresponding authors on reasonable request.
References
Campbell BCV, Khatri P (2020) Stroke. Lancet 396(10244):129–142. https://doi.org/10.1016/s0140-6736(20)31179-x
Tsivgoulis G, Katsanos AH, Sandset EC, Turc G, Nguyen TN, Bivard A, Fischer U, Khatri P (2023) Thrombolysis for acute ischaemic stroke: current status and future perspectives. Lancet Neurol 22(5):418–429. https://doi.org/10.1016/s1474-4422(22)00519-1
Wang H, Gaur U, Xiao J, Xu B, Xu J, Zheng W (2018) Targeting phosphodiesterase 4 as a potential therapeutic strategy for enhancing neuroplasticity following ischemic stroke. Int J Biol Sci 14(12):1745–1754. https://doi.org/10.7150/ijbs.26230
Jiang RQ, Li QQ, Sheng R (2023) Mitochondria associated ER membranes and cerebral ischemia: molecular mechanisms and therapeutic strategies. Pharmacol Res 191:106761. https://doi.org/10.1016/j.phrs.2023.106761
Khan MM, Paez HG, Pitzer CR, Alway SE (2023) The therapeutic potential of mitochondria transplantation therapy in neurodegenerative and neurovascular disorders. Curr Neuropharmacol 21(5):1100–1116. https://doi.org/10.2174/1570159x05666220908100545
Guo S, Wehbe A, Syed S, Wills M, Guan L, Lv S, Li F, Geng X, Ding Y (2023) Cerebral glucose metabolism and potential effects on endoplasmic reticulum stress in stroke. Aging Dis 14(2):450–467. https://doi.org/10.14336/ad.2022.0905
Larrea D, Pera M, Gonnelli A, Quintana-Cabrera R, Akman HO, Guardia-Laguarta C, Velasco KR, Area-Gomez E et al (2019) MFN2 mutations in Charcot-Marie-Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum Mol Genet 28(11):1782–1800. https://doi.org/10.1093/hmg/ddz008
Filadi R, Greotti E, Pizzo P (2018) Highlighting the endoplasmic reticulum-mitochondria connection: focus on Mitofusin 2. Pharmacol Res 128:42–51. https://doi.org/10.1016/j.phrs.2018.01.003
Jiang S, Nandy P, Wang W, Ma X, Hsia J, Wang C, Wang Z, Niu M et al (2018) Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Mol Neurodegener 13(1):5. https://doi.org/10.1186/s13024-018-0238-8
Martorell-Riera A, Segarra-Mondejar M, Muñoz JP, Ginet V, Olloquequi J, Pérez-Clausell J, Palacín M, Reina M et al (2014) Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death. Embo J 33(20):2388–2407. https://doi.org/10.15252/embj.201488327
Zeng KW, Wang JK, Wang LC, Guo Q, Liu TT, Wang FJ, Feng N, Zhang XW et al (2021) Small molecule induces mitochondrial fusion for neuroprotection via targeting CK2 without affecting its conventional kinase activity. Signal Transduct Target Ther 6(1):71. https://doi.org/10.1038/s41392-020-00447-6
Xu BT, Li MF, Chen KC, Li X, Cai NB, Xu JP, Wang HT (2023) Mitofusin-2 mediates cannabidiol-induced neuroprotection against cerebral ischemia in rats. Acta Pharmacol Sin 44(3):499–512. https://doi.org/10.1038/s41401-022-01004-3
Safe S, Jin UH, Hedrick E, Reeder A, Lee SO (2014) Minireview: Role of orphan nuclear receptors in cancer and potential as drug targets. Mol Endocrinol 28(2):157–172. https://doi.org/10.1210/me.2013-1291
Fuhrmann G, Chung AC, Jackson KJ, Hummelke G, Baniahmad A, Sutter J, Sylvester I, Schöler HR et al (2001) Mouse germline restriction of Oct4 expression by germ cell nuclear factor. Dev Cell 1(3):377–387. https://doi.org/10.1016/s1534-5807(01)00038-7
Wang Y, Wan X, Hao Y, Zhao Y, Du L, Huang Y, Liu Z, Wang Y et al (2019) NR6A1 regulates lipid metabolism through mammalian target of rapamycin complex 1 in HepG2 cells. Cell Commun Signal 17(1):77. https://doi.org/10.1186/s12964-019-0389-4
Liu X, Fan Z, Li Y, Li Z, Zhou Z, Yu X, Wan J, Min Z et al (2020) microRNA-196a-5p inhibits testicular germ cell tumor progression via NR6A1/E-cadherin axis. Cancer Med 9(23):9107–9122. https://doi.org/10.1002/cam4.3498
Cai Z, He X, Liu S, Bai Y, Pan B, Wu K (2022) Linear ubiquitination modification of NR6A1 by LUBAC inhibits RIPK3 kinase activity and attenuates apoptosis of vascular smooth muscle cells. J Biochem Mol Toxicol 36(8):e23091. https://doi.org/10.1002/jbt.23091
Tan P, Xue T, Wang Y, Hu Z, Su J, Yang R, Ji J, Ye M et al (2022) Hippocampal NR6A1 impairs CREB-BDNF signaling and leads to the development of depression-like behaviors in mice. Neuropharmacology 209:108990. https://doi.org/10.1016/j.neuropharm.2022.108990
Shao B, Cheng Y, Jin K (2012) Estrogen, neuroprotection and neurogenesis after ischemic stroke. Curr Drug Targets 13(2):188–198. https://doi.org/10.2174/138945012799201702
Selvaraj UM, Zuurbier KR, Whoolery CW, Plautz EJ, Chambliss KL, Kong X, Zhang S, Kim SH et al (2018) Selective nonnuclear estrogen receptor activation decreases stroke severity and promotes functional recovery in female mice. Endocrinology 159(11):3848–3859. https://doi.org/10.1210/en.2018-00600
Li J, Siegel M, Yuan M, Zeng Z, Finnucan L, Persky R, Hurn PD, McCullough LD (2011) Estrogen enhances neurogenesis and behavioral recovery after stroke. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 31(2):413–425. https://doi.org/10.1038/jcbfm.2010.181
Merchenthaler I, Dellovade TL, Shughrue PJ (2003) Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci 1007:89–100. https://doi.org/10.1196/annals.1286.009
Lilley E, Stanford SC, Kendall DE, Alexander SPH, Cirino G, Docherty JR, George CH, Insel PA et al (2020) ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. Br J Pharmacol 177(16):3611–3616. https://doi.org/10.1111/bph.15178
Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y et al (2019) Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177(5):1262-1279.e1225. https://doi.org/10.1016/j.cell.2019.03.032
Xu B, Qin Y, Li D, Cai N, Wu J, Jiang L, Jie L, Zhou Z et al (2020) Inhibition of PDE4 protects neurons against oxygen-glucose deprivation-induced endoplasmic reticulum stress through activation of the Nrf-2/HO-1 pathway. Redox Biol 28:101342. https://doi.org/10.1016/j.redox.2019.101342
Li M, Xu B, Li X, Li Y, Qiu S, Chen K, Liu Z, Ding Y et al (2022) Mitofusin 2 confers the suppression of microglial activation by cannabidiol: insights from in vitro and in vivo models. Brain Behav Immun 104:155–170. https://doi.org/10.1016/j.bbi.2022.06.003
Xu B, Wang T, Xiao J, Dong W, Wen HZ, Wang X, Qin Y, Cai N et al (2019) FCPR03, a novel phosphodiesterase 4 inhibitor, alleviates cerebral ischemia/reperfusion injury through activation of the AKT/GSK3β/ β-catenin signaling pathway. Biochem Pharmacol 163:234–249. https://doi.org/10.1016/j.bcp.2019.02.023
Xu B, Xu J, Cai N, Li M, Liu L, Qin Y, Li X, Wang H (2021) Roflumilast prevents ischemic stroke-induced neuronal damage by restricting GSK3β-mediated oxidative stress and IRE1α/TRAF2/JNK pathway. Free Radic Biol Med 163:281–296. https://doi.org/10.1016/j.freeradbiomed.2020.12.018
Hao S, Huang H, Ma RY, Zeng X, Duan CY (2023) Multifaceted functions of Drp1 in hypoxia/ischemia-induced mitochondrial quality imbalance: from regulatory mechanism to targeted therapeutic strategy. Mil Med Res 10(1):46. https://doi.org/10.1186/s40779-023-00482-8
Ma XX, Xie HY, Hou PP, Wang XJ, Zhou W, Wang ZH (2023) Nuclear factor erythroid 2-related factor 2 is essential for low-normobaric oxygen treatment-mediated blood-brain barrier protection following ischemic stroke. Mol Neurobiol. https://doi.org/10.1007/s12035-023-03767-0
Wang H, Wang X, Xu X, Kyba M, Cooney AJ (2016) Germ cell nuclear factor (GCNF) represses Oct4 expression and globally modulates gene expression in human embryonic stem (hES) cells. J Biol Chem 291(16):8644–8652. https://doi.org/10.1074/jbc.M115.694208
Villa RF, Ferrari F, Moretti A (2018) Post-stroke depression: mechanisms and pharmacological treatment. Pharmacol Ther 184:131–144. https://doi.org/10.1016/j.pharmthera.2017.11.005
Gebara E, Zanoletti O, Ghosal S, Grosse J, Schneider BL, Knott G, Astori S, Sandi C (2021) Mitofusin-2 in the nucleus accumbens regulates anxiety and depression-like behaviors through mitochondrial and neuronal actions. Biol Psychiat 89(11):1033–1044. https://doi.org/10.1016/j.biopsych.2020.12.003
Liu Y, Qiao Y, Pan S, Chen J, Mao Z, Ren K, Yang Y, Feng Q et al (2023) Broadening horizons: the contribution of mitochondria-associated endoplasmic reticulum membrane (MAM) dysfunction in diabetic kidney disease. Int J Biol Sci 19(14):4427–4441. https://doi.org/10.7150/ijbs.86608
Jiang M, Yu S, Yu Z, Sheng H, Li Y, Liu S, Warner DS, Paschen W et al (2017) XBP1 (X-box-binding protein-1)-dependent O-GlcNAcylation is neuroprotective in ischemic stroke in young mice and its impairment in aged mice is rescued by thiamet-G. Stroke 48(6):1646–1654. https://doi.org/10.1161/strokeaha.117.016579
Wang Z, Li X, Spasojevic I, Lu L, Shen Y, Qu X, Hoffmann U, Warner DS et al (2021) Increasing O-GlcNAcylation is neuroprotective in young and aged brains after ischemic stroke. Exp Neurol 339:113646. https://doi.org/10.1016/j.expneurol.2021.113646
Gu P, Morgan DH, Sattar M, Xu X, Wagner R, Raviscioni M, Lichtarge O, Cooney AJ (2005) Evolutionary trace-based peptides identify a novel asymmetric interaction that mediates oligomerization in nuclear receptors. J Biol Chem 280(36):31818–31829. https://doi.org/10.1074/jbc.M501924200
Wang Y, Zhang Y, Dai X, Liu Z, Yin P, Wang N, Zhang P (2015) NR6A1 couples with cAMP response element binding protein and regulates vascular smooth muscle cell migration. Int J Biochem Cell Biol 69:225–232. https://doi.org/10.1016/j.biocel.2015.10.026
Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G et al (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413(6852):179–183. https://doi.org/10.1038/35093131
Hu L, Guo Y, Song L, Wen H, Sun N, Wang Y, Qi B, Liang Q et al (2022) Nicotinamide riboside promotes Mfn2-mediated mitochondrial fusion in diabetic hearts through the SIRT1-PGC1α-PPARα pathway. Free Radic Biol Med 183:75–88. https://doi.org/10.1016/j.freeradbiomed.2022.03.012
Funding
This work was supported by the Science and Technology Projects of Guangzhou (Grant Nos. 2024A04J4016, 2024A03J0965, 202201010748, 202201010163, and 2023A03J0960) and the National Natural Science Foundation of China (Grant No. 82371364).
Author information
Authors and Affiliations
Contributions
Yuanyuan Wang and Bingtian Xu conceived the study and designed the experiments. Honghao Wang, Haitao Wang, and Jiangping Xu provided experimental facilities and helped conceive the study. Bingtian Xu, Zhuhe Liu, Kechun Chen, Qian Zhao, Haixia Wen, and Jingfang Lin performed the experiments and analyzed the data. Bingtian Xu, Zhuhe Liu, and Yuanyuan Wang wrote this manuscript. Yuanyuan Wang revised this manuscript. All authors have agreed to final version of this manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
All animal experiments were conducted in accordance with the guidelines approved by the Laboratory Animal Ethics Committee of Southern Medical University (approval number: SMUL2022195).
Consent to Participate
Not applicable.
Consent for Publication
All authors approved the final manuscript and the submission.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, B., Liu, Z., Chen, K. et al. Mitofusin 2 Mediates the Protective Effect of NR6A1 Silencing Against Neuronal Injury in Experimental Stroke Models. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04466-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04466-0