
Abstract
Astrocytic necroptosis plays an essential role in the progression and regression of neurological disorders, which contributes to the neuroinflammation and disrupts neuronal regeneration and remyelination of severed axons. Electroacupuncture (EA), an effective therapeutic efficacy against spinal cord injury (SCI), has been proved to reduce neuronal cell apoptosis, inhibit inflammation, and prompt neural stem cell proliferation and differentiations. However, there have been few reports on whether EA regulate astrocytic necroptosis in SCI model. To investigate the effects of EA on astrocytic necroptosis and the mechanisms involved in the inhibition of astrocytic necroptosis after SCI in mice by EA, 8-week-old female C57BL/6 mice were subjected to SCI surgery and randomly divided into EA and SCI groups. Mice receiving sham surgery were included as sham group. “Jiaji” was selected as points for EA treatment, 10 min/day for 14 days. The in vitro data revealed that EA treatment significantly improved the nervous function and pathological changes after SCI. EA also reduced the number of GFAP/P-MLKL, GFAP/MLKL, GFAP/HMGB1, and Iba1/HMGB1 co-positive cells and inhibited the expressions of IL-6, IL-1β, and IL-33. The results indicate a significant reduction in inflammatory reaction and astrocytic necroptosis in mice with SCI by EA. Additionally, the expressions of RIP1, MLKL, and TLR4, which are associated with necroptosis, were found to be downregulated by EA. In this study, we confirmed that EA can inhibit neuroinflammation by reducing astrocytic necroptosis through downregulation of RIP1/MLKL/TLR4 pathway in mice with SCI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spinal cord injury (SCI) is a devastating disease with an estimated incidence of 250,000 to 500,000 every year and a catastrophic impact on patients’ quality of life [1]. SCI includes primary injury produced by the impact and secondary injury which occur after the immediate impact. Secondary injury is complex and includes disruption of the blood–spinal cord barrier, hemorrhage, edema, vasospasm, oxidative stress, ischemia, and reperfusion injury, which in turn could lead to neuronal and glial apoptosis and necrosis [2].
In the pathological process of SCI, cell necrosis is the core and most important step. Astrocyte death disrupts the protection of neurons by glial cells and is the primary source of the danger-associated molecular patterns (DAMPs) in the context of SCI [3,4,5,6]. High mobility group box1 (HMGB1) is one such DAMP that contributes to the inflammatory milieu, followed by neuroinflammation and disrupts neuronal regeneration and remyelination of severed axons [7, 8].
Cell necrosis was once considered a passive form of cell death. Recent studies, however, have shown that a new type of cell death known as necroptosis, which mediated by specific death signals [9, 10]. Astrocytes are the most numerous glial cells in the central nervous system (CNS) and the primary cells that undergo necroptosis after SCI [11]. Following the necroptosis of a large number of astrocytes, there is extensive infiltration of inflammatory cells and subsequent neuroinflammation [12], neuronal loss, axonal degeneration, and collapse of the myelin sheath [11]. Receptor-interacting protein kinase 1/3 (RIP1/3) and mixed-spectrum kinase structural domain-like proteins (MLKL) [13] are the predominant signaling pathways that regulate necroptosis. Toll-like receptor (TLR) signaling could lead to polyubiquitination of RIP and MLKL in the necroptotic complex [14]. Direct activation of necroptosis in both caspase-inactivated and caspase-deficient conditions could be initiated by the interaction of the RHIM domains in TRIF and RIP; in fibroblasts, this pathway proceeds independent of RIP1 or its kinase activity; however, it remains dependent on MLKL downstream of RIP3 kinase [15, 16]. Therefore, inhibiting the RIP1/MLKL/TLR4 pathway to decrease necroptosis has been explored as a potential therapeutic strategy in the treatment of SCI.
Many previous researches have found that electroacupuncture (EA) could improve SCI and its complications [17,18,19]. It has been proved that EA delivered to “Jiaji” acupoints attenuates apoptosis by upregulating the expression of anti-apoptotic proteins, inhibiting endoplasmic reticulum stress factors [20], moderating M1/M2 polarization in macrophage [21], and regulating cellular autophagy by elevating LC3-IIb and decreasing P62 protein levels in cells to advance autophagic flow [22]. The underlying mechanisms of EA are highly complex [23], and there are few researches about EA on astrocytic necroptosis. Hence, the current study sought to examine the effects of EA at “Jiaji” on astrocytic necroptosis and explore the mechanisms of EA inhibiting astrocytic necroptosis in SCI model mice.
Materials and Methods
Animals
Female C57BL/6 mice (8 weeks of age; from the Experimental Animal Center of Chongqing Medical University) were housed in standard cages in a specific pathogen-free facility at 22–24 °C and relative humidity of 65–75% under a 12-h light/dark cycle, with free access to standard rodent chow and water. All experiments were approved by the Institutional Animal Care and Use Committer of Chongqing Medical University and carried out in strict accordance with “Guiding Opinions on the Treatment of Experimental Animals” (2006) from the Ministry of Science and Technology of the People’s Republic of China.
Experimental Design
The overall study consisted of two parts. In part one, effects of EA treatment on hind limb motor function, neuroinflammation, and astrocytic necroptosis were examined in a mouse model of SCI. Briefly, mice were subjected to SCI and then randomized to receive EA treatment or not (n = 24/group). Twenty-four mice receiving sham surgery were included as an additional control. Hind limb motor function was assessed using the Basso mouse scale (BMS) on days 1, 7, and 14. Compound action potential amplitude (CAPA) and nerve conduction velocity (NCV) were measured by electrophysiology on day 14. Then mice were sacrificed to obtain spinal cord tissue for a variety of assays to assess the severity of injury, inflammatory infiltration, western blot, and transcriptome analysis of relevant molecules.
Part two of the study was designed to investigate the mechanisms of EA effects. Briefly, mice were subjected to SCI and then randomized to receive vehicle, EA treatment, necrostatin-1 s (Nec-1), or EA + Nec-1 (n = 12/group). Twelve mice receiving sham surgery and vehicle injection were included as an additional control. Mice were sacrificed to obtain spinal cord tissue for immunofluorescence and western blot.
Surgical Procedure
SCI was performed as described previously [24]. Briefly, mice were anesthetized with sodium pentobarbital (80 mg/kg, intraperitoneal injection). A 1-cm longitudinal incision was made in the posterior midline to expose the spinous process, lamina, and transverse processes at the T12-L2 levels. An aneurysm clip was used to compress the spinal cord for 15 s with 20 × g pressure. The surgical site was irrigated with 0.9% NaCl prior to closure using standard sutures. After surgery, the bladder was manually massaged twice a day to assist urination until the mice regained voluntary urination. Prophylactic penicillin was given at a dose of 100 mg/kg, twice a day, until voluntary urination. Mice in the sham control group were subjected to the same procedure, including removal of the vertebral plates, but the spinal cord was not compressed.
EA Treatment
EA was delivered to 4 sites (bilateral sides at T12 and L2, 5 mm from the midline of the spine, known as Jiaji) [25]. Stainless steel needles (0.25 mm × 13 mm, Jiangsu Medical Instruments Inc., China) were inserted into the acupoints with a depth of 3–5 mm below the skin. Then, the needle handles were linked to the output terminals of an electronic acupuncture instrument (SDZ-II, Suzhou Medical Products, Suzhou, China), which offered a pattern of parse-dense waves (60 Hz for 1.05 s and 2 Hz for 2.85 s, alternately). The current was adjusted to a level that induced a slight twitch in the hind limbs and was typically below 5 μA. Each daily session lasted for 10 min, and the treatment lasted for 14 consecutive days.
Nec-1 Treatment
Nec-1 (necrostatin-1, No.S8037, selleck) was dissolved in 10% dimethyl sulfoxide (DMSO), and then 40% polyethylene glycol (PEG) and 50% normal saline were added to achieve a final concentration of 2.5 mg/ml. The treatment lasted for 30 consecutive days, at a daily dose of 1.8 mg/kg (intraperitoneal injection).
Hind Limb Motor Function
Motor function of the hind limbs was assessed using the Basso mouse scale (BMS) in an open field on days 1, 7, and 14 by two independent investigators who were blinded to group allocation. The scores of BMS range from 0 to 9 points (0 points mean complete paralysis, and 9 points mean completely normal) and are based on observation of the frequency, range of motion, coordination, rear ankle joint activities, touch of the soles of the feet and insteps, position of the feet, stability of the trunk, and position of the tail as previously described [26]. Each session lasted for 5 min, and the scores by two investigators were averaged.
Electrophysiology
Mice were anesthetized with sodium pentobarbital (30 mg/kg, intraperitoneal injection). An incision was made at the site of original incision on the right posterior lateral femur. Tissues were separated to expose the sciatic nerve. A stimulating electrode was placed on the nerve trunk at 3 mm from the inferior edge of the pear-shaped muscle. A recording electrode was placed on the gastrocnemius muscle at 10 mm above the ipsilateral lateral ankle joint. A 4-mA current with 1-Hz frequency was used for stimulation. CAP and NCV were measured with a BL-420N analysis system.
Tissue Preparation
Mice were deeply anesthetized and perfused transcardially with 0.01-M phosphate-buffered saline (PBS) followed by 4% paraformaldehyde in 0.01-M PBS. A 1-cm segment of the spinal cord with the injury epicenter (L1) in the middle was harvested and fixed in 4% paraformaldehyde in 0.1-M PBS for 24 h at 4 ℃. The specimen was dehydrated using a graded ethanol series, permeabilized with xylene, and embedded in paraffin before preparation of tissue Sections (6 μm) for histological analysis.
Hematoxylin–Eosin Staining
Hematoxylin–eosin (HE) staining was conducted using a standard protocol. Stained sections were observed under a light microscope to examine pathology of spinal cord tissue, including tissue edema, tissue cavities, and inflammatory cell infiltration.
Immunofluorescence Assay
For immunofluorescence staining, the tissue sections were placed in a repair cassette filled with EDTA antigen repair buffer (PH 8.0) for antigen retrieval in a microwave oven; serum covered for 30 min and incubated overnight at 4 °C with a primary antibody as follows: GFAP antibody (ab7260; Abcam; 1:1000), Iba1 antibody (ab178846; Abcam; 1:1000), and the next day with HMGB1 antibody (10,829–1-AP; Proteintech; 1:1000), MLKL antibody (21,066–1-AP; Proteintech; 1:1000), and P-MLKL antibody (ab196436; Abcam; 1:1000). The nuclei were counterstained with 4′6-diamidino-2-phenylindole (DAPI) and sealed with an anti-fluorescence quencher. Fluorescence microscopy was performed at 400 × , and 3–4 fields of view were randomly selected for each section. Image-Pro Plus was used to analyze and calculate the proportion of cells co-expressing Iba1/HMGB1, GFAP/HMGB1, GFAP/MLKL, and GFAP/P-MLKL.
Enzyme-Linked Immunosoribent Assay
The serum concentration of IL-6, IL-1β, and IL-33 was measured using serial dilution with ELISA kits (AF2163-A, AF2040-A, AF30395-A, Afantibody, China).
RNA-Seq Analysis
Total RNA was extracted from the spinal cord samples using TRIzol reagents (Invitrogen), reverse transcribed into cDNA and amplified using PCR amplification prior to sequencing using the Illumina NovaSeq6000 gene sequencer. Raw sequencing data were filtered to yield high-quality sequencing data. HiSat2 was used to obtain mapped data (reads). Expression levels of genes and transcripts were analyzed using RSEM. Differentially expressed genes were defined as |log2FC|≥ 2 (as determined with DESeq2) and adjusted P < 0.05. The GO database (http://www.geneontology.org/) and KEGG database (http://www.genome.jp/kegg/) were used to analyze the function and the pathways of the differentially expressed genes.
Western Blot Assay
Spinal cord specimen was homogenized and centrifuged to extract the total protein. Samples were subjected to 12% SDS-PAGE gel electrophoresis, transferred to a PVDF membrane, and incubated with a primary antibody against RIP1 (17,519–1-AP; Proteintech; 1:1000), RIP3 (17,563–1-AP; Proteintech; 1:1000), MLKL (21,066–1-AP; Proteintech; 1:1000), P-MLKL (ab196436; Abcam; 1:1000), HMGB1 (10,829–1-AP; Proteintech; 1:1000), TLR4 (19,811–1-AP; Proteintech; 1:1000), MyD88 (bs-1047R; Bioss; 1:1000), β-actin (bs-1047R; Bioss; 1:1000) antibodies, or GAPDH (ab181602; Abcam; 1:1000) overnight at 4℃ and then with an goat-anti-rabbit IgG H&L/HRP antibody (bs-0295G-HRP; Bioss; 1:10,000) for 1 h. The bands were visualized using a chemiluminescence method, captured by IMAGE STUDIO imaging system, and analyzed by ImageJ. Results were standardized using the internal control. Protein concentration was determined using a BCA method.
Co-immunoprecipitation Assay
Co-immunoprecipitation assay (CO-IP) was performed as previously described [25]. Briefly, 300–500 μl tissue lysate was incubated with 0.5–2 μg antibody against RIP1 (sc-133102; Santa Cruz; 1:1000), K63 (ab179434; Abcam; 1:1000), or GAPDH (ab181602; Abcam; 1:1000) for 3 h at 4 °C. Fifty microliter Protein G agarose beads (Beyotime) was then added and incubated overnight. After washing, samples were boiled for 10 min in sample‐loading buffer and then subjected to SDS-PAGE (5–15%) and analyzed by western blot.
Statistical Analysis
Data are presented as mean ± standard deviation (SD) and analyzed using one-way ANOVA followed by the Tukey–Kramer test for post hoc pairwise comparisons. Motor function (BMS score) was analyzed with analysis of variance for repeated measures. P < 0.05 was considered statistically significant. All statistical analyses were performed using SPSS 22.0 (IBM, Armonk USA).
Results
Effects of EA on Behavioral and Pathological Changes
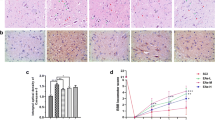
The baseline BMS score did not differ among the sham control, the SCI group, and the EA group (Fig. 1a–c). BMS score after the treatment was significantly higher in the EA versus the SCI group.

EA treatment improved the hindlimb function and attenuated pathological changes in SCI mice. a Hindlimb function was evaluated in mice before SCI and at 1, 7, and 14 days after SCI. Main effect and 2-way interaction were assessed. b BMS score at 1, 7, and 14 days after SCI (n = 8/group). c Electrophysiology on the 14th day after SCI and representative action potential. d, e Quantitative analysis of CAPA and NCV (n = 4/group). f Representative HE staining (scale bar = 20 μm). EA, electroacupuncture; SCI, spinal cord injury; BMS, Basso mouse scale; CAPA, compound action potential amplitude; NCV, nerve conduction velocity; HE, hematoxylin–eosin. Black arrows indicate normal neurons; blue arrows indicate inflammatory cytokines; red arrows indicate tissue cavities. Data are mean ± SD. **P < 0.01 vs. sham group; #P < 0.05, ##P < 0.01 vs. SCI group
Representative electrophysiological recordings are shown in Fig. 1d. Both CAPA and NCV were significantly improved by EA treatment (Fig. 1e, P < 0.01 versus SCI group). Representative images of HE staining are shown in Fig. 1f. Damage to spinal cord tissue by SCI was prominent and included structural disorder, tissue cavities, and inflammatory infiltration. EA treatment attenuated these changes.
Effects of EA on Neuroinflammation
Iba1-positive microglia and GFAP-positive astrocyte in the sham control group were shuttle-shaped and in the branched resting state. In the SCI group, however, Iba1-positive cells and GFAP-positive cells were round in shape, contained large cytosomes and thick protrusions, and appeared to be a highly branched activation type. Iba1-positive microglia and GFAP-positive astrocyte in the EA group were mostly at the resting state (Fig. 2a and b). EA treatment decreased the number of Iba1/HMGB1 co-positive microglia and GFAP/HMGB1 co-positive astrocyte (P < 0.05 versus the SCI group, Fig. 2c and d). In addition, EA treatment also reduced HMGB1 versus the SCI group (P < 0.01, Fig. 2e and f).

EA treatment decreased neuroinflammation in SCI mice. a Representative immunofluorescence staining of Iba1/HMGB1 (scale bar = 20 μm). b Representative immunofluorescence staining of GFAP/HMGB1. c Quantitative analysis of relative Iba1/HMGB1 by immunofluorescence. d Quantitative analysis of relative GFAP/HMGB1 by immunofluorescence. e, f Western blot analysis of HMGB1. g, h, i Serum IL-6, IL-1β, and IL-33. EA, electroacupuncture; SCI, spinal cord injury; DAPI, 4′6-diamidino-2-phenylindole; Iba-1, microglia; HMGB1, high mobility group box1; GFAP, glial fibrillary acidic protein. All data are mean ± SD (n = 6/group). * P < 0.05, ** P < 0.01 vs. sham group; # P < 0.05, ## P < 0.01 vs. SCI group
Serum concentrations of IL-6, IL-1β, and IL-33 were significantly higher in the SCI group versus the sham control; these changes were attenuated by EA treatment (P < 0.05, Fig. 2g–i).
Effects of EA on Astrocytic Necroptosis
Representative immunofluorescent images of GFAP/MLKL and GFAP/P-MLKL colocalization are shown in Fig. 3a and b; representative western blot of MLKL and P-MLKL are shown in Fig. 3e. The number of GFAP/MLKL and GFAP/P-MLKL co-positive cells and the expression of MLKL and P-MLKL were significantly higher in the SCI versus the sham control; EA treatment attenuated these changes (P < 0.05, Fig. 3c–d, Fig. 3f–g).

Necroptosis of astrocytes in the spinal cord. a Representative immunofluorescence staining of GFAP/MLKL (scale bar = 20 μm). b Representative immunofluorescence staining of GFAP/P-MLKL (scale bar = 20 μm). c Quantitative analysis of relative GFAP/MLKL by immunofluorescence. d Quantitative analysis of relative GFAP/P-MLKL by immunofluorescence. e, f, g Western blot of MLKL and P-MLKL in the spinal cord. EA, electroacupuncture; SCI, spinal cord injury; DAPI, 4′6-diamidino-2-phenylindole; MLKL, mixed-spectrum kinase structural domain-like proteins; P-MLKL, phosphorylation of MLKL; GFAP, glial fibrillary acidic protein. All data are mean ± SD (n = 6/group). ** P < 0.01 vs. sham group; # P < 0.05, ## P < 0.01 vs. SCI group
GO and KEGG Enrichment Analyses
The GO enrichment analysis showed that EA treatment significantly altered the cellular component (CC), binding in molecular function (MF) and cellular process in biological process (BP) in the spinal cord (Fig. 4a and c). EA treatment also altered the cellular process, a sequence of biological tasks accomplished via one or more ordered assemblies of molecular functions (Fig. 4b and d). KEGG pathway enrichment analysis showed the TLR signaling pathway expression was significantly affected in both the SCI group and the EA group (Fig. 4e and f). Western blot analysis confirmed an increased expression of TLR4 and MyD88 in the SCI group and attenuation of the changes by EA treatment (P < 0.05, Fig. 5a–c).

RNA-Seq analysis and bioinformatics analysis. a Gene analysis by GO annotations analysis between the SCI and sham control groups. b KEGG enrichment analysis gene analysis between the SCI and sham control groups. c GO annotations analysis between the EA and SCI groups. d KEGG enrichment analysis between the EA and SCI groups. e KEGG pathway enrichment analysis between the SCI and sham control groups. f KEGG pathway enrichment analysis between the EA and SCI groups. n = 8/group. GO annotation analysis column chart: the upper horizontal axis indicates the GO term; the horizontal axis indicates the level of significance of enrichment; and the vertical axis indicates the number of genes/transcripts on the comparison for that GO term, corresponding to the height of the column. By default, the top 10 enrichment results are displayed if P < 0.05. KEGG enrichment analysis column chart: the horizontal axis represents the pathway name, and the vertical axis represents the ratio of the rich factor (the ratio of the number of genes/transcripts enriched in the pathway (sample number) to the number of annotated genes/transcripts (background number). By default, the top 10 enrichment results are displayed if P < 0.05

Western blot analysis of the TLR4/MyD88 and RIP1/ RIP3 signaling pathway. a, b, c TLR4/MyD88. d, e, f RIP1/RIP3. g RIP1/K63 ubiquitination. EA, electroacupuncture; SCI, spinal cord injury; TLR4, toll-like receptor 4; MyD88, recombinant myeloid differentiation factor 88; RIP1/3, receptor-interacting protein kinase 1/3. All data are mean ± SD (n = 6/group). * P < 0.05, ** P < 0.01 vs. sham group; # P < 0.05, ## P < 0.01 vs. SCI group
The expression of RIP1, RIP3, MLKL, and P-MLKL was increased after SCI; these changes were attenuated by EA treatment (Fig. 5d–f and Fig. 3e–f). Co-IP analysis revealed a significantly increased expression of the K63-type polyubiquitin chain by the EA treatment (Fig. 5g).
Inhibitory Effects of EA Versus Nec-1 on Necroptosis
EA treatment significantly reduced astrocyte necroptosis, with a similar effect to that obtained with Nec-1 treatment. EA treatment and Nec-1 treatment or a combination of both could reduce the GFAP/P-MLKL co-positive cells (Fig. 6a). The expression of RIP1, RIP3, and P-MLKL detected by western blot was significantly lower in mice receiving EA or Nec-1 treatment and a further decrease in mice receiving a combination treatment with EA and Nec-1 than either EA or Nec-1 alone (P < 0.01, Fig. 6b–e).

Effects of necrostatin-1 s on RIP1/3 activation. a Representative immunofluorescence staining of GFAP/P-MLKL (scale bar = 20 μm). b, c, d, e Western blot analysis of RIP1, RIP3, and P-MLKL. EA, electroacupuncture; SCI, spinal cord injury; Nec-1 + EA, necrostatin-1 s combine with EA treatment. DAPI, 4′6-diamidino-2-phenylindole; GFAP, glial fibrillary acidic protein; MLKL, mixed-spectrum kinase structural domain-like proteins; P-MLKL, phosphorylation of MLKL; RIP1/3, receptor-interacting protein kinase 1/3. All data are mean ± SD (n = 6/group). *** P < 0.001 vs. sham group; ### P < 0.001 vs. SCI group; ^^^ P < 0.001 vs. Nec-1 group; & P < 0.001 vs. EA group
Discussion
In our previous study, EA at “Jiaji” in a mouse model for SCI downregulated HMGB1, a critical component of DAMPs after necroptosis, and TLR4, a key factor in the TLR pathway [27]. This observation was associated with decreased inflammatory responses and improved neurologic function. The current study showed that EA could attenuate astrocytic necroptosis and neuroinflammation; such effects were associated with downregulation of the RIP1/MLKL/TLR4 signaling pathway.
Astrocytes are critical in maintaining homeostasis in the CNS and play an integral nutritional, structural, and metabolic role in the development and physiology of the CNS [28, 29]. A previous in vitro study showed when astrocytes lose their normal cellular function, and reactive astrocytes proliferate after SCI [30,31,32], thus forming a physical barrier around the injury site to prevent the ingrowth of regenerating axons [33]. In addition, necrotic glial cells trigger an inflammatory cascade and excessive stress on astroglial cells around the central area of SCI [34]. Necroptosis is a type of cell death mediated by a unique molecular pathway that does not overlap with conventional apoptosis [35]. The mechanisms underlying necroptosis in response to different stimuli, including tumor necrosis factor, T cell receptors, interferons, and virus-associated molecules, are highly complex [36] and may involve the critical upstream markers RIP1 and RIP3 and executioners MLKL and P-MLKL [37]. RIP1/3 and MLKL are recruited and form a complex (necrosomes) [38], a crucial mediator of downstream necroptosis. Necrosomes form an oligomeric, membrane-associated channel-like structure that disrupts cell membrane integrity and lysis [39, 40], ultimately inducing necroptosis and causing the release of DAMPs [41], including HMGB1 [42] and IL-33 [43], which in turn activate the immune system and generate a series of inflammatory cascades [44]. In necroptosis, RIP1 and RIP3 interact and bind to form necrosomes through the shared RHIM structural domain to promote necroptosis [45]. The process of necroptosis is tightly controlled by the ubiquitination of RIP1 in necrosome [38, 46]. Ubiquitination changes the conformation of proteins and enhances their adaptability to complex environmental variations and participating in regulating various cellular processes, such as signal transduction and protein transport [47]. Ubiquitination at the Lys 377 site in the intermediate domain promotes the degradation of RIP1. The attachment of K63 at Lys 377 activates the NF-κB pathway and pro-cell survival genes, promoting cell survival [48].
In the study, we found elevated expression levels of RIP1, RIP3, MLKL, and P-MLKL in the RIP signaling pathway upon SCI, and significant attenuation of SCI-induced changes by EA treatment. These findings suggested (1) astrocytes did undergo necroptosis, and (2) EA attenuated SCI-induced necroptosis in astrocytes. Co-IP assay indicated direct interaction between RIP1 and K63 polyubiquitin chain and increased interaction by EA treatment. Together with decreased expression of RIP1, these findings suggest that EA may promote the ubiquitination of K63-type and RIP1 to inhibit the RIP pathway, ultimately inhibiting necroptosis.
RIP1 and RIP3 are implicated in regulating pattern recognition receptor pathways, particularly the TLRs [35]. TLRs are membrane-associated receptors located on the cell surface (TLR1, TLR2, TLR4, TLR5, and TLR6) or in the endocytic compartment (TLR3, TLR7, TLR8, and TLR9) that recognize characteristic molecules of pathogens [49]. The regulatory role of RIP in the TLRs pathway is most clearly defined in the TLR4 signaling pathway, which is also involved in the linkage of lysine at position 377 of RIP1 to polyubiquitin chains, mediating the interaction between RIP1 and RIP3 to produce necrosome and triggering the activation of MLKL [39]. In the current study, we found increased expression of TLR4 and MyD88 upon SCI and attenuation of SCI-induced changes by EA treatment, suggesting that the downregulation of TLR4/MyD88 signaling pathway expression may underlie the effects of EA on RIP ubiquitination and subsequently on astrocytic necroptosis.
Neuroinflammation is characterized by sustained microglia over-activation and is closely associated with necroptosis [50]. DAMPs are a group of nuclear and cytoplasmic proteins that initiate and amplify innate immune pathways [51]. HMGB1 is the main constitutively expressed natural DAMPs released upon necroptosis [52]. Blocking HMGB1 could attenuate neuroinflammation induced by microglia over-activation [53]. Astrocytic necroptosis also promotes neuroinflammation after injury [54]. In the current study, large numbers of HMGB1-Iba + and HMGB1-astrocytic+ were observed after SCI; EA treatment reduced the number of HMGB1-glial+ cells. SCI-induced elevation of serum IL-6, IL-1β, and IL-33 was decreased by EA treatment in the current study, demonstrating that EA treatment reduced neuroinflammation and alleviated the infiltration of pro-inflammatory factors in the injury site which relates to limit necroptosis in astrocytes in the core tissue of SCI, ultimately promoting functional recovery of the spinal cord.
Our finding found the combination treatment with EA and Nec-1 (an inhibitor of necroptosis) could significantly reduce GFAP/P-MLKL co-positive cells and inhibit the expression of RIP1, RIP3, and P-MLKL in the SCI mice. It suggests the combination treatment is a better curative effect than monotherapy, and the former would be an effective strategy for clinical treatment of SCI. In addition, the data also indicated the underlying mechanisms of EA improving SCI is related with the inhibition of the cellular necroptosis. In the future research, further work is needed to use agonists to verify the effect of EA on necroptosis and crucial mediators, which will help us to identify deeply therapeutic targets for SCI.
Conclusions
EA promoted nervous function recovery and signal transmission along neuronal axons in a mouse model for SCI, possibly via limiting the over-activation of microglia and mitigating astrocytic necroptosis and neuroinflammation by regulating the RIP1/MLKL/TLR4 signaling pathway (a schematic summary of the mechanisms is shown in Fig. 7).

A schematic diagram of the mechanisms that likely underlie the effects of EA on astrocytic necroptosis and neuroinflammation
Data Availability
All data are contained within the manuscript.
References
Spinal cord injury. Available online: https://www.who.int/en/news-room/fact-sheets/detail/spinal-cord-injury. Accessed 19 Nov 2013
Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T (2005) In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med 11:572–577. https://doi.org/10.1038/nm1229
Liu Z, Yao X, Jiang W, Li W, Zhu S, Liao C, Zou L, Ding R, Chen J (2020) Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-kappaB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflammation 17:90. https://doi.org/10.1186/s12974-020-01751-2
Liu H, Zhang J, Xu X, Lu S, Yang D, Xie C, Jia M, Zhang W et al (2021) SARM1 promotes neuroinflammation and inhibits neural regeneration after spinal cord injury through NF-kappaB signaling. Theranostics 11:4187–4206. https://doi.org/10.7150/thno.49054
Lindahl H, Bryceson YT (2021) Neuroinflammation associated with inborn errors of immunity. Front Immunol 12. https://doi.org/10.3389/fimmu.2021.827815
Rice T, Larsen J, Rivest S, Yong VW (2007) Characterization of the early neuroinflammation after spinal cord injury in mice. J Neuropathol Exp Neurol 66:184–195. https://doi.org/10.1097/01.jnen.0000248552.07338.7f
Tran AP, Warren PM, Silver J (2018) The biology of regeneration failure and success after spinal cord injury. Physiol Rev 98:881–917. https://doi.org/10.1152/physrev.00017.2017
Thadathil N, Nicklas EH, Mohammed S, Lewis TL Jr, Richardson A, Deepa SS (2021) Necroptosis increases with age in the brain and contributes to age-related neuroinflammation. Geroscience 43:2345–2361. https://doi.org/10.1007/s11357-021-00448-5
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517:311–320. https://doi.org/10.1038/nature14191
Sepand MR, Aliomrani M, Hasani-Nourian Y, Khalhori MR, Farzaei MH, Sanadgol N (2020) Mechanisms and pathogenesis underlying environmental chemical-induced necroptosis. Environ Sci Pollut Res Int 27:37488–37501. https://doi.org/10.1007/s11356-020-09360-5
Fan H, Zhang K, Shan L, Kuang F, Chen K, Zhu K, Ma H, Ju G, Wang Z (2016) Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol Neurodegener 11:14. https://doi.org/10.1186/s13024-016-0081-8
Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH 3rd, Tait SWG, Martinez J, Gale M et al (2017) RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell 169(301–313). https://doi.org/10.1016/j.cell.2017.03.011
Salvadores N, Court FA (2020) The necroptosis pathway and its role in age-related neurodegenerative diseases: will it open up new therapeutic avenues in the next decade? Expert Opin Ther Targets 24:679–693. https://doi.org/10.1080/14728222.2020.1758668
Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S et al (2015) RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 6:6282. https://doi.org/10.1038/ncomms7282
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW et al (2013) Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288:31268–31279. https://doi.org/10.1074/jbc.M113.462341
He S, Liang Y, Shao F, Wang X (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A 108:20054–20059. https://doi.org/10.1073/pnas.1116302108
Dai N, Tang C, Liu H, Huang S (2021) Effect of electroacupuncture on inhibition of inflammatory response and oxidative stress through activating ApoE and Nrf2 in a mouse model of spinal cord injury. Brain Behav 11:e2328. https://doi.org/10.1002/brb3.2328
Tan C, Yang C, Liu H, Tang C, Huang S (2021) Effect of Schwann cell transplantation combined with electroacupuncture on axonal regeneration and remyelination in rats with spinal cord injury. Anat Rec (Hoboken) 304:2506–2520. https://doi.org/10.1002/ar.24721
Chen Y, Wu L, Shi M, Zeng D, Hu R, Wu X, Han S, He K et al (2022) R Electroacupuncture inhibits NLRP3 activation by regulating CMPK2 after spinal cord injury. Front Immunol 13:788556. https://doi.org/10.3389/fimmu.2022.788556
Sun Z, Li J, Yin H, Zeng X, Li Q, Li Z (2019) Review on acupuncture promoting the recovery of nerve function after spinal cord injury and the mechanism of related signal pathway. China J Tradit Chin Med Pharm 34; 5291–5296, CNKI:SUN:BXYY.0.2019–11–081
David S, Kroner A (2011) Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci 12:388–399. https://doi.org/10.1038/nrn3053
Dai P, Huang S, Tang C, Dai N, Zhao H, Tan Y, Yang Y, Tao C (2021) Effects of electroacupuncture at "Jiaji" (EX-B2 ) on autophagy and endoplasmic reticulum stress in spinal cord injury mice. Zhenci Yanjiu Acupunct Res 46; 41–48. https://doi.org/10.13702/j.1000-0607.200229
Jiang K, Sun Y, Chen X (2022) Mechanism underlying acupuncture therapy in spinal cord injury: a narrative overview of preclinical studies. Front Pharmacol 13:875103. https://doi.org/10.3389/fphar.2022.875103
Paterniti I, Esposito E, Cuzzocrea S (2018) An in vivo compression model of spinal cord injury. Methods Mol Biol 1727:379–384. https://doi.org/10.1007/978-1-4939-7571-6_29
Guo Y, Guo X (2021) The experimental guidance of experimental acupuncture and moxibustion. China Traditional Chinese Medicine Publishing House, Beijing, pp 52–57
Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG (2006) Basso Mouse scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23:635–659. https://doi.org/10.1089/neu.2006.23.635
Zhao H, Huang S, Tang C, Dai N, Dai P, Tan Y (2021) Influence of electroacupuncture on locomotor function and expression of spinal HMGB 1 and TLR4 in mice with spinal cord injury. Acupuncture Research 46; 259–265. https://doi.org/10.13702/j.1000-0607.200490
Allen NJ, Eroglu C (2017) Cell biology of astrocyte-synapse interactions. Neuro 96(697):708. https://doi.org/10.1016/j.neuron.2017.09.056
Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH (2012) Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 26:891–907. https://doi.org/10.1101/gad.188326.112
Kobayashi NR, Fan DP, Giehl KM, Bedard AM, Wiegand SJ, Tetzlaff W (1997) BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1-tubulin mRNA expression, and promote axonal regeneration. J Neurosci 17:9583–9595
Abbott NJ (2002) Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat 200:629–638. https://doi.org/10.1046/j.1469-7580.2002.00064.x
Acosta MT, Gioia GA, Silva AJ (2006) Neurofibromatosis type 1: new insights into neurocognitive issues. Curr Neurol Neurosci Rep 6:136–143. https://doi.org/10.1007/s11910-996-0036-5
Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV (2013) Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33:12870–12886. https://doi.org/10.1523/JNEUROSCI.2121-13.2013
Khoury MK, Gupta K, Franco SR, Liu B (2020) Necroptosis in the pathophysiology of disease. Am J Pathol 190:272–285. https://doi.org/10.1016/j.ajpath.2019.10.012
Liu L, Tang Z, Zeng Y, Liu Y, Zhou L, Yang S, Wang D (2021) Role of necroptosis in infection-related, immune-mediated, and autoimmune skin diseases. J Dermatol 48:1129–1138. https://doi.org/10.1111/1346-8138.15929
Newton K, Manning G (2016) Necroptosis and inflammation. Annu Rev Biochem 85:743–763. https://doi.org/10.1146/annurev-biochem-060815-014830
Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, Zhang Y, Ma H, Huang D, Li W, Xia Z, Han J (2013) Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem 288:16247–16261. https://doi.org/10.1074/jbc.M112.435545
Zhang J, Yang Y, He W, Sun L (2016) Necrosome core machinery: MLKL. Cell Mol Life Sci 73:2153–2163. https://doi.org/10.1007/s00018-016-2190-5
Witt A, Vucic D (2017) Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Diffe 24:1160–1171. https://doi.org/10.1038/cdd.2017.33
Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, Li Y, Lu Q et al (2019) TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol Cell 75(457–468). https://doi.org/10.1016/j.molcel.2019.05.022
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG et al (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16:55–65. https://doi.org/10.1038/ncb2883
Murao A, Aziz M, Wang H, Brenner M, Wang P (2021) Release mechanisms of major DAMPs. Apoptosis 26:152–162. https://doi.org/10.1007/s10495-021-01663-3
Shlomovitz I, Erlich Z, Speir M, Zargarian S, Baram N, Engler M, Edry-Botzer L, Munitz A et al (2019) Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. FEBS J286:507–522. https://doi.org/10.1111/febs.14738
Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, Meng Q, Yu X et al (2020) Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol 13:110. https://doi.org/10.1186/s13045-020-00946-7
Menon MB, Gropengiesser J, Fischer J, Novikova L, Deuretzbacher A, Lafera J, Schimmeck H, Czymmeck N et al (2017) p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol 19:1248–1259. https://doi.org/10.1038/ncb3614
Simoes Eugenio M, Faurez F, Kara-Ali GH, Lagarrigue M, Uhart P, Bonnet MC, Gallais I, Com E et al (2021) TRIM21, a new component of the TRAIL-induced endogenous Necrosome Complex. Front Mol Biosci 8:645134. https://doi.org/10.3389/fmolb.2021.645134
Zhang Q, Jia Q, Gao W, Zhang W (2022) The role of deubiquitinases in virus replication and host innate immune response. Front Microbiol 13:839624. https://doi.org/10.3389/fmicb.2022.839624
Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P (20007) RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death Differ 14;400–410. https://doi.org/10.1038/sj.cdd.4402085
Wang XW, Wang JX (2013) Pattern recognition receptors acting in innate immune system of shrimp against pathogen infections. Fish Shellfish Immunol 34:981–989. https://doi.org/10.1016/j.fsi.2012.08.008
Koper MJ, Van Schoor E, Ospitalieri S, Vandenberghe R, Vandenbulcke M, von Arnim CAF, Tousseyn T, Balusu S et al (2020) Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol 139:463–484. https://doi.org/10.1007/s00401-019-02103-y
Land WG (2021) Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome-with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun 22:141–160. https://doi.org/10.1038/s41435-021-00140-w
Land WG (2015) The role of damage-associated molecular patterns in human diseases: part I - promoting inflammation and immunity. Sultan Qaboos Univ Med J 15:e9–e21
Fonken LK, Frank MG, Kitt MM, D’Angelo HM, Norden DM, Weber MD, Barrientos RM, Godbout JP et al (2016) The alarmin HMGB1 mediates age-induced neuroinflammatory priming. J Neurosci 36:7946–7956. https://doi.org/10.1523/JNEUROSCI.1161-16.2016
Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT et al (2018) Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359:1269–1273. https://doi.org/10.1126/science.aal3589
Funding
This work was supported by the National Natural Science Foundation of China (Grant numbers: 81403466 and 81273870), the Natural Science Foundation Project of CQ CSTC (Grant numbers: cstc2021jcyj-msxmX0203), and the joint Project of CQ CSTC and Health Commission of Chongqing (Grant numbers: ZY201802026, 2021ZY023890).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Hongdi Zhao and Xiaoqin Zong performed the whole experiments and molecular studies. Hongdi Zhao and Xiaoqin Zong contributed equally to this work. Long Li performed the molecular biology study and drafted the manuscript. Na Li and Chunlei Liu performed the molecular biology. Wanchao Zhang and Juan Li performed data analysis. Siqin Huang funded and conceived the study and participated in the study design and revised the manuscript. Cheng Yang participated in the study design. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
This study was performed in line with “Guiding Opinions on the Treatment of Experimental Animals” (2006) from the Ministry of Science and Technology of the People’s Republic of China. Approval was granted by the Institutional Animal Care and Use Committer of Chongqing Medical University.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, H., Zong, X., Li, L. et al. Electroacupuncture Inhibits Neuroinflammation Induced by Astrocytic Necroptosis Through RIP1/MLKL/TLR4 Pathway in a Mouse Model of Spinal Cord Injury. Mol Neurobiol 61, 3258–3271 (2024). https://doi.org/10.1007/s12035-023-03650-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03650-y