
Abstract
C1q/tumor necrosis factor-related protein-3 (CTRP3) had shown its angiogenesis and enhancement of mitochondrial biogenesis properties in the treatment of myocardial infarction, but its potential roles in cerebral ischemic stroke had not been fully understood. This study aimed to clarify the underlying mechanism of how CTRP3 regulated mitochondrial functions in hippocampal neuronal cells (HPPNCs) after oxygen-glucose deprivation (OGD)/reoxygenation (R) treatment. Results showed that impeded CTRP3 expression and weakened viability were detected in OGD/R treated HPPNCs. CTRP3 showed its ability to enhance the viability and inhibited apoptosis of HPPNCs after OGD/R treatment and it could also promote the mitochondrial biogenesis and physiological functions. Silencing of PGC-1α partially abolished the protective function of CTRP3 on mitochondria and CTRP3 mediated the expression of PGC-1α via the AMPK/SIRT1-PGC-1α pathway. These findings provided information that CTRP3 prevented mitochondria from OGD/R injury through activating the AMPK/SIRT1-PGC-1α pathway. Our study suggested that CTRP3 might have the potential to become an emerging protective agent applied in the reperfusion treatment of ischemic stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke has become the most common cerebral vascular disease and the second most dangerous killer worldwide in this decade following myocardial infarction with rapidly increasing incidence [1]. To treat acute ischemic stroke, reperfusion therapy such as surgical thrombectomy is the most popular approach to solve the occlusion of cerebral blood vessel and rescue the nerve tissue ischemia and infarction [2]. Besides, the application of thrombolytic agent recombinant tissue plasminogen activator (rtPA) can dissolve the blood clot which also restores blood flow to the brain [3]. However, at this moment, only a fraction of patients can benefit from the reperfusion therapy and the current therapeutic methods only focused on the reperfusion but not targeted brain tissues. Besides, lots of studies using animals or oxygen-glucose deprivation/reperfusion (OGD/R) models of acute ischemic stroke introduce neuroprotectant uric acid which can decrease the acute ischemic brain injury and reduce the rate of complications in preclinical models [4]. Some in vitro studies tried to block the inflammation process thus saving the neurons to limit the ischemic damage and functional impairments [5] or activation of molecular pathways to enhance angiogenesis in the brain for functional neuronal repairment [6]. Therefore, there is an urgent requirement for a more complete understanding of the underlying mechanisms associated with ischemic stroke at the molecular level and the discovery and development of molecular approaches involved in the treatment of cerebral ischemic stroke.
Mitochondria are a type of special cellular organelle exerts energy production function. Their numbers are particularly prominent in most of the human cells including neurons and gliocytes which need energy for their normal functioning. Mitochondria play crucial roles in adenosine triphosphate (ATP) synthesis and mediation of calcium homeostasis and its function can be affected by reactive oxygen species (ROS) and ischemia which contributes to the generation of ROS. In ischemic stroke, the occlusion of blood vessels and insufficient oxygen supply to neurons will trigger tissue necrosis, disrupt the mitochondria, release of pro-apoptotic proteins and also impair the phosphorylation process involved in the mitochondrial functions thus disturbing ATP supply and breaking the metabolism and homeostasis [7]. Under ischemic conditions, mitochondria will also produce ROS to directly damage the lipids, proteins and DNA in the cells. Likewise, ROS can trigger a variety of molecular signaling pathways involved in apoptosis [8]. After reperfusion, the overproduction of ROS after ischemic stroke will overstress the anti-oxidant capacity of mitochondria and triggers the DNA damage of the mitochondria, disruption of calcium balance, neuronal inflammation and even apoptosis of neurons [9]. Thus the protection of mitochondrial function is a vital determinant of neuronal survival in the treatment of cerebral ischemic stroke.
Peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) is the key mediator of mitochondrial biogenesis and energy metabolism. A study has reported that overexpression of PGC-1α increases the number of mitochondria in mice [10]. PGC-1α acts as a master transcription factor for mitochondrial biogenesis which can activate the downstream transcription factors called the nuclear respiratory factors (NRF) 1 and 2 to strengthen the transcription of mitochondrial genes, then the activated NRF-1 and NRF-2 induce the gene encoding transcription factor A (TFAM) expression to accelerate the transcription, translation and replication machinery of mitochondrial genes. And these transcriptional factors are in turn activated directly or indirectly by PGC-1α which can be influenced by extrinsic factors such as oxygen level and energy deprivation [11]. Moreover, sirtuin 1 (SIRT1) is a NAD-dependent deacetylase that functionally interacts with PGC-1α through post-transcriptional modification to control the mitochondrial biogenesis. Evidence has shown that mutilated expression of SIRT1 downregulates PGC-1α level and abates mitochondrial biogenesis function [12]. Furthermore, the activated AMP-activated protein kinase (AMPK) can also trigger the phosphorylation of PGC-1α which is involved in mitochondrial function. Research has suggested that the AMPK/SIRT1-PGC-1α pathway is activated by adiponectin to stimulate the mitochondrial gene expression [13]. Besides that, the activation of AMPK/SIRT1/PGC1α by swimming can suppress neuronal apoptosis and inflammation in aging hippocampus [14] and in the rats model of neonatal hypoxic-ischemic encephalopathy, AMPK/Sirt1/PGC-1α pathway can be activated by ghrelin to suppress neuronal apoptosis by attenuating oxidative stress [15]. However, less is known about how the AMPK/SIRT1-PGC-1α pathway is working in hippocampal neurons during cerebral ischemia.
C1q/tumor necrosis factor-related protein (CTRP) 3 is an adipokine that belongs to the CTRP family which expresses in a variety of cells such as adipocytes, myocytes and neurons. It takes part in the regulation of cell proliferation, apoptosis, inflammatory process and internal metabolism [16] and it shows its antiapoptotic, proangiogenic, and cardioprotective abilities in vivo [17]. For instance, research has reported that CTRP3 attenuates cardiac impairment and induces angiogenesis in the mice with myocardial infarction [18] and protects the brain from the injury caused by intracerebral hemorrhage in mice [19]. A study has proposed that CTRP3 plays a neuroprotective role against anesthesia-induced cognitive dysfunction via regulation of the AMPK/SIRT1 and PI3K/AKT signaling pathways [20]. In addition, in the hippocampus of mice model with depression induced by chronic unpredictable mild stress (CUMS) and lipopolysaccharide (LPS)-treated microglial cells, CTRP3 exerts anti-apoptotic and anti-inflammatory effects through regulating p38 and JNK signaling pathways [21]. Another study has shown that CTRP3 promotes mitochondrial biogenesis in cardiomyocytes of mice through activation of the AMPK/PGC-1α pathway [22]. Currently, the role of CTRP3 in cerebral ischemic stroke has not been studied. Therefore, this research aims to investigate the molecular mechanism of how CTRP3 plays a role in the hippocampal neurons in cerebral ischemic stroke and build up the connection between CTRP3 and AMPK/SIRT1-PGC-1α pathway.
Material and Methods
Hippocampal Neuronal Cell Culture and OGD/R Model
The HPPNCs were purchased from Bluefbio (China) and maintained in our lab. Briefly, the cells were cultivated in DMEM (Procell, China) containing 10% fetal bovine serum (Procell, China) with 1% penicillin and streptomycin (Procell, China) and kept in a humidified incubator (QQ-80A-II, Beyotime, China) supplemented with 5% CO2 at 37 °C.
The cerebral ischemic stroke cell model was built up by the OGD/R treatment of HPPNCs cells. The culture medium was replaced by a sugar-free medium and the cells were cultivated under an oxygen-deficient environment (3% O2, 92% N2, 5% CO2) for different periods (0, 1, 2, 4, 6 h) and then the cells were transferred back to the normoxic environment (95% air, 5% CO2) with a complete DMEM medium for 24 h reperfusion. The cells maintained in the complete medium under the normoxic condition were used as control.
Cells Transfection
To overexpress CTRP3 in the HPPNCs, a lentiviral vector carrying CTRP3 cDNA (Lenti-CTRP3) or control (Lenti-NC) was constructed and provided by Genepharma (China) was selected. The cells were cultured with 10 µl viral vector containing 1 × 109 genomic copies of the CTRP3 gene or Lenti-NC according to the manufacturer’s suggestions. 48 h after incubation, the transfected cells were harvested.
To knock down the intrinsic PGC-1α expression in the HPPNCs, siRNA targeted PGC-1α was designed and synthesized by GenePharma (China). The cells were transfected with 50 nmol/L siR-PGC-1α or si-NC through Entranster-R4000 (Engreen Biosystem, China) as the recommended instructions. The siRNA sequences used were: siR-PGC-1α sense: CCAAGACUCUAGACAACUATT; siR-PGC-1α anti-sense: UAGUUGUCUAGAGUCUUGGTT. si-NC sense: UUCUCCGAACGUGUCACGUTT; and si-NC anti-sense: ACGUGACACGUUCGGAGAATT. The transfection efficiency was verified 48 h after transfection following the isolation of total RNA.
QRT-PCR
The RNAeasy Animal RNA Isolation kit (Beyotime, China) was used to extract the total RNA. cDNA was converted from mRNA through the application of the BeyoRT cDNA Synthesis kit (Beyotime, China). QRT-PCR was executed on QuantStudio 7 Flex (Thermo Fisher, USA) using the BeyoFast SYBR Green qPCR Mix kit (Beyotime, China). The primers were listed in Table 1. Quantification of RNA levels was normalized with β-actin by \(2^{-{\Delta\Delta\text{C}_{\text{T}}}}\) cycle threshold method [22].
Western Blot
The total proteins were extracted by the Cell lysis buffer for Western and IP (Beyotime, China) from the HPPNCs 48 h after transfection. After separation by 10% SDS-PAGE, the samples were transferred onto the PVDF membrane (Beyotime, China). 5% non-fat milk (Beyotime, China) was applied for membrane blocking and the membrane was incubated with primary mouse antibodies against CTRP3 (1:5000), BCL-2 (1:500), Bax (1:500), NRF-1 (1:1000), NRF-2 (1:1000), TFAM (1:1000), PGC-1α (1:1000), p-PGC-1α (1:1000) and β-actin (1:500) (Beyotime, China) for 12 h at 4 °C. The membrane was rinsed and the horseradish peroxidase-conjugated anti-mouse IgG secondary antibody (1:1000; Beyotime, China) was added and incubated for 1 h. After washing, the bands were stained by the chemiluminescence kit (Beyotime, China) and visualized. The relative protein expression levels were calculated using Image J 1.53b (NIH, USA).
CCK-8 Assay
The HPPNCs were seeded into 96-well plates with a density of 2 × 103 cells per well and incubated for different periods (0, 12, 24, 48, 72 h) at 37 °C with 5% CO2. The cell viability was assessed using the CCK-8 assay kit (Beyotime, China) according to the manufacturer’s protocol. The OD values were obtained through using the Varioskan LUX microplate reader (Thermo Fisher, USA) under a wavelength of 450 nm with a background control as the blank. The cell survival ratio was expressed as the percentage of the control.
Apoptotic analysis by flow cytometry
The HPPNCs were collected 48 h after transfection and rinsed three times with PBS (Beyotime, China) followed by fixation with 75% ethanol (Beyotime, China) at 4 °C for 12 h. After resuspension in PBS containing RNase A (Beyotime, China), the cells were incubated with propidium iodide (Beyotime, China) for 1 h. Propidium iodide was a fluorescent dye and it was able to pass through the membrane of dead cells to intercalate with cellular DNA and emit red fluorescence which could be detected by flow cytometer [23]. The apoptotic analysis was performed using the CytoFLEX Flow Cytometer (Beckman Coulter, USA).
Evaluation of Mitochondrial DNA
The genome DNA of mitochondria was isolated and purified through using the TIANamp Genomic DNA Kit (Tiangen, China). After DNA quantification using a Varioskan LUX microplate reader (Thermo Fisher, USA), the mitochondrial DNA copy number was determined by the quantitative PCR systems (ProFlex PCR, Thermo Fisher, USA) through the relative number of tRNALeu to genome β-2-microglobulin which was introduced by Venegas et al. [24]. The primers used were: tRNALeu forward: CACCCAAGAACAGGGTTTGT; reverse: TGGCCATGGGTATGTTGTTA. β-2-microglobulin forward: TGCTGTCTCCATGTTTGATGTATCT; reverse: TCTCTGCTCCCCACCTCTAAGT. The procedure was strictly according to Venegas’s methods. In order to calculate the mitochondrial DNA content, the difference in mean CT values between the tRNALeu and β-2-microglobulin termed ΔCT were calculated as ΔCT = (β-actin CT) − (CytBCT) and mitochondrial DNA copy number = \(2 \times 2^{\Delta\text{C}_{\text{T}}}\). \(2 \times 2^{\Delta\text{C}_{\text{T}}}\) = mitochondrial DNA content. And the relative mitochondrial DNA content = mitochondrial DNAsample/mitochondrial DNAcontrol.
Detection of Mitochondrial Membrane Potential
The mitochondria were extracted from the HPPNCs and purified using the Cell Mitochondria Isolation Kit (Beyotime, China). Then the detection of mitochondrial membrane potential was achieved using the Mitochondrial Membrane Potential Assay Kit with JC-1 (Beyotime, China). Briefly, 0.1 ml of 100 µg mitochondria was added into 0.9 ml JC-1 staining solution. The higher membrane potential would cause JC-1 aggregation in the matrix of mitochondria and produced red fluorescence whereas JC-1 would behave as a monomer in the matrix and showed green fluorescence when the membrane potential was low. The fluorescent signal was detected using the Varioskan LUX microplate reader (Thermo Fisher, USA) under the wavelengths of 485 nm excitation and 590 nm emission and normalized with internal control according to the supplier’s protocol.
Evaluation of ATP Level
The ATP level of HPPNCs was measured using the ATP Assay Kit (Beyotime, China) after the cells were lysed using the lysis buffer in this reagent kit. The standard curve was made following the provider’s instructions and the ATP level was positively proportional to the firefly luciferase which was measured by GloMax 20/20 Luminometer (Promega, USA). The ATP level = [(absorbancesample − absorbancecontrol)/(absorbancestandard − absorbanceblank)] × the concentration of standard × dilution factor/the protein concentration of the sample.
Detection of ROS and H2O2 Levels
The generation of ROS was a major contributor to neuronal damage following ischemia. It consisted of highly reactive charged oxygen free radicals, hydroxyl radical and non-radical oxidants [25]. The total ROS level of HPPNCs was estimated using the Reactive Oxygen Species Assay Kit (Beyotime, China). DCFH-DA could pass through the plasma membrane and be hydrolyzed to DCFH which could not pass through the plasma membrane. The ROS inside the cell could oxidize DCFH to produce DCF and show fluorescence. In details, the samples were incubated with DCFH-DA probe in a humidified incubator (QP-160II, Biobase, China) at 37 °C for 20 min. ROS could oxidize DCFH to DCF which exerts fluorescent activities. The fluorescence intensity was detected through Varioskan LUX microplate reader (Thermo Fisher, USA) and it was monitored at 488 nm excitation and 525 nm emission. The results were normalized with the internal reference Rosup provided in the kit. Because SOD could catalyze and increase the production of H2O2 and O2. The mitochondrial H2O2 level was measured by the MitoPY1 fluorescent probe (Tocris, UK) and the detailed procedures had been described by Dickinson et al. [26]. The reaction of MitoPY1 with H2O2 triggered a fluorescence increase by its conversion to MitoPY1ox, which possessed one major absorption band at 510 nm and enhanced emission. Therefore, the MitoPY1 fluorescence intensity was detected at 510 nm excitation and 528 nm emission using Varioskan LUX microplate reader (Thermo Fisher, USA).
Examination of SOD Activity
The antioxidant enzyme superoxide dismutase (SOD) catalyzed the dismutation of reactive charged oxygen free radicals into H2O2. The SOD activity was checked using the Total Superoxide Dismutase Assay Kit with WST-8 (Beyotime, China) according to the provider’s instructions. In general, WST-8 could react with negatively charged oxygen free radicals (O2−) which was catalyzed by xanthine oxidase to produce formazan. Because SOD catalyzed the disproportionation of O2− to inhibit the production of formazan, the activity of SOD should be negatively proportional to the formazan. Briefly, the HPPNCs were washed with iced PBS and incubated with SOD solution for cell lysis. After centrifugation at 37 °C under 12,000×g for 5 min, the supernatant was collected. Subsequently, 151 µl supernatant, 8 µl WST-8, 1 µl enzyme was mixed well and sent for absorbent detection. The absorbance was detected using the Varioskan LUX microplate reader (Thermo Fisher, USA) under the wavelength of 450 nm. The results were normalized with the internal standard provided in the kits.
Immunofluorescent Assay
To evaluate the expression of mitochondrial morphology, the harvested HPPNCs were fixed in 4% formaldehyde (Beyotime, China) for 15 min, followed by PBS rinsing three times. After that, PBS, 5% FBS (Beyotime, China) and 0.3% Triton X-100 (Beyotime, China) were applied for cell blocking for 1 h and the cells were stained with rabbit anti-Tom 20 (1:200) antibody (1:500, CST, USA) for 12 h. Next, after washing the samples three times with PBS, the secondary goat anti-rabbit IgG antibody conjugated to Alexa Fluor 488 (1:500, Beyotime, China) was added to the samples and incubated for 2 h. At last, the cells were observed under the fluorescence inverted microscope (Olympus CKX53, Japan) and the mitochondria with fragmented patterns were termed as the mitochondrial fission.
Immunoprecipitation
The immunoprecipitation assay was performed using the Immunoprecipitation Kit (Sangon, China). In details, the HPPNCs were harvested and lysed by the lysis buffer, after centrifugation at 10,000×g for 5 min, then the supernatant was collected and incubated with Protein A/G Plus Agarose and the reagents for 1 h at 4 °C. After another centrifugation step of 6000×g for 5 min, the supernatant was collected and 5 µg/mL anti-PGC-1α or anti-acetyl lysine antibody (GeneTex, USA) was added and incubated for 12 h. Then the samples were incubated and rotated with Protein A/G beads for 2 h at 4 °C. After washing 3 times with PBS and resuspension, the proteins were analyzed through western blot assay which had been described above.
Statistical Analysis
All experiments were independently conducted in triplicate and repeated three times. Data were expressed as mean ± SD. Statistical analyses were conducted using SPSS 25.0 (SPSS Inc, USA) and Graphpad Prism 8.3.0 (Graphpad LLC, USA). Student’s t-test or one-way analysis of variance (ANOVA) was used to determine the statistical differences between groups under different conditions. P < 0.05 was considered to indicate a statistically significant difference.
Results
The Decreased CTRP3 Expression in OGD/R Treated HPPNCs
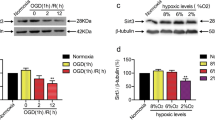
At first, we established the OGD/R model in vitro. CCK-8 assay was performed to evaluate the viability of HPPNCs to choose the optimum time of OGD treatment and the results indicated that the cell viability was decreased with an increase of OGD/R treatment time. The cell viability after 4 h OGD treatment was 58% whereas the viability after 6 h OGD was 45% (Fig. 1a). Therefore 4 h was chosen as the optimum OGD/R treatment time. Then the expression of CTRP3 in HPPNCs cells was examined by qRT-PCR and western blot. The outcomes indicated that OGD/R treated cells expressed less CTRP3 at both mRNA and protein levels (Fig. 1b, c).

The decreased CTRP3 expression in OGD/R treated HPPNCs cells. a The cell viability was detected by CCK-8 assay. b The mRNA expression of CTRP3 was determined by qRT-PCR. c The protein expression of CTRRP3 was checked by western blot. *P < 0.05, **P < 0.01
CTRP3 Increased the Viability and Inhibited the Apoptosis of OGD/R Cells
To investigate the effect of CTRP3 on HPPNCs undergone ischemic stroke, we overexpressed CTRP3 using lentiviral expression vectors in OGD/R treated and untreated HPPNCs. QRT-PCR and western blot were performed to check the transfection efficiency. The results indicated that the CTRP3 mRNA and protein expressions in OGD/R + Lenti-CTRP3 and Lenti-CTRP3 groups were significantly elevated compared with OGD/R and control groups, respectively (Fig. 2a, b). Subsequently, the result of CCK-8 assay showed that OGD/R treatment significantly decreased the cell viability of HPPNCs compared with the untreated HPPNCs and overexpression of CTRP3 could enhance the viability of HPPNCs after OGD/R treatment (Fig. 2c) and it also suppressed the apoptosis of OGD/R cells which was suggested by the results of flow cytometry (Fig. 2d). After that, the outcomes of western blot also verified the results of flow cytometry which demonstrated that OGD/R treatment downregulated the expression of Bcl-2 but upregulated the expression of Bax. Then the Bcl-2 expression was increased but the Bax expression was decreased by overexpression of CTRP3 in the OGD/R + Lenti-CTRP3 group compared with the OGD/R and OGD/R + Lenti NC groups (Fig. 2e). These results proved that CTRP3 could enhance the viability and protect the HPPNCs from apoptosis after OGD/R treatment.

CTRP3 increased the viability and inhibited the apoptosis of OGD/R cells. a The mRNA expression of CTRP3 was determined by qRT-PCR. b The protein expression of CTRP3 was checked by western blot. c The cell viability was detected by CCK-8 assay. d Cell apoptosis was evaluated by flow cytometry. e The expressions of Bax and BCL-2 were examined by western blot. *P < 0.05, **P < 0.01
CTRP3 Promoted the Mitochondrial Biogenesis and Physiological Functions
A previous study found that CTRP3 had shown the enhancement of mitochondrial biogenesis properties in the treatment of myocardial infarction [22]. Firstly, we explored the expression levels of genes and proteins involved in the biogenesis of mitochondria in OGD/R treated and untreated HPPNCs. The results indicated that OGD/R treatment could decrease the expression of NRF-1, NRF-2 and TFAM at both mRNA (Fig. 3a) and protein levels (Fig. 3b) compared with the control group and its effects in the OGD/R treated HPPNCs could be counteracted by CTRP3. In addition to this, CTRP3 rescued the expression of mitochondrial DNA which was downregulated by OGD/R treatment in HPPNCs (Fig. 3c). Besides, CTRP3 overexpression could also overcome the influences caused by OGD/R treatment to increase the mitochondrial membrane potential (Fig. 3d) and ATP (Fig. 3e) level in OGD/R treated HPPNCs. Moreover, it was also indicated by the immunofluorescent assay that the mitochondrial morphology was tubular and tightly connected in the control, Lenti-NC, Lenti-CTRP3 groups whereas the mitochondrial fission was observed and their elongated network changed to small and globular structures scattered in the neurons in OGD/R, OGD/R + Lenti NC groups, and Lenti-CTRP3 partly redeemed the mitochondrial fragmentation caused by OGD/R (Fig. 3f). Moreover, the addition of CTRP3 could diminish the ROS and H2O2 levels (Fig. 3g, h) and upregulate the SOD activity (Fig. 3i) to neutralize the effects caused by OGD/R treatment in HPPNCs. These results proved that CTRP3 had the protective roles in mitochondrial biogenesis and the physiological functions of HPPNCs.

CTRP3 promoted the mitochondrial biogenesis. a The mRNA expressions of mitochondrial genes were verified by qRT-PCR. b The protein expressions of mitochondrial genes were tested by western blot. c The evaluation of mitochondrial DNA level by a reagent kit. d The test of mitochondrial membrane potential by a reagent kit. e The determination of the ATP level by a reagent kit. f The mitochondrial morphology and fission were checked by immunofluorescent assay. Scale bars: 50 µm. g The ROS level was tested by a reagent kit. h The H2O2 level was tested by MitoPY1 fluorescent probe. i The SOD activity was determined by a reagent kit. *P < 0.05, **P < 0.01
Inhibition of PGC-1α Counteracted the Protective Effects of CTRP3
Since PGC-1α was a key factor in mitochondrial biogenesis, we examined the effect of CTRP3 on PGC-1α expression. The western blot analysis showed that the expression of PGC-1α was decreased in OGD/R treated HPPNCs compared with the untreated cells, and overexpression of CTRP3 enhanced PGC-1α expression in the OGD/R treated HPPNCs (Fig. 4a) which revealed that PGC-1α expression was mediated by the regulatory effect of CTRP3 on mitochondrial biogenesis in HPPNCs. Furthermore, si-PGC-1α and CTRP3 were co-transfected into HPPNCs and the cells underwent OGD/R treatment. The results of western blot indicated that OGD/R treatment downregulated PGC-1α expression and this impact would be partially reversed by CTRP3 transfection. And the addition of siR-PGC-1α suppressed the expression of PGC-1α in siR-PGC-1α group (Fig. 4b). Subsequently, mitochondrial biogenesis-related genes (NRF-1, NRF-2, TFAM) expressions were examined using qRT-PCR and western blot. And the results showed that in the OGD/R + Lenti-CTRP3 + si-PGC-1α group, si-PGC-1α transfection repealed CTRP3-induced the upregulated expressions of NRF-1, NRF-2 and TFAM at both mRNA (Fig. 4c) and protein levels (Fig. 4d) compared with the OGD/R + Lenti-CTRP3 + si-NC group. Afterward, the transfection of siR-PGC-1α could also impede the mitochondrial DNA level in the OGD/R + Lenti-CTRP3 + siR-PGC-1α group (Fig. 4e). Furthermore, knockdown of PGC-1α diminished the mitochondrial membrane potential (Fig. 4f) and ATP (Fig. 4g) level inducted by CTRP3. The ROS and H2O2 levels were increased in the OGD/R + Lenti-CTRP3 + si-PGC-1α group compared with the OGD/R + Lenti-CTRP3 group (Fig. 4h, i). At the same time, in the OGD/R + Lenti-CTRP3 + si-PGC-1α group, the SOD activity mediation by CTRP3 was suppressed by si-PGC-1α compared with the OGD/R + Lenti-CTRP3 group (Fig. 4j). These results proposed that knockdown of PGC-1α hampered the mitochondrial biogenesis and it negatively affected the protective function of CTRP3 in HPPNCs.

Inhibition of PGC-1α counteracted the protective effects of CTRP3. a The protein expression of PGC-1α was checked by western blot. b The protein expression of PGC-1α was checked by western blot. c The mRNA expressions of mitochondrial genes were verified by qRT-PCR. d The protein expressions of mitochondrial genes were tested by western blot. e The evaluation of mitochondrial DNA level by a reagent kit. f The test of mitochondrial membrane potential by a reagent kit. g The determination of the ATP level by a reagent kit. h The ROS level was tested by a reagent kit. i The H2O2 level was tested by MitoPY1 fluorescent probe. j The SOD activity was determined by a reagent kit. *P < 0.05, **P < 0.01
CTRP3 regulated PGC-1α expression via the control of AMPK and Sirt1
To further investigate the molecular mechanism, we detected AMPK and Sirt1 expressions which were the upstream transcription factors of PGC-1α. As shown by the results Fig. 5a, the OGD/R treatment resulted in the downregulation of p-AMPK and Sirt1 compared with the control group, and CTRP3 overexpression improved p-AMPK and Sirt1 expressions in the OGD/R treated HPPNCs. To further verify whether AMPK and Sirt1 expressions were controlled by the effect of CTRP3 on PGC-1α, the 10 µM AMPK inhibitor AraA which was a competitive inhibitor blocked the phosphorylation of AMPK and also inhibited its expression and 10 µM Sirt1 inhibitor EX-527 which could close the NAD+ binding site of SIRT1 and reduce SIRT1 expression were added into OGD/R treated HPPNCs transfected with Lenti-CTRP3, respectively. The results of western blot subsequently demonstrated that the addition of AraA hampered AMPK, p-AMPK and PGC-1α expressions, meanwhile decreased the expression of Sirt1. EX-527 diminished the expression levels of Sirt1 and PGC-1α which were increased by overexpression of CTRP3 (Fig. 5b). And the expression level of phosphorylated PGC-1α was attenuated by AraA but EX-527 had no effect on p-PGC-1α expression. (Fig. 5b). These outcomes proved that knockdown of AMPK abated the expressions of p-PGC-1α. Subsequently, the results of immunoprecipitation revealed that the EX-527 significantly inhibited the deacetylation of PGC-1α induced by CTRP3 (Fig. 5c). Afterward, the upregulated levels of NRF-1, NRF-2 and TFAM induced by CTRP3 overexpression were reduced by the addition of AraA and EX-527 (Fig. 5d). Last but not least, the addition of AraA and EX-527 weakened the effect of CTRP3 on mitochondrial functions in OGD/R treated HPPNCs (Fig. 5e–i). These results above interpreted that CTRP3 mediated the expression of PGC-1α through the regulation of AMPK and Sirt1 in OGD/R treated HPPNCs.

CTRP3 regulated PGC-1α expression via the control of AMPK and Sirt1. a The protein expressions of AMPK, p-AMPK and Sirt1 were tested by western blot. b The protein expressions of AMPK, p-AMPK, Sirt1, PGC-1α and p-PGC-1α were checked by western blot. c The deacetylation of PGC-1α was evaluated by immunoprecipitation. d The protein expressions of mitochondrial genes were tested by western blot. e The test of mitochondrial membrane potential by a reagent kit. f The determination of the ATP level by a reagent kit. g The ROS level was tested by a reagent kit. h The H2O2 level was tested by MitoPY1 fluorescent probe. i The SOD activity was determined by a reagent kit. *P < 0.05, **P < 0.01
Discussion
Cerebral ischemic stroke had high morbidity and the recovery of blood and oxygen supply timely to the ischemic brain tissue was crucial for patients’ survival. Although reperfusion therapy was the most common approach applied clinically in the treatment of cerebral ischemic stroke, the damages caused by reperfusion such as calcium overload, oxidative stress, inflammatory responses and neuronal apoptosis could not be ignored [27]. OGD/R treatment was a classic and widely used method that could mimic the ischemia/reperfusion (I/R) injury in vitro [28]. In our study, after OGD/R treatment, the hippocampal neuronal cells showed lower viability compared with normal neurons and a study designed by Wang et al. showed that OGD/R treatment could trigger neuronal autophagic cell death [29]. Bax was a pro-apoptotic protein whereas BCL-2 was an anti-apoptotic protein, both of them were related to the intrinsic mitochondrial apoptotic program, regulated the permeability of the mitochondrial membrane, the opening of the channels and release of cytochrome c to control apoptosis [28]. Here, we found that OGD/R triggered mitochondrial fission which was consistent with Zhou et al.’s report [30]. OGD/R also upregulated the expression level of Bax but downregulated the expression level of BCL-2. CTRP3 was an adipokine that owned crucial roles in metabolic regulatory properties. The other studies raised that the CTRP3 level could be reduced by depression [21] and myocardial dysfunction [31]. We found in this study that CTRP3 expression was diminished in the OGD/R treated hippocampal neuron cells HPPNCs compared with untreated cells. In addition, CTRP3 showed its neuroprotective effects to overcome cognitive dysfunction [20] and prevented mesenchymal stem cells from hypoxia-induced apoptosis through the phosphoinositide 3-kinase (PI3K)/Akt pathway [32].
Mitochondria played vital roles in maintaining cellular energy production and homeostasis, cerebral ischemia led to the deterioration of the hippocampal neuronal mitochondria [33]. The extensive ROS during cerebral ischemic stroke would trigger mitochondrial fission and dysfunction, which subsequently contributed to characteristic pathological changes in the hippocampal neurons and brain [34]. In this study, we proposed that overexpression of CTRP3 enhanced the viability with downregulated apoptosis of HPPNCs, ameliorated mitochondrial fission and improved the mitochondrial biogenesis and functions in OGD/R treated HPPNCs which were consistent with the results of the study conducted by Zhang et al., which suggested that CTRP3 could promote mitochondrial biogenesis in cardiomyocytes [22]. These ideas pointed out that CTRP3 might have potential beneficial roles for the regulation of mitochondria in ischemic stroke.
PGC-1α served as a transcriptional co-activator for the mediation of mitochondrial biogenesis. A study had proved that silencing of endogenous PGC-1α would impede the expressions of mitochondrial-related genes NRF-1 and NRF-2 thus reducing the expressions of downstream nuclear-encoded mitochondrial genes such as cytochrome c (cyt c) and TFAM in human neuroblastoma cells [35]. In our study, we also verified that knockdown PGC-1α decreased the expressions of NRF-1, NRF-2 and TFAM at both mRNA and protein levels which was consistent with Koh et al.’s findings [36]. The expression level of PGC-1α could be strengthened by CTRP3 in vascular smooth muscle cells to ameliorate the energy production function of mitochondria [37]. Our research conducted on hippocampal neuronal cells firstly presented that the CTRP3 could improve the expression of PGC-1α and knockdown of PGC-1α partially neutralized the beneficial effects brought by CTRP3. Besides, AMPK was an enzyme centrally controlled of intracellular energy balance and mitochondrial homeostasis. Its main role was to phosphorylate the downstream target molecules to enhance ATP production and decrease ATP consumption [38]. Research paper had proposed that AMPK phosphorylated PGC-1α to overcome ischemic tolerance in cardiac myocytes [39]. The AMPK-PGC-1α also protected the astrocyte glutathione system against oxidative and metabolic injury after retinal ischemia/reperfusion [40]. In addition, Zhang et al. pointed out that AMPK could be phosphorylated by CTRP3 to ameliorate mitochondrial dysfunction in cardiomyocytes [22] which was consistent with the results of this present study which conducted on hippocampal neuronal cells. Besides this, the activated AMPK would upregulate the expression of SIRT1 which was a histone deacetylase [41]. Deacetylation of PGC-1α by SIRT1 could promote the transcription of mitochondrial genes and enhance mitochondrial functioning [42]. Here, we found that SIRT1 deacetylated PGC-1α to enhance mitochondrial biogenesis in hippocampal neuron cells.
Taken together, we presented in this research for the first time with compelling evidence that CTRP3 was a novel activator of AMPK, AMPK could directly phosphorylate PGC-1α or enhance SIRT1 expression thus promoting PGC-1α deacetylation (Fig. 6). In conclusion, our data provided a neuronal preventative method against cerebral ischemia and reperfusion and indicated a rational direction for further in vivo research.

The schematic model for the molecular working mechanism of CTRP3
Data Availability
The datasets used during the present study are available from the corresponding author upon reasonable request.
Abbreviations
- CTRP3:
-
C1q/tumour necrosis factor-related protein-3
- OGD/R:
-
Oxygen-glucose deprivation/reoxygenation
- HPPNCs:
-
Hippocampal neuronal cells
- PGC-1α:
-
Peroxisome proliferator-activated receptor γ coactivator-1α
- ATP:
-
Adenosine triphosphate
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- rtPA:
-
Recombinant tissue plasminogen activator
- NRF:
-
Nuclear respiratory factors
- TFAM:
-
Gene encoding transcription factor A
- SIRT1:
-
Sirtuin 1
- AMPK:
-
AMP-activated protein kinase
- CTRP3:
-
C1q/tumor necrosis factor-related protein 3
References
Feigin VL, Krishnamurthi RV, Parmar P, Norrving B, Mensah GA, Bennett DA, Barker-Collo S, Moran AE, Sacco RL, Truelsen T, Davis S, Pandian JD, Naghavi M, Forouzanfar MH, Nguyen G, Johnson CO, Vos T, Meretoja A, Murray CJ, Roth GA (2015) Update on the global burden of ischemic and hemorrhagic stroke in 1990–2013: tthe GBD 2013 study. Neuroepidemiology 45(3):161–176. https://doi.org/10.1159/000441085
Shi L, Rocha M, Leak RK, Zhao J, Bhatia TN, Mu H, Wei Z, Yu F, Weiner SL, Ma F (2018) A new era for stroke therapy: integrating neurovascular protection with optimal reperfusion. 38(12):2073–2091. https://doi.org/10.1177/0271678x18798162
Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67(2):181–198. doi:https://doi.org/10.1016/j.neuron.2010.07.002
Becker BF (1993) Towards the physiological function of uric acid. Free Radic Biol Med 14(6):615–631. doi:https://doi.org/10.1016/0891-5849(93)90143-i
Shekhar S, Cunningham MW, Pabbidi MR, Wang S, Booz GW, Fan F (2018) Targeting vascular inflammation in ischemic stroke: recent developments on novel immunomodulatory approaches. Eur J Pharmacol 833:531–544. https://doi.org/10.1016/j.ejphar.2018.06.028
Zou Y, Hu J, Huang W, Ye S, Han F, Du J, Shao M, Guo R, Lin J, Zhao Y, Xiong Y, Wang X (2020) Non-mitogenic fibroblast growth factor 1 enhanced angiogenesis following ischemic stroke by regulating the sphingosine-1-phosphate 1 pathway. Front Pharmacol 11:59. https://doi.org/10.3389/fphar.2020.00059
Yang JL, Mukda S, Chen SD (2018) Diverse roles of mitochondria in ischemic stroke. Redox Biol 16:263–275. doi:https://doi.org/10.1016/j.redox.2018.03.002
Niizuma K, Yoshioka H, Chen H, Kim GS, Jung JE, Katsu M, Okami N, Chan PH (2010) Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim Biophys Acta 1802(1):92–99. doi:https://doi.org/10.1016/j.bbadis.2009.09.002
Ham PB 3, Raju R (2017) Mitochondrial function in hypoxic ischemic injury and influence of aging. Progress Neurobiol 157:92–116. https://doi.org/10.1016/j.pneurobio.2016.06.006
Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP (2004) Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res 94(4):525–533. https://doi.org/10.1161/01.res.0000117088.36577.eb
Scarpulla RC (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88(2):611–638. https://doi.org/10.1152/physrev.00025.2007
Chuang YC, Chen SD (2019) Sirtuin 1 regulates mitochondrial biogenesis and provides an endogenous neuroprotective mechanism against seizure-induced neuronal cell death in the hippocampus following status epilepticus. Int J Mol Sci. https://doi.org/10.3390/ijms20143588
Huang B, Cheng X, Wang D, Peng M, Xue Z, Da Y, Zhang N, Yao Z, Li M, Xu A, Zhang R (2014) Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1α signaling. Oncotarget 5(13):4732–4745. doi:https://doi.org/10.18632/oncotarget.1963
Lin JY, Kuo WW, Baskaran R, Kuo CH, Chen YA, Chen WS, Ho TJ, Day CH, Mahalakshmi B, Huang CY (2020) Swimming exercise stimulates IGF1/PI3K/Akt and AMPK/SIRT1/PGC1α survival signaling to suppress apoptosis and inflammation in aging hippocampus. Aging 12(8):6852–6864. doi:https://doi.org/10.18632/aging.103046
Huang J, Liu W, Doycheva DM, Gamdzyk M, Lu W, Tang J, Zhang JH (2019) Ghrelin attenuates oxidative stress and neuronal apoptosis via GHSR-1α/AMPK/Sirt1/PGC-1α/UCP2 pathway in a rat model of neonatal HIE. Free Radic Biol Med 141:322–337. doi:https://doi.org/10.1016/j.freeradbiomed.2019.07.001
Li Y, Wright GL, Peterson JM (2017) C1q/TNF-related protein 3 (CTRP3) function and regulation. Compr Physiol 7(3):863–878. https://doi.org/10.1002/cphy.c160044
Yi W, Sun Y, Yuan Y, Lau WB, Zheng Q, Wang X, Wang Y, Shang X, Gao E, Koch WJ, Ma XL (2012) C1q/tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation 125(25):3159–3169. doi:https://doi.org/10.1161/circulationaha.112.099937
Sun B, Ding Y, Jin X, Xu S, Zhang H (2019) Long non-coding RNA H19 promotes corneal neovascularization by targeting microRNA-29c. Biosci Rep. https://doi.org/10.1042/BSR20182394
Wang S, Zhou Y, Yang B, Li L, Yu S, Chen Y, Zhu J, Zhao Y (2016) C1q/tumor necrosis factor-related protein-3 attenuates brain injury after intracerebral hemorrhage via AMPK-dependent pathway in rat. Front Cell Neurosci 10:237. https://doi.org/10.3389/fncel.2016.00237
Yang LH, Xu YC, Zhang W (2020) Neuroprotective effect of CTRP3 overexpression against sevoflurane anesthesia-induced cognitive dysfunction in aged rats through activating AMPK/SIRT1 and PI3K/AKT signaling pathways. Eur Rev Med Pharmacol Sci 24(9):5091–5100. doi:https://doi.org/10.26355/eurrev_202005_21202
Meng J, Wang DM, Luo LL (2019) CTRP3 acts as a novel regulator in depressive-like behavior associated inflammation and apoptosis by meditating p38 and JNK MAPK signaling. Biomed Pharmacother [Biomedecine pharmacotherapie] 120:109489. https://doi.org/10.1016/j.biopha.2019.109489
Zhang CL, Feng H, Li L, Wang JY, Wu D, Hao YT, Wang Z, Zhang Y (1861) Wu LL (2017) globular CTRP3 promotes mitochondrial biogenesis in cardiomyocytes through AMPK/PGC-1α pathway. Biochim Biophys Acta Gen Subj 1:3085–3094. https://doi.org/10.1016/j.bbagen.2016.10.022
Krämer CE, Wiechert W, Kohlheyer D (2016) Time-resolved, single-cell analysis of induced and programmed cell death via non-invasive propidium iodide and counterstain perfusion. Sci Rep 6:32104. https://doi.org/10.1038/srep32104
Venegas V, Halberg MC (2012) Measurement of mitochondrial DNA copy number. Methods Mol Biol (Clifton, NJ) 837:327–335. https://doi.org/10.1007/978-1-61779-504-6_22
Gill T, Levine AD (2013) Mitochondria-derived hydrogen peroxide selectively enhances T cell receptor-initiated signal transduction. J Biol Chem 288(36):26246–26255. doi:https://doi.org/10.1074/jbc.M113.476895
Dickinson BC, Chang CJ (2008) A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc 130(30):9638–9639. doi:https://doi.org/10.1021/ja802355u
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22(9):391–397. https://doi.org/10.1016/s0166-2236(99)01401-0
Juurlink BH, Hertz L (1993) Ischemia-induced death of astrocytes and neurons in primary culture: pitfalls in quantifying neuronal cell death. Brain Res Dev Brain Res 71(2):239–246. https://doi.org/10.1016/0165-3806(93)90175-a
Wang G, Wang T, Hu Y, Wang J, Wang Y, Zhang Y, Li F, Liu W, Sun Y, Yu B, Kou J (2020) NMMHC IIA triggers neuronal autophagic cell death by promoting F-actin-dependent ATG9A trafficking in cerebral ischemia/reperfusion. Cell death Dis 11(6):428. https://doi.org/10.1038/s41419-020-2639-1
Zhou X, Wang HY, Wu B, Cheng CY, Xiao W, Wang ZZ, Yang YY, Li P, Yang H (2017) Ginkgolide K attenuates neuronal injury after ischemic stroke by inhibiting mitochondrial fission and GSK-3β-dependent increases in mitochondrial membrane permeability. Oncotarget 8(27):44682–44693. doi:https://doi.org/10.18632/oncotarget.17967
Wei WY, Ma ZG, Zhang N, Xu SC, Yuan YP, Zeng XF, Tang QZ (2018) Overexpression of CTRP3 protects against sepsis-induced myocardial dysfunction in mice. Mol Cell Endocrinol 476:27–36. https://doi.org/10.1016/j.mce.2018.04.006
Hou M, Liu J, Liu F, Liu K, Yu B (2014) C1q tumor necrosis factor-related protein-3 protects mesenchymal stem cells against hypoxia- and serum deprivation-induced apoptosis through the phosphoinositide 3-kinase/Akt pathway. Int J Mol Med 33(1):97–104. doi:https://doi.org/10.3892/ijmm.2013.1550
Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC (2018) Neuronal cell death. Physiol Rev 98(2):813–880. https://doi.org/10.1152/physrev.00011.2017
Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cerebral Blood Flow Metab 21(1):2–14. https://doi.org/10.1097/00004647-200101000-00002
Ye Q, Chen C, Si E, Cai Y, Wang J, Huang W, Li D, Wang Y, Chen X (2017) Mitochondrial effects of PGC-1alpha silencing in MPP(+) treated human SH-SY5Y neuroblastoma cells. Front Mol Neurosci 10:164. https://doi.org/10.3389/fnmol.2017.00164
Koh JH, Hancock CR, Terada S, Higashida K, Holloszy JO, Han DH (2017) PPARβ is essential for maintaining normal levels of PGC-1α and mitochondria and for the increase in muscle mitochondria induced by exercise. Cell Metabol 25(5):1176-1185.e1175. https://doi.org/10.1016/j.cmet.2017.04.029
Feng H, Wang JY, Zheng M, Zhang CL, An YM, Li L, Wu LL (2016) CTRP3 promotes energy production by inducing mitochondrial ROS and up-expression of PGC-1α in vascular smooth muscle cells. Exp Cell Res 341(2):177–186. https://doi.org/10.1016/j.yexcr.2016.02.001
Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19(2):121–135. https://doi.org/10.1038/nrm.2017.95
Quan N, Wang L, Chen X, Luckett C, Cates C, Rousselle T, Zheng Y, Li J (2018) Sestrin2 prevents age-related intolerance to post myocardial infarction via AMPK/PGC-1α pathway. J Mol Cell Cardiol 115:170–178. doi:https://doi.org/10.1016/j.yjmcc.2018.01.005
Guo X, Jiang Q, Tuccitto A, Chan D, Alqawlaq S, Won GJ, Sivak JM (2018) The AMPK-PGC-1α signaling axis regulates the astrocyte glutathione system to protect against oxidative and metabolic injury. Neurobiol Dis 113:59–69. doi:https://doi.org/10.1016/j.nbd.2018.02.004
Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127(6):1109–1122. doi:https://doi.org/10.1016/j.cell.2006.11.013
Chandrasekaran K, Anjaneyulu M, Choi J, Kumar P, Salimian M, Ho CY, Russell JW (2019) Role of mitochondria in diabetic peripheral neuropathy: influencing the NAD(+)-dependent SIRT1-PGC-1α-TFAM pathway. Int Rev Neurobiol 145:177–209. https://doi.org/10.1016/bs.irn.2019.04.002
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
J.G. and T.Q. performed the experiments, analyzed data and wrote the paper. W.W. conceived and designed the experiments and revised the manuscript. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gao, J., Qian, T. & Wang, W. CTRP3 Activates the AMPK/SIRT1-PGC-1α Pathway to Protect Mitochondrial Biogenesis and Functions in Cerebral Ischemic Stroke. Neurochem Res 45, 3045–3058 (2020). https://doi.org/10.1007/s11064-020-03152-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-03152-6