
Abstract
Tau protein plays a pivotal role in the central nervous system (CNS), participating in microtubule stability, axonal transport, and synaptic communication. Research interest has focused on studying the role of post-translational tau modifications in mitochondrial failure, oxidative damage, and synaptic impairment in Alzheimer’s disease (AD). Soluble tau forms produced by its pathological cleaved induced by caspases could lead to neuronal injury contributing to oxidative damage and cognitive decline in AD. For example, the presence of tau cleaved by caspase-3 has been suggested as a relevant factor in AD and is considered a previous event before neurofibrillary tangles (NFTs) formation.
Interestingly, we and others have shown that caspase-cleaved tau in N- or C- terminal sites induce mitochondrial bioenergetics defects, axonal transport impairment, neuronal injury, and cognitive decline in neuronal cells and murine models. All these abnormalities are considered relevant in the early neurodegenerative manifestations such as memory and cognitive failure reported in AD. Therefore, in this review, we will discuss for the first time the importance of truncated tau by caspases activation in the pathogenesis of AD and how its negative actions could impact neuronal function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tau protein is a microtubule-associated protein (MAP) that contributes to neuronal function by participating in microtubule dynamics and stability [1, 2]. Tau undergoes numerous post-translational modifications such as phosphorylation, glycosylation, nitration, methylation, and proteolytic cleavage, among others [reviewed in 3]. For a long time, tau hyperphosphorylation has been proposed as the main neurotoxic factor in AD [3-5]. This tau form promotes the formation of neurofibrillary tangles (NFTs) that accumulate in neuropil threads and neuritic plaques in AD [6]. However, nowadays has been proposed that proteolytic cleavage of tau by caspases could play a vital role in the genesis and progression of AD [7-9]. Furthermore, several studies have suggested that caspase-3-cleaved tau contributes to NFT formation and neurodegeneration in the late stages of AD and aging [7]. Current reports suggest that this tau form is an early toxic component in AD and is a crucial protagonist in neuronal dysfunction [10].
Tau is a substrate for caspases cleaving this protein’s C-terminus or N-terminus regions [11-13]. These proteolytic processes generate tau fragments that induce aberrant neuronal functions, including synaptic impairment, cell death, neurodegeneration [14-16], and cognitive loss [17]. Interestingly, evidence has shown that caspase-3-cleaved tau contributes to neuronal damage [18-20]. More importantly, caspase-3-cleaved tau promotes mitochondrial injury, which is also considered an important event in the pathogenesis of AD [21-23]. Mitochondrial function plays an essential role in neuronal function [24-27], and several abnormalities in these organelles have been described in AD [28]. In this context, reports showed that truncated tau by caspase-3 induces defects in mitochondrial dynamics, transport, and bioenergetics [29, 30]. However, how cleavage of tau by caspases leads to mitochondrial dysfunction and consequently participates in the onset and progression of AD remains poorly understood. Therefore, in this review, we will discuss evidence supporting the toxic effects of truncated tau by caspases against mitochondrial and neuronal function and its role in the pathogenesis of AD.
The Physiological Role of Tau in Neurons
Tau has been studied for its role in tauopathies such as AD [23, 29, 31]. Tau is widely expressed in neurons, being more abundant in axons [32], although studies have shown their presence in synaptic zones [33]. During development, tau is distributed in neurons, while in the maturation process, tau is enriched in axons [34, 35]. The MAPT gene encodes tau on human chromosome 17q21 [34, 36]. Tau gene contains 16 exons producing six isoforms of tau where alternative mRNA splicing of exons 2 and 3 produces isoforms with no (0N with no exons 2 and 3), one (1N, with no exon 3) or two (2N, with exons 2 and 3) amino-terminals inserts in the central nervous system (CNS) [34, 37]. Instead, exons 10 form isoforms with three (3N, with no exons 10) or four (4N, with exons 10) microtubule-bindings regions resulting in tau of 352 to 441 amino acids depending on the immature and mature brain [37].
Tau interacts with microtubules, specifically with tubulin (a central component of microtubules in the neuronal cytoskeletal) by the C-terminal domain [34], and this process is modulated through the phosphorylation state of tau [38]. In this context, the tau function is regulated by its phosphorylated and dephosphorylated states induced by several kinases and phosphatases, respectively [39]. Three categories of protein kinases phosphorylate tau: (a) second-messenger-activated kinases, including protein kinase C (PKC), protein kinase A (PKA), and Ca2+/calmodulin-dependent protein kinase II (CAMKII); (b) Ser/Pro-directed kinases such as Mitogen-activated-protein-kinase (MAPK), Glycogen synthase kinase-3 beta (GSK-3β), Cyclin-dependent kinase 2 (cdk2), and Cyclin-dependent kinase 5 (cdk5); and (c) other tau-directed kinases such as Ser-626, casein kinases, and DNA-dependent protein kinases [38, 39].
It is well understood that tau stabilizes and polymerizes microtubules which are vital for the axonal transport of organelles, proteins, and lipids by anterograde and retrograde movement through motor proteins such as kinesin (anterograde transport) and dynein (retrograde transport) [40, 41]. Axonal transport is pivotal to neuronal function, and abnormalities in this process induced by pathological forms of tau could trigger synaptic dysfunction and neuronal death [42].
Tau plays a vital role in synaptic function, being involved in the long terminal depression (LTD) process by its post-synaptic compartment localization in the hippocampus [43]. For example, Briner et al. described an interesting role of tau in N-methyl-D-aspartate (NMDA)-mediated synaptic function in hippocampal neurons [41]. This mechanism is related to the interaction between tau, the Fyn protein, a non-receptor tyrosine-protein kinase member of the Src family, the post-synaptic density protein 95 (PSD95), and the NMDA receptors [44]. Here, microtubule stability and assembly are regulated by a proline-rich region of tau that contains PXXP motifs to promote interaction with Fyn [34, 37]. This interactive link promotes a complex formation composed of PSD95-NMDA receptor-Fyn-tau, where tau regulates this communication in post-synaptic zones, enhancing the synaptic process [45]. In pathological conditions, tau can be modified by promoting tau disassembly from microtubules reducing PSD95-NMDA receptor-Fyn-tau complex formation, and the latter affects post-synaptic NMDA localization and LTD activity [41-43, 45].
On the other hand, our previous work has suggested an exciting role of tau in regulating hippocampal mitochondrial function [44]. We showed that genetic ablation of tau reduced oxidative stress, improved mitochondrial function, and prevented cognitive decline in young and aged mice, indicating that tau plays an important role in regulating neuronal metabolism [46].
Tau undergoes post-translational modifications such as glycosylation, nitration, methylation, prolyl-isomerization, glycation, phosphorylation, and proteolytic cleavage [34]. Nevertheless, tau can be hyperphosphorylated and cleaved in the neurodegenerative context contributing to AD [8, 21, 23, 29]. Furthermore, several findings showed that tau is a substrate of several proteases, including caspases 2, 3, 6, and 9 [12, 13, 17, 47]. Interestingly, this evidence has shown that caspase induces cleavage of tau in the early stages of AD, which precedes NFT formation [7]. Complementary, several reports have demonstrated that proteolytic cleavage of tau contributes to its aggregation and, finally, to NFT formation [47-49], which is observed in the late stages of AD [50]. Therefore, in the following sections, we will discuss how tau could be proteolytically cleavaged and how these toxic soluble forms may contribute to the pathogenesis of AD.
Soluble and Insoluble Tau Modifications
Deposits of misfolded proteins, including tau, are considered a hallmark in AD [51, 52]. The etiology of how soluble tau becomes insoluble has been primarily studied, highlighting the contribution of these forms in NFT formation [53]. As tau progresses to its final insoluble state (NFT), it first passes through a soluble state in which tau undergoes several conformational changes, including dimeric, oligomeric, and fibrillar states [54]. In this context, in vitro studies have shown that NFT formation consists of previous steps such as dimerization, multimerization, oligomerization, and finally, a fibrillar structure formation which fractions are insoluble in detergents such as sarkosyl [55-57]. NFT (or insoluble state of tau) has been proposed as a protective or compensatory mechanism when soluble toxic tau forms are present [54, 58].
Additionally, studies have documented that N- and C-terminal truncation of tau is an early event in tau aggregation, inducing a soluble tau stage to sarkosyl, which later promotes tangle formation in transgenic rats expressing human truncated tau [59]. Concordantly, other studies suggested that neurons from the hippocampal Cornus ammonia (CA) 1 region of AD patients containing NFT can survive 15 to 25 years [60], indicating that the NFT presence is not related to neuronal loss in AD [60]. Importantly, Santa Cruz and collaborators have presented interesting evidence studying the role of NFT formation and the cognitive and neurodegenerative changes present during AD [61]. These studies showed that PHF-1 tau aggregates (hyperphosphorylated tau epitope of NFT) are not correlated with cognitive and memory impairment in AD murine model [61]. However, the presence of soluble tau induced neurotoxicity and cognitive decline, demonstrating the importance of pathological soluble forms of tau in the early stages of AD [61]. Complementary, other studies using P301L transgenic mice model for frontotemporal dementia showed that the tangle-bearing neurons (confirmed by PHF-1 antibody) were Arc (reflect electrophysiological neuronal response) positive compared to soluble tau mice that presented a reduction in Arc levels and neuronal loss [15]. Therefore, this evidence strongly suggests that soluble tau can be the primary toxic agent against neuronal function and cognitive performance in the early stages of AD.
Interestingly, the study of soluble tau forms such as caspase-cleaved tau has caused great interest since its presence has been associated with several neuronal abnormalities observed in AD. The following section will discuss caspase’s function and how its actions can negatively affect tau protein.
How Is Tau Cleaved?
Caspases are proteases with a well-identified role in programmed cell death, apoptosis, and inflammation [62, 63]. Several studies have explored the non-apoptotic role of caspases in the toxic proteolytic of different proteins, including tau, which contributes to AD onset [7, 64, 65]. This section will briefly discuss caspases’ contribution to the tau proteolytic process in the brain (Fig. 1).

Caspase activity modifies tau structure and induces neuronal damage in AD. (A) Caspases are involved in apoptotic activity. Here, cytochrome C is released from mitochondrial intermembrane space and binds to apoptosis protease-activating factor-1 (Apaf-1), which then promotes caspase-9 and caspase-3 activation leading to apoptosis. (B) Accumulative evidence has shown that caspases present a non-apoptotic activity, inducing cleave of tau protein. In this context, different caspases could cleave different tau regions; caspase-6 cleave N-terminal D13, and caspase-2, 9, and 3 cleave D314, D315, and AspD421, tau residues, respectively. More importantly, several studies have reported that cleaved tau by caspases induced neuronal impairment, whereas caspase-3-cleaved tau has been suggested to have an essential role in aging and the early stages of AD. Also, caspase-3-cleaved tau is accumulated in neurites zones, which could affect synaptic function
Caspase Role
Caspases are a proteases family responsible for hydrolyzing cysteine-dependent peptide bond residues [66] and are defined by their different activities [67]. Furthermore, David and collaborators categorized the caspase family by its apoptotic role: caspases 3, 6, 7, 8, 9; and inflammation role: caspases 1, 4, 5, 12 [67]. Also, other studies grouped caspase family by caspase initiators: caspases 2, 8, 9, 10; effector caspases: caspases 3, 6, 7; and inflammatory caspases: caspases 1, 4, 5, 11, 12 [68]. Furthermore, caspases are activated by intrinsic cell pathways where mitochondria actively participate in the apoptotic process [69] by cytochrome C release [70]. This pathway is activated by oxidative stress [71], DNA damage [72], accumulation/aggregation of unfolded proteins [73], and hypoxia [74], among others. Briefly, cytochrome C is released from mitochondrial intermembrane space into the cytosol, which binds to the apoptosis protease-activating factor-1 adapter molecule (Apaf-1) [75]. Then, Apaf-1 will generate the caspase-9 activation, which promotes the activation of caspase-3 and 7 to induce apoptosis (Fig. 1) [75, 76].
Apoptotic cascade and consequent caspase-9 activation can be modulated by mitochondrial dysfunction [77-80]. Mitochondrial injury produced by ROS production is linked to several types of cellular damage, including hypoxia and unfolded protein aggregations [73, 74, 81, 82]. In addition, ROS-induced oxidative damage will affect mitochondrial permeability, realizing Cyt C, which starts an apoptotic cascade and caspase-9 activation [81, 82]. Interestingly, several reports have documented that soluble forms of tau-induced ROS overproduction lead to mitochondrial failure and, consequently, to neuronal damage induced by caspase activation [77, 81, 82]. This evidence indicates that mitochondrial dysfunction can be considered an upstream event in the caspase activation process.
Interestingly, caspases are also involved in non-apoptotic functions, specifically caspase-3, which participates in synaptic plasticity [78]. First, reports have demonstrated an increased caspase-3 expression in CNS [79, 80]. Also, Bravarenko and Cols showed by electrophysiological experiments that blockage of caspase-3 by z-DEVD-fmk prevented the long-term potential (LTPs) process suggesting a physiological role of caspase-3 in synaptic plasticity [83]. In addition, other studies presented that caspase-3 activity is observed in the post-synaptic zone of zebrafish’s auditory forebrain, which is necessary for memory and learning performance [84]. However, additional research has highlighted the contribution of proteolytical caspase-3 activity on tau modifications during aging and neurodegenerative diseases such as AD [10, 17, 47, 80, 85].
Tau as a Substrate for Caspase Activity
Nowadays, it is well documented that caspases exert apoptosis activity and can cleave different proteins in aspartate residues, including tau [86]. Importantly, Zhao et al. have identified that the proteolytic process of tau by caspases could be an event mediated by apoptosis (pro-apoptotic protein) [87]. An increase in apoptosis levels was correlated to an increase in caspase-3 activity and the cleavage of tau at D421, which is known as truncated tau by caspase-3 [87]. Additionally, apoptosis overexpression increased the cleavage of tau, affecting synaptic function, whereas apoptosis inhibition reduced caspase-3-cleaved tau production and synaptic failure [87]. Furthermore, several cleaved tau forms have been shown in five brain regions, including the AD brain’s entorhinal cortex, prefrontal cortex, motor cortex, and hippocampus (Fig. 1) [88]. Interestingly, the most prominent cleaved tau forms found were truncated in the C-terminal in these AD patients [88]. Complementary, in vitro studies have demonstrated that the deletion in the first 150 aa and the last 50 aa of tau is involved in its pathologic activity promoting self-aggregation [89], which was an age-dependent effect [90].
Effector caspases depend on the cleavage at aspartate residues, where caspase-3 has an essential role in the proteolytic process in this tau region observed in AD [47, 68]. In this context, tau is an essential substrate for caspase-3, leading to the soluble monomeric state of tau and its aggregation in AD [91]. Also, several reports have indicated that tau is cleaved by caspase-3 at the carboxy-terminus residue aspartic acid (Asp421) [92, 93]. Furthermore, in vitro studies in SH-SY5Y cells subjected to UV-irradiation treatment showed an increase in caspase-3-cleaved tau levels and NFTs whose expression was inhibited by zVAD-fmk ((carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone) pan-caspase inhibitor) treatment [47].
On the other hand, microtubule organization is essential to translate different cargoes in neurons maintaining their activity-dependent function [94, 95]. For example, a study demonstrated that truncated tau in Asp421 destabilizes microtubule activity in the mouse brain and HEK (human embryonic kidney) cells [96]. Here, studies using buffer PIPES (Piperazine-N, N′-bis(2-ethanesulfonic acid) and centrifugation showed that the interaction between truncated tau by caspase-3 and cytoskeleton reduced full-length tau/cytoskeleton binding inducing the disassembling of tau from microtubules [96].
Truncated Tau Impairs Neuronal Function
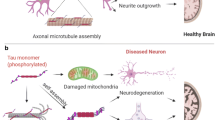
Synaptic plasticity is an active process linked to neurological modifications in the cytoskeleton involved in learning and memory [97]. Defects in cognitive and memory performance have been linked to AD [47, 98]. Studies have shown that soluble tau forms promote aggregation [57, 99], neuronal loss, and cognitive deficiencies [61]. More importantly, evidence proposes soluble tau as a toxic agent, while insoluble tau aggregates may protect neuronal cells [100]. In this context, it was thought that NFTs formed principally by aggregates of hyperphosphorylated tau at PHF-1 residues (Ser 396/Thr 404) causing synaptic and cognitive impairment in AD; however, current reports suggest that other actors, including caspase-3-cleaved tau, may contribute to this process (Fig. 2) [47].

Caspase-3-cleaved tau promotes mitochondrial dysfunction and synaptic injury. (A) In normal/physiological conditions, tau is enriched in axons (B), promoting microtubule stability and contributing to anterograde and retrograde transport of cargos such as mitochondria. (C) Mitochondria are essential organelles for neuronal activity by supplying ATP, regulating calcium levels, and recycling neurotransmitters. However, these processes are harmed in AD. (D) Additionally, caspase-3 activity has been involved in the onset and progression of AD. (E) Caspase-3-cleaved tau induces microtubule destabilization leading to tangle formation, which is observed in the late stages of AD. (F) Caspase-3-cleaved tau promotes mitochondrial dysfunction, such as ROS increase, mitochondrial transport damage, ATP loss, and calcium deregulation, where mitochondrial permeability transition pore (mPTP) could play a pivotal role. (G) Mitochondrial injury induced by caspase-3-cleaved tau could lead to synaptic failure
Tau has been observed in synaptic compartments of the AD brain, playing a pivotal role in the defects of neuronal communication [101, 102]. Furthermore, several studies have demonstrated that truncated tau is detected in pre- and post-synaptic zones affecting neuronal function [16, 103]. Also, expression of human truncated tau at N- and C-terminals (encompassing three repeats (amino acid (aa) 151–391; line SHR24)) affected cytoskeletal proteins in the pre- and post-synaptic compartments in transgenics rats (heterozygous transgenic male rats expressing human N- and C-terminally truncated tau) [104]. Furthermore, truncated tau increased β-tubulin expression, reduced synaptophysin, slightly increased bassoon (synaptic vesicle clustering protein) expression levels, and significantly reduced the number of synaptic vesicles compared to wild-type rats [104]. Accordingly, using flow cytometry and immunofluorescence analyses, Sokolow and collaborators showed that truncated tau by caspase-3 was accumulated in synaptosomes from AD patients [16]. More importantly, these authors showed that caspase-3-cleaved tau was the most abundant in synaptic zones of AD patients [16].
On the other hand, using electrophysiology analysis, Loon et al. showed that overexpression of caspase-3-cleaved tau reduced long-term potentiation (LTP) in older C57BL/6 J mice compared to the age-matched control group without caspase-3-cleaved tau expression [105]. These effects also triggered a neuronal loss in the entorhinal cortex and hippocampus observed by NeuN immunoreactivity [105]. In the same context, synaptic impairment and neurodegeneration were studied in a murine model (embryos and positive F0 mice) that expressed caspase-3-cleaved tau at developmental levels [106]. Immunohistochemical assays revealed that the expression of caspase-3-cleaved tau reduced synaptophysin (synaptic vesicle protein) levels in an age/dependent manner in the hippocampal CA3 region [106]. In addition, synaptic proteins such as PSD95, N-methyl-D-aspartic receptor 1(NR1), and 2B (NR2B) were decreased in the hippocampus of 3- and 6-month-old mice expressing caspase-3-cleaved tau [106]. Complementary, these changes in synaptic density were associated with the impairment of spatial working memory observed in 1-month-old mice that expressed caspase-3-cleaved tau worsened age-dependently [106]. Interestingly, these observations were recently corroborated by our group in which caspase-3-cleaved tau expression reduced dendritic filopodia and synaptic vesicles number in hippocampal neurons from mice and rats [107].
As aforementioned, caspase-3 has been abundantly found in the post-mortem brain of AD patients in the frontal, temporal, parietal, and cerebellar cortex [108]. At the same time, caspase-3 has been observed in post-synaptic density, which is significantly increased in the cingulate cortex and hippocampus of the AD brain [109]. Although caspase-3 is vital to induce proper neuronal development, LTP, and consequent neuronal plasticity [110], its overactivation is critical to trigger neuronal injury [47]. For example, analysis of hippocampal tissue in C57BL/6 senescent mice (27 months old) showed high levels of caspase-3-cleaved tau compared to young mice (2 months old) [111]. These findings were associated with decreased interaction between this tau form and microtubules and reduced transport cargos promoting dendritic atrophy in hippocampal CA1 neurons [111].
Also, caspase-6 contributes to the tau-cleaved process, although its neurotoxic effect remains under discussion [112, 113]. Cleavage of tau by caspase-6 is produced at D13 N-terminal tau (TauΔcasp6) [13]. Caspase-6-cleaved tau has been observed in shorted neurites and soma from neurons studied in post-mortem AD brains [114]. Interestingly, increasing levels of caspase-6-cleaved tau have been found in hippocampal tissue from aging patients compared to young brains. Also, these changes were associated with reduced cognitive performance in aging patients [115]. Complementary, caspase-6-cleaved tau levels were observed in cerebrospinal fluid (CSF) of non-cognitively impaired (NCI), mild cognitive impairment (MCI), and AD patients, and they were correlated with neurocognitive and neuropathologic states being increased as cognitive performance and disease worsen [116].
Tau is also cleaved by caspase-2 at Asp314, generating Δtau314 [117]. Furthermore, amino-terminal tau fragments cleaved by caspase-2 induce synaptic impairment and memory loss [112, 118]. For example, in hippocampal culture neurons, the expression of Δtau314 promotes the spreading of tau into dendritic zones, while dendritic spines of wild-type neurons rarely showed the presence of tau [118], suggesting that caspase activity contributes to tau accumulation in post-synaptic zones. Concordantly, measurements of miniature excitatory post-synaptic currents (mEPSCs) in culture hippocampal neurons with Δtau314 expression showed a significant reduction in mEPSCs amplitude and frequency [116]. Also, these studies showed a decrease in Glur1 (subunit of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor) expression [118], which was enough to induce neuronal and cognitive damage [118]. Finally, it was also observed that caspase-9 has proteolytic activity on tau at Asp315, where this overactivation is documented in AD brains [12]. In addition, several studies indicate that caspase-9 is involved in NFT formation, which was observed in both hippocampus and frontal cortex [12]. Notably, it has been reported that caspase-9 activity could trigger dystrophia neuronal, synaptic plasticity failure, and memory loss from FDDKI mice (a mice model that develops synaptic and memory impairment by loss of BRI2 protein, which regulates amyloid precursor protein (APP) processing) [12, 119].
Therefore, caspases-induced tau modifications result in neurotoxic effects associated with synaptic loss and severe cognitive impairment. More importantly, the analysis of caspase-3-cleaved tau has generated a significant impact since its presence is detrimental to neuronal function and the onset and progression of AD, which we will discuss in the next section.
Caspase-3-Cleaved Tau Is an Early Hallmark of AD
AD is a neurodegenerative disorder representing the most common form of dementia clinically characterized by cognitive deficits such as learning and memory impairment, spatial disorientation, and behavioral disabilities [120]. In addition, AD is characterized by synaptic loss [121], oxidative damage [122], and neuroinflammation [123], where tau protein has been mainly postulated as a critical candidate to induce these effects [124, 125]. Accumulative evidence has suggested that NFTs are the main histopathologic hallmarks in AD [3, 126, 127]. However, it was demonstrated that the aggregation and insoluble tau state is not required to trigger the onset and memory impairment shown in AD [128]. In contrast, caspase-3-cleaved tau has raised interest in AD research since studies have demonstrated that this soluble tau form is considered an early event in tangle formation promoting neuronal injury [129]. For example, Kang and collaborators showed that PHF-1 tau is cleaved by caspase-3 during apoptosis-inducing neuronal damage in post-mortem tissue of AD patients and primary cortical neurons [19]. Here, cortical neurons were treated with staurosporine (100 nM) and calyculin A (10 nM) (apoptosis inducers), inducing cleavage of PHF-1 tau at 8 h by activation of caspase-3, which was inhibited by zDEVD (inhibitor of caspase-3) treatment [19]. More importantly, fragments of PHF-1 tau were observed from post-mortem brains of AD patients associated with increased caspase-3 and pro-caspase-3 levels accompanied by a 70% of colocalization between PHF-1 tau-positive neurons and caspase-3 [19]. These results suggest a crucial role of caspase-3 activity in the cleavage of hyperphosphorylated tau (Fig. 2) [64].
In the same context, the cleaved tau at C-terminal, specifically in AspD421 residue, by caspase-3 is considered an essential contributor to AD onset and progression [48, 130]. Evidence has shown a significative immunoreactivity of caspase-3-cleaved tau within the hippocampal CA1 region and entorhinal cortex in post-mortem brain from AD patients compared to control age-matched brain samples [64]. Also, Loon et al. demonstrated that 4- and 12-month-old mice (C57BL/6 J mice) expressing caspase-3-cleaved tau showed cognitive abnormalities such as spatial and recognition memory impairment, short-term work memory failure, and long-term potentiation (LTP) impairment in CA1 region by electrophysiologic analyses of field excitatory post-synaptic potentials (fEPSPs) [105]. Also, other studies reported that stable expression of caspase-3-cleaved tau in immortalized cortical neurons induced cell viability loss observed by an increase in lactate dehydrogenase (LDH) release compared to cells expressing full-length tau [20]. Furthermore, Conze and collaborators demonstrated that caspase-3-cleaved tau reduced axonal transport contributing to synaptic impairment [111]. First, they observed that 27-month-old mice (C57BL/6 J mice) showed increased caspase-3-cleaved tau levels compared to age-match mice [111]. In addition, PC12 cells (catecholamine cells positive for norepinephrine and dopamine) expressing caspase-3-cleaved tau showed a reduced number, velocity, and speed of axonal cargos mitochondria and synaptic vesicles, which also reduced dendritic length [111]. Therefore, the accumulation of caspase-3-cleaved tau negatively affects neuronal transport and synaptic morphology, triggering cognitive damage and neurodegeneration (Fig. 2).
On the other hand, caspase-6-cleaved tau has also been proposed to promote AD progression [131]. Increased caspase-6 expression levels were observed in adult human neurons compared to the fetal brain [108, 132]. Active caspase-6 and caspase 6-cleaved tau are abundant in neurites plaques (NPS), neuropil threads (NPTs), NFT, and pre-tangles of AD brain [113], suggesting that caspase-6 is activated from early stages of tangle formation [111]. According to this, Albrecht and collaborators showed that active caspase-6 was observed in the hippocampus from MCI and AD brains [131]. However, caspase-6-cleaved-tau was significantly increased in severe AD post-mortem brains compared to age-match control and MCI brain samples [131]. Other studies have correlated increased caspase-6 activity with memory loss in the hippocampus and entorhinal cortex during aging [115, 133]. Importantly, caspase-6-cleaved tau has been observed in different hippocampal regions from AD cases, such as CA1, CA2, CA3, and CA4 [116]. In this context, LeBlanc and collaborators showed an increase in caspase-6 activity and caspase-6-cleaved tau levels in the CA1 region of the aged human brain, which was correlated with semantic and declarative memory impairment [134]. Although previous evidence has demonstrated a possible implication of caspase-6-cleaved tau in the pathogenesis of AD, nowadays has been discussed whether an increase in caspase-6 activity can be considered a key element to induced pathological modifications of tau in AD. For example, caspase-6 fails to induce tau hyperphosphorylation, aggregation, cognitive deficit, and neuroinflammation from transgenic mice expressing cleaved tau by caspase-6 [112]. Also, mice expressing CAMKII-Cre-dependent hTau and hCasp6 in the cortical region and hippocampal CA1 pyramidal did not show cognitive and locomotor decline accompanied by no changes in dendritic spines and inflammatory response [112].
Altogether, this evidence demonstrates that the cleavage of tau by caspase-6 could be associated with clinical signs of AD. However, more studies are necessary to understand its definitive role in this disease.
Caspase-2- and 9-cleaved tau have been found in AD brain patient samples [12, 135]. In this context, caspase-9 activation has been observed in the entorhinal cortex and hippocampus of AD cases and was colocalized with AT8 increased levels (indicates tau hyperphosphorylation in Ser202/Thr305 sites), and with oxidative damage observed by an increase in anti-8oxodG (8-Oxo-2′-deoxyguanosine) levels (oxidative damage indicator for either DNA or RNA) [12]. Also, caspase-2-cleaved tau levels were elevated in aged brain subjects with cognitive deficits [118]. In addition, the infusion of anti-caspase-2 morpholino oligonucleotides during 28 days in lateral ventricles of rTg4510 mice (mice model that expresses a form of tau containing the P301L mutation linked to frontotemporal dementia) reduced caspase-2 cleavages of tau and, consequently, reverted cognitive impairment [118]. Accordingly, caspase-2 expression levels were increased in AD- and MCI-diagnosed brain samples [135].
Nevertheless, it was recently observed that caspase-2 mRNA levels are not elevated in both MCI and AD samples, which were not associated with a reduction in synaptosome-associated protein ((SNAP-25) protein complex responsible for pre-synaptic membrane fusion of synaptic vesicles) mRNA levels [136]. Therefore, the caspase-2 and 9-cleaved tau role in AD is still elusive.
Contribution of Caspase-3-Cleaved Tau to Mitochondrial Dysfunction in AD
Mitochondria are pivotal organelles to neuronal function, supplying energy and contributing to redox balance and calcium regulation, all essential for synaptic plasticity [137]. In addition, axonal mitochondrial transport is also crucial to neuronal function, which is regulated by motor proteins involved in mitochondrial attachment to microtubules and allows the arrival of these organelles to zones with high energy demands where synapses occur [137, 138].
Previously, we highlighted that the cleaved tau by caspases triggers neuronal dysfunction observed in AD cases and several murine models. However, to this date, how truncated tau is involved in mitochondrial impairment in AD has not been entirely understood. Therefore, we will discuss pivotal evidence demonstrating how caspase-3-cleaved tau at D421 is involved in mitochondrial and synaptic damage in AD (Fig. 2).
Mitochondrial Dysfunction Is a Critical Event in AD
Neuronal cells present a high energetic demand for synaptic function, regulating mitochondrial shape as a necessary process [139-142]. Mitochondrial shape (characterized by fusion and fission process) and its localization are regulated by a synaptic activity where its density and number increase in the dendritic compartment [139]. In this context, specialized GTPases proteins controlling the fusion of the outer mitochondrial membrane (OMM) are the mitofusins 1 and 2 (Mfn1/2), while optic atrophy protein 1 (Opa1) controls the fusion of the inner mitochondrial membrane (IMM) [143]. Also, Dynamin-related protein 1 (DRP1) is a cytosolic protein recruited into mitochondria by the Fis1 protein promoting mitochondrial fission [143]. Mitochondrial dynamics are negatively regulated during AD observing abnormal mitochondrial distribution and synaptic failure [144, 145]. For example, recent studies showed that mitochondrial genes involved in the regulation of mitochondrial morphology and movement, such as Mfn2, DNML1, Opa1, and Fis1 genes (commanding mitochondrial morphology) and GAPDH (mitochondrial transport), are negatively modified in AD brains [144]. Also, complementary studies in primary neurons from APP AD mice showed a reduction of Drp1, Mfn2, Mfn1, and Opa-1 protein levels; however, Fis1 protein levels were significantly increased. These results were correlated with diminished mitochondrial density and dendritic spine numbers from primary culture neurons [145]. Additionally, Mankzak and collaborators showed adverse changes in mitochondrial dynamics genes in AD brain samples [146]. Here, Drp1 and Fis1 mRNA expression was upregulated, whereas Mfn1, Mfn2, and Opa1 mRNA expression was downregulated in early-stage AD patients [146]. Also, other studies demonstrated an abnormal regulation of mitochondrial dynamics and transport using primary neurons from transgenic mice Tg2576 (mouse model expressing human amyloid precursor protein) [147]. Notably, hippocampal neurons showed mitochondrial fragmentation produced by decreased Mfn1 and Opa1 levels and reduced mitochondrial number and anterograde transport [147].
During neuronal activity, ATP is generated by the action of electron transport chain (ETC) complexes, I–II and III (located at IMM), which pump protons into mitochondrial intermembrane space to activate ATP synthase (complex V) [140-142]. In addition, ROS are produced mainly by mitochondrial complexes I and III, which are balanced by antioxidant defenses produced by mitochondria [148-150]. On the other hand, mitochondria regulate cytosolic calcium levels by mitochondrial calcium uniporter (MCU), mitochondrial permeability transition pore (mPTP), and Na+/Ca+ and H+/Ca+ exchangers [151-154]. These mitochondrial events are essential to neuronal communication regulating dendritic spines and vesicle recycling [155, 156].
Mitochondrial dysfunction has been strongly associated with the onset and progression of AD, where ATP loss, ROS overproduction, and calcium deregulation were observed [157-160]. ETC activity, mitochondrial complexes protein expression, and consequently, ATP production were reduced in AD [161]. These results were accompanied by decreased glycolytic enzyme expression and decreased pyruvate dehydrogenase (PDH) activity [161]. Concordantly, other studies have reported a reduction in cerebral glucose utilization in AD patients by measuring 2[18F] fluoro-2-deoxy-D-glucose (18F-FDG) metabolism by positron emission tomography (PET) [162]. Furthermore, hippocampal tissue of 12-month-old 3xTg-AD mice, a mouse model that expresses three mutations associated with AD: APP, P301L, and presenilin 1, showed a reduction in PDH and complex IV proteins levels with a concomitant mitochondrial respiratory capacity impairment [160]. Also, studies using P301L mutant mice, a mouse model that showed accumulation of hyperphosphorylated tau, showed a decrease in complexes I and V activities, mitochondrial respiratory capacity, and ATP production [163]. Furthermore, analysis of AD brain tissue shows a reduction in mRNA expression of glycolysis, tricarboxylic acid (TCA) cycle, and OXPHOS protein components [164]. Decreased mitochondrial membrane potential (ΔΨm) has also been considered a vital sign of mitochondrial injury observed in the brain cortex of APP mice [165]. ROS overproduction is also reported in different brain regions such as the cortex, hippocampus, striatum, and amygdala [166], triggering oxidative stress and, finally, synaptic failure in Aβ APP-Swedish mutation (AβPPsw) mice [122]. These deleterious mitochondrial defects are shown in AD brains observing downregulation of complex I, III, and IV gene expression, affecting OXPHOS activity and consequently inducing ΔΨm depolarization and ATP loss [167].
Mitochondrial calcium handling defects have been associated with neuronal dysfunction in AD [168, 169]. In this context, mPTP contributes to mitochondrial calcium uptake, whose activity is regulated by cyclophilin-D (CypD) [170]. Interestingly, hippocampal neurons treated with cyclosporin A (CsA), an inhibitor of mPTP opening, significantly prevent calcium overload, mitochondrial dysfunction, and excitotoxicity [171]. In addition, brain mitochondria have been observed to be sensible to mPTP opening in AD, leading to mitochondrial swelling, mitochondrial cristae disruption, ΔΨm decay, and ATP loss [172]. Increased CypD levels have been observed in the AD brain and are associated with deregulating mitochondrial dynamics [146]. Furthermore, Du and collaborators showed that CypD contributes to mitochondrial bioenergetics defects (ROS, ATP, and calcium regulation) and, consequently, to synaptic injury and cognitive damage in AD mice model overexpressing a mutant human form of APP (mAPP, J-20 mice) [173]. Interestingly, CypD deletion rescue mitochondrial bioenergetic failure and neuronal function by regulating calcium concentration [173].
Mitochondrial dysfunction is an important event promoting the onset and progression of AD. Notably, solid reports suggest that proteolytic cleavage of tau by caspase-3, generating caspase-3-cleaved tau, induces mitochondrial defects, which will be discussed in the next section.
Mitochondrial Impairment Induced by Caspase-3-Cleaved Tau
Mitochondrial function impairment has been widely reported in AD pathology, where proteolytic cleavage of tau could be an important contributor to these abnormalities [19, 21, 29]. Interestingly, we showed that genetic suppression of tau improved mitochondrial bioenergetics and dynamics in tau knock-out (−/−) mice[46]. Also, we observed that homozygous tau knock-out (tau −/−) mice improved hippocampal memory performance, increased Mfn2 levels, and decreased Fis1 levels, reduced lipid peroxidation, and nitrosylated protein levels, and increased ATP levels [46]. More importantly, tau (−/−) mice showed a reduction in mRNA and expression of CypD levels compared to age-matched mice (18 months old) [174], suggesting that CypD could play a key role in mitochondrial dysfunction induced by tau. Complementary studies of Lopes et al. demonstrated that tau (−/−) mice prevent dendritic dystrophy, improved vesicle recycling, and increased levels of ETC subunits such as complex I, III, IV, and V when tau (−/−) mice were subjected to stress [175].
Mitochondria are dynamic organelles that move, fusion, and divide and whose actions play a role in neuronal function through synapses [176]. In AD, mitochondrial dynamics are significantly altered, where reports showed that mitochondrial fragmentation is increased and the fusion process is reduced, leading to morphologic changes [159] and dynamic process imbalance [177, 178]. In this context, increased expression of Drp1 has been related to mitochondrial fragmentation and neurotoxicity promoted by tau pathology [179]. In this context, partial reduction of Drp1 has been proposed as a therapeutic target in AD since it improves cognitive performance and reverts neuronal plasticity injury in a transgenic tau mouse model [179]. Complementary, Fis1 levels have increased while Mfn1/2 and Opa1 expression is significantly reduced in hippocampal neurons from AD mice models [145]. Also, Opa1deficiencies contribute to dendritic spines loss and reduced expression of synaptic proteins such as synaptophysin (pre-synaptic protein) and PSD95 (post-synaptic protein) in the hippocampus of Opa1 (+/−) mice which were associated with memory loss [180]. Previously, our group demonstrated that Opa1 and mitochondrial fusion is affected by the expression of caspase-3-cleaved tau in immortalized cortical neurons, rat hippocampal neurons, and wild-type and tau (−/−) mice cultured neurons [21, 181]. Also, the expression of caspase-3-cleaved tau enhances mitochondrial dynamics abnormalities (fragmentation) induced by Aβ treatment [181] (Fig. 2).
Mitochondrial transport and distribution are pivotal to the synaptic process, including vesicle recycling [182], exocytosis/endocytosis [155, 183], dendritic spines formation [184], and neuronal plasticity [185]. Mitochondrial transport is commanded by motor proteins such as kinesin (anterograde transport), dynein (retrograde transport) [186], and trafficking kinesin-binding (TRAK) 1/2, known as mitochondrial adaptor protein [187], which interacts with the heavy kinesin chains domain [188, 189]. Interestingly, aggregation of tau induced a decrease in mitochondrial velocity, reduced mitochondrial population in the neuronal process [190, 191], and decreased mitochondrial motor protein expression in rat neuroblastoma 2a (N2a) cells [192]. Furthermore, mitochondrial transport was significantly affected by caspase-3-cleaved tau expression in hippocampal neurons from tau (−/−) knock-out mice and rats [30]. Likewise, rat/mice hippocampal neurons expressing cleaved tau by caspase-3 decrease the number of moving mitochondria in the axonal and dendritic processes without affecting mitochondrial velocity [193]. Concordantly, expression of caspase-3-cleaved tau significantly promoted mitochondrial accumulation in neuronal soma compared to hippocampal neurons transfected with full-length tau [30].
Interestingly, further studies showed that caspase-3-cleaved tau affects the function of mitochondrial adaptor protein TRAK2 affecting mitochondrial transport [30]. In this context, TRAK2 is pivotal to mitochondrial localization in synaptic terminals since its genetic deletion has shown a reduced mitochondrial location in the neuronal process [194]. Furthermore, our group showed that caspase-3-cleaved tau reduced the expression of TRAK2 in immortalized cortical neurons and hippocampal neurons, affecting its axonal distribution and inducing TRAK2 accumulation in neuronal soma [30]. Therefore, these findings strongly suggest that truncated tau at D421 by caspase-3 affects mitochondrial dynamics and transport, where abnormalities in Opa1 and TRAK2 could play a role in AD (Fig. 2).
Studies of our group determine that caspase-3-cleaved tau expression impairs mitochondrial bioenergetics, harming neuronal survival [20, 22, 81]. Also, the expression of caspase-3-cleaved tau enhanced mitochondrial membrane potential (ΔΨm) loss and increased superoxide production induced by Aβ treatment [181]. Surprisingly, the expression of phosphorylated tau (Ser396/404) did not show any summative effects against the ΔΨm levels induced by Aβ treatment in hippocampal neuronal culture; however, caspase-3-cleaved tau reduced ΔΨm and increased superoxide production compared to neurons expressing full-length tau and hyperphosphorylated tau (PHF-1) [193]. Likewise, expression of caspase-3-cleaved tau promoted a decrease in ATP levels compared to cortical neurons expressing full-length tau along with ROS overproduction and the impairment of the Nrf2-dependent antioxidant pathway [81]. Interestingly, sulforaphane treatment (activator of the Nrf2 pathway) reduced ROS levels and reversed mitochondrial damage increasing ATP levels and preventing ΔΨm loss induced by caspase-3-cleaved tau [81] (Fig. 2).
Mitochondrial impairment induced by caspase-3-cleaved tau could be produced by activating mPTP [21, 107]. Caspase-3-cleaved tau expression reduces the influx of mitochondrial calcium, induces mitochondrial depolarization, and compromises mitochondrial membrane integrity [21]. Notably, the inhibition of mPTP opening by CsA prevented mitochondrial calcium influx and rescued all mitochondrial abnormalities induced by truncated tau by caspase-3 [21]. In addition, CsA treatment reverted mitochondrial fragmentation and integrity and increased ΔΨm, which were affected by cleavage of tau at D421 [21]. Importantly, we recently showed that CsA prevented mitochondrial dysfunction, ROS increase, and ATP loss induced by caspase-3-cleaved tau [107]. Also, CsA treatment reduced dendritic spine loss and prevented synaptic vesicle impairment produced by caspase-3-cleaved tau in mice hippocampal neurons [107]. Altogether, these findings suggest a novel role of mPTP on mitochondrial injury induced by caspase-3-cleaved tau (Fig. 2).
However, further studies are needed to determine the mechanism involved in mitochondrial damage induced by caspase-3.
Tau Pathology in Other Diseases
AD onset correlates to chronic diseases such as diabetes mellitus [195]. Briefly, hyperglycemia is one of the main symptoms of diabetes that correlates with cognitive decline [196] observed in AD. Furthermore, accumulative evidence has shown that hyperglycemia and diabetes could also be risk factors for AD [197, 198]. In this context, hippocampal neurons exposed to hyperglycemia showed accumulation of Aβ, increased hyperphosphorylated tau levels, oxidative stress, and mitochondrial dysfunction [197, 199-202]. Additionally, increased lipid peroxidation was observed in the hippocampus of diabetic rats [203].
Furthermore, reports have suggested that diabetes and AD are associated with abnormal brain structure, mitochondrial dysfunction, and reduced energy metabolism by ATP loss, reduced Δψm, and oxidative stress [204]. More importantly, other studies showed that diabetic mice induced by streptozotocin contributed to the cleavage of tau by caspase-3 and tau hyperphosphorylation in the brain [205]. Also, caspase-3 activity was increased in primary cortical neuronal cultures treated with glucose [205]. Furthermore, other studies using the streptozotocin diabetic mice model showed increased caspase-3-cleaved tau and tau hyperphosphorylated levels in the cortex and hippocampus [206]. Additionally, Latina and collaborators showed that cleavage of tau (66–81 aa of N-terminal) observed in streptozotocin-diabetic mice model contributed to hippocampal recognition and spatial memory loss, oxidative stress, reduced complex IV activity, and decreased mitochondrial respiratory capacity [207].
Altogether, these findings suggested that diabetes could contribute to the onset and progression of AD. However, future research must elucidate how hyperglycemia or diabetic models could promote proteolytic cleavage of tau and mitochondrial dysfunction.
Conclusions
In this review, we discussed evidence showing an essential role of truncated tau by caspases in neuronal injury and mitochondrial dysfunction. Tau is a MAP that contributes to microtubule stability, transports cargo such as mitochondria, and influences neuronal activity. Tau presents different post-translational modifications where proteolytic cleavage by caspases has been involved in the pathogenesis of AD (Fig. 1). Importantly, accumulated evidence has demonstrated that caspase-3-cleaved tau promotes tau aggregation and induces NFT formation. Furthermore, caspase-3-cleaved tau appears in the early stages of AD, modifying mitochondrial dynamics, transport, and bioenergetic functions affecting neuronal communication. In this context, caspase-3-cleaved tau is observed in synaptic zones, and its presence has been associated with reduced vesicle release, dendritic spines loss, and synaptic failure. More importantly, current evidence suggests that caspase-3-cleaved tau can be associated with mPTP opening, increasing ROS production, inducing calcium overload, and promoting mitochondrial dysfunction (Fig. 2).
Finally, truncated tau could be essential to synaptic plasticity impairment through the harmful modifications of mitochondrial function and pathological tau aggregation observed in AD.
Data Availability
N/A.
Abbreviations
- AA :
-
Amino acids
- AD :
-
Alzheimer’s disease
- AMPA :
-
α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- Apaf-1 :
-
Apoptosis protease-activating factor-1 adapter molecule
- Asp421 :
-
Asparginine 421
- AT8 :
-
Marker of tau hyperphosphorylation in Ser202/Thr305 sites
- ATP :
-
Adenosine triphosphate
- CA :
-
Cornus ammonis
- CAMKII :
-
Ca2+/calmodulin-dependent protein kinase II
- Cdk2 :
-
Cyclin-dependent kinase 2
- Cdk5 :
-
Cyclin-dependent kinase 5
- CNS :
-
Central nervous system
- CsA :
-
Cyclosporine A
- DNA :
-
Deoxyribonucleic acid
- DRP1 :
-
Dynamin-related protein 1
- fEPSPs :
-
Field excitatory post-synaptic potentials
- GSK-3β :
-
Glycogen synthase kinase-3 beta
- HEK :
-
Human embryonic kidney cells
- IMM :
-
Inner mitochondrial membrane
- LDH :
-
Lactate dehydrogenase
- LTP :
-
Long-term potentiation
- LTD :
-
Long terminal depression
- MAP :
-
Microtubule-associated protein
- MAPK :
-
Mitogen-activated protein kinase
- MCI :
-
Mild cognitive impairment
- MCU :
-
Mitochondrial calcium uniporter
- mEPSCs :
-
Miniature excitatory post-synaptic currents
- MFN :
-
Mitofusins
- mPTP :
-
Mitochondrial permeability transition pore
- N2a :
-
Neuroblastoma 2a
- NCI :
-
Non-cognitively impaired
- NFTs :
-
Neurofibrillary tangles
- NMDA :
-
N-Methyl-D-aspartate
- NPS :
-
Neurites plaques
- NPTs :
-
Neuropil threads
- NR1 :
-
N-Methyl-D-aspartic receptor 1
- NR2B :
-
N-Methyl-D-aspartic receptor 2
- Nrf2 :
-
Nuclear factor (erythroid-derived 2)-like 2
- OMM :
-
Outer mitochondrial membrane
- Opa1 :
-
Optic atrophy protein 1
- OXPHOS :
-
Oxidative phosphorylation
- PIPES :
-
Piperazine-N,N′-bis(2-ethane sulfonic acid)
- PKA :
-
Protein kinase A
- PKC :
-
Protein kinase C
- PSD95 :
-
Post-synaptic density protein 95
- ROS :
-
Reactive oxygen species
- SNAP-25 :
-
Synaptosome-associated protein
- TRAK :
-
Trafficking kinesin-binding
- zVAD-fmk :
-
Carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone
- 8oxodG :
-
8-Oxo-2′-deoxyguanosine
- ΔΨm :
-
Mitochondrial membrane potential
References
Kadavath H, Hofele RV, Biernat J et al (2015) Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc National Acad Sci 112:7501–7506. https://doi.org/10.1073/pnas.1504081112
Mietelska-Porowska A, Wasik U, Goras M et al (2014) Tau protein modifications and interactions: their role in function and dysfunction. Int J Mol Sci 15:4671–4713. https://doi.org/10.3390/ijms15034671
Yen S, Liu W-K, Hall FL et al (1995) Alzheimer neurofibrillary lesions: molecular nature and potential roles of different components. Neurobiol Aging 16:381–387. https://doi.org/10.1016/0197-4580(95)00022-7
Drummond E, Pires G, MacMurray C et al (2020) Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 143:awaa223. https://doi.org/10.1093/brain/awaa223
Wang J-Z, Wang Z-H, Tian Q (2014) Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer’s disease. Neurosci Bull 30:359–366. https://doi.org/10.1007/s12264-013-1415-y
Braak H, Braak E, Grundke-Iqbal I, Iqbal K (1986) Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett 65:351–355. https://doi.org/10.1016/0304-3940(86)90288-0
de Calignon A, Fox LM, Pitstick R et al (2010) Caspase activation precedes and leads to tangles. Nature 464:1201–1204. https://doi.org/10.1038/nature08890
Kanno T, Tsuchiya A, Nishizaki T (2014) Hyperphosphorylation of tau at Ser396 occurs in the much earlier stage than appearance of learning and memory disorders in 5XFAD mice. Behav Brain Res 274:302–306. https://doi.org/10.1016/j.bbr.2014.08.034
del Alonso AC, Zaidi T, Novak M et al (2001) Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc National Acad Sci 98:6923–6928. https://doi.org/10.1073/pnas.121119298
Bolós M, Pallas-Bazarra N, Terreros-Roncal J et al (2017) Soluble tau has devastating effects on the structural plasticity of hippocampal granule neurons. Transl Psychiat 7:1267. https://doi.org/10.1038/s41398-017-0013-6
Gamblin TC, Chen F, Zambrano A et al (2003) Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc National Acad Sci 100:10032–10037. https://doi.org/10.1073/pnas.1630428100
Rohn TT, Rissman RA, Davis MC et al (2002) Caspase-9 Activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol Dis 11:341–354. https://doi.org/10.1006/nbdi.2002.0549
Horowitz PM, Patterson KR, Guillozet-Bongaarts AL et al (2004) Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s Disease. J Neurosci 24:7895–7902. https://doi.org/10.1523/jneurosci.1988-04.2004
Ozcelik S, Sprenger F, Skachokova Z et al (2016) Co-expression of truncated and full-length tau induces severe neurotoxicity. pdf. Nature. https://doi.org/10.1038/mp.2015.228
Fox LM, William CM, Adamowicz DH et al (2011) Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. pdf. OXFORD Academic. https://doi.org/10.1097/nen.0b013e318220a658
Sokolow S, Henkins KM, Bilousova T et al (2015) Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J Neurochem 133:368–379. https://doi.org/10.1111/jnc.12991
Liu P, Smith BR, Montonye ML et al (2020) A soluble truncated tau species related to cognitive dysfunction is elevated in the brain of cognitively impaired human individuals. Sci Rep-uk 10:3869. https://doi.org/10.1038/s41598-020-60777-x
Biundo F, d’Abramo C, Tambini MD et al (2008) Abolishing tau cleavage by caspases at Aspartate421 causes memory:synaptic plasticity deficits and pre-pathological tau alterations. pdf. Trans Psych Nat. https://doi.org/10.1038/tp.2017.165
Kang HJ, Yoon WJ, Moon GJ et al (2005) Caspase-3-mediated cleavage of PHF-1 tau during apoptosis irrespective of excitotoxicity and oxidative stress: an implication to Alzheimer’s disease. Neurobiol Dis 18:450–458. https://doi.org/10.1016/j.nbd.2004.12.004
Matthews-Roberson TA, Quintanilla RA, Ding H, Johnson GVW (2008) Immortalized cortical neurons expressing caspase-cleaved tau are sensitized to endoplasmic reticulum stress induced cell death. Brain Res 1234:206–212. https://doi.org/10.1016/j.brainres.2008.07.111
Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GVW (2009) Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer’disease. J Biol Chem 284:18754–18766. https://doi.org/10.1074/jbc.m808908200
Quntanilla RA, Tapia-Monsalves C (2020) The role of mitochondrial impairment in Alzheimer’s disease neurodegeneration: the tau connection. Curr Neuropharmacol 18:1076–1091. https://doi.org/10.2174/1570159x18666200525020259
Pérez MJ, Jara C, Quintanilla RA (2018) Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front Neurosci-switz 12:441. https://doi.org/10.3389/fnins.2018.00441
Javadov S, Kozlov AV, Camara AKS (2020) Mitochondria in health and diseases. pdf. MDPI. https://doi.org/10.3390/cells9051177
Morio B, Panthu B, Bassot A, Rieusset J (2021) Role of mitochondria in liver metabolic health and diseases. Cell Calcium 94:102336. https://doi.org/10.1016/j.ceca.2020.102336
Nguyen BY, Ruiz-Velasco A, Bui T et al (2019) Mitochondrial function in the heart: the insight into mechanisms and therapeutic potentials. Brit J Pharmacol 176:4302–4318. https://doi.org/10.1111/bph.14431
de Caldeira DAF, Weiss DJ, Rocco PRM et al (2021) Mitochondria in focus: from function to therapeutic strategies in chronic lung diseases. Front Immunol 12:782074. https://doi.org/10.3389/fimmu.2021.782074
Martin LJ (2010) Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharm 3:839–915. https://doi.org/10.3390/ph3040839
Pérez MJ, Vergara-Pulgar K, Jara C et al (2018) Caspase-cleaved tau impairs mitochondrial dynamics in Alzheimer’s disease. Mol Neurobiol 55:1004–1018. https://doi.org/10.1007/s12035-017-0385-x
Quintanilla RA, Tapia-Monsalves C, Vergara EH et al (2020) Truncated tau induces mitochondrial transport failure through the impairment of TRAK2 protein and bioenergetics decline in neuronal cells. Front Cell Neurosci 14:175. https://doi.org/10.3389/fncel.2020.00175
Briel N, Pratsch K, Roeber S et al (2021) Contribution of the astrocytic tau pathology to synapse loss in progressive supranuclear palsy and corticobasal degeneration. Brain Pathol 31:e12914. https://doi.org/10.1111/bpa.12914
Binder LI, Frankfurter A, Rebhun LI (1985) The distribution of tau in the mammalian central nervous system pdf. J Cell Biol. https://doi.org/10.1083/jcb.101.4.1371
Hanger DP, Goniotaki D, Noble W (2019) synaptic localization of tau. Adv Exp Med Biol. https://doi.org/10.1007/978-981-32-9358-8_9
Tapia-Rojas C, Cabezas-Opazo F, Deaton CA et al (2018) It’s all about tau. Prog Neurobiol 175:54–76. https://doi.org/10.1016/j.pneurobio.2018.12.005
Takemura R, Kanai Y, Hirokawa N (1991) In situ localization of tau mRNA in developing rat brain. Neuroscience 44:393–407. https://doi.org/10.1016/0306-4522(91)90064-u
Neve RL, Harris P, Kosik KS et al (1986) Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol Brain Res 1:271–280. https://doi.org/10.1016/0169-328x(86)90033-1
Götz J, Halliday G, Nisbet RM (2019) Molecular pathogenesis of the tauopathies pdf. Ann Rev. https://doi.org/10.1146/annurev-pathmechdis-012418-012936
Johnson GVW, Stoothoff WH (2004) Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci 117:5721–5729. https://doi.org/10.1242/jcs.01558
Billingsley ML, Kincaid RL (1997) Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J 323:577–591. https://doi.org/10.1042/bj3230577
Goedert M, Jakes R (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. Embo J 9:4225–4230. https://doi.org/10.1002/j.1460-2075.1990.tb07870.x
Chen Q, Zhou Z, Zhang L et al (2012) Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem Int 60:233–242. https://doi.org/10.1016/j.neuint.2011.12.013
Mandelkow E-M, Stamer K, Vogel R et al (2003) Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging 24:1079–1085. https://doi.org/10.1016/j.neurobiolaging.2003.04.007
Kimura T, Whitcomb DJ, Jo J et al (2014) Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philosophical Trans Royal Soc B Biol Sci 369:20130144. https://doi.org/10.1098/rstb.2013.0144
Briner A, Götz J, Polanco JC (2020) Fyn kinase controls tau aggregation in vivo. Cell Reports 32:108045. https://doi.org/10.1016/j.celrep.2020.108045
Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A et al (2012) Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-d-aspartate receptor-dependent tau phosphorylation*. J Biol Chem 287:32040–32053. https://doi.org/10.1074/jbc.m112.401240
Jara C, Aránguiz A, Cerpa W et al (2018) Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol 18:279–294. https://doi.org/10.1016/j.redox.2018.07.010
Means JC, Gerdes BC, Kaja S et al (2016) Caspase-3-dependent proteolytic cleavage of tau causes neurofibrillary tangles and results in cognitive impairment during normal aging. Neurochem Res 41:2278–2288. https://doi.org/10.1007/s11064-016-1942-9
Jarero-Basulto JJ, Luna-Muñoz J, Mena R et al (2013) Proteolytic cleavage of polymeric tau protein by caspase-3: implications for Alzheimer disease. J Neuropathol Exp Neurol 72:1145–1161. https://doi.org/10.1097/nen.0000000000000013
Zhang Q, Zhang X, Sun A (2009) Truncated tau at D421 is associated with neurodegeneration and tangle formation in the brain of Alzheimer transgenic models. Acta Neuropathol 117:687–697. https://doi.org/10.1007/s00401-009-0491-6
Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR et al (2005) Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging 26:1015–1022. https://doi.org/10.1016/j.neurobiolaging.2004.09.019
Mroczko B, Groblewska M, Litman-Zawadzka A (2019) The role of protein misfolding and tau oligomers (TauOs) in Alzheimer’s disease (AD). Int J Mol Sci 20:4661. https://doi.org/10.3390/ijms20194661
He H, Liu Y, Sun Y, Ding F (2021) Misfolding and self-assembly dynamics of microtubule-binding repeats of the Alzheimer-related protein tau. J Chem Inf Model 61:2916–2925. https://doi.org/10.1021/acs.jcim.1c00217
Kovacech B, Novak M (2010) Tau truncation is a productive posttranslational modification of neurofibrillary degeneration in Alzheimer’s disease.pdf. Curr Alzheimer Res 7:708–716. https://doi.org/10.2174/156720510793611556
Kopeikina KJ, Hyman BT, Spires-Jones TL (2013) Soluble forms of tau are toxic in Alzheimer’s disease pdf. Trans Neurosci 3:223–233. https://doi.org/10.2478/s13380-012-0032-y
Barghorn S, Mandelkow E (2002) Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments †. Biochemistry-us 41:14885–14896. https://doi.org/10.1021/bi026469j
Sahara N, Maeda S, Murayama M et al (2007) Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci 25:3020–3029. https://doi.org/10.1111/j.1460-9568.2007.05555.x
Patterson KR, Remmers C, Fu Y et al (2011) Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease*. J Biol Chem 286:23063–23076. https://doi.org/10.1074/jbc.m111.237974
Schneider A, Biernat J, von Bergen M et al (1999) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments †. Biochemistry-us 38:3549–3558. https://doi.org/10.1021/bi981874p
Zilka N, Filipcik P, Koson P et al (2006) Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. Febs Lett 580:3582–3588. https://doi.org/10.1016/j.febslet.2006.05.029
Morsch R, Simon W, Coleman PD (1999) Neurons may live for decades with neurofibrillary tangles.pdf. Journal neuropathology Experimental Neurology 58:188–197. https://doi.org/10.1097/00005072-199902000-00008
SantaCruz K, Lewis J, Spires T et al (2005) Tau suppression in a neurodegenerative mouse model improves memory function. J Frankl Inst 309:476–481. https://doi.org/10.1126/science.1113694
Eldadah BA, Faden AI (2000) Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotraum 17:811–829. https://doi.org/10.1089/neu.2000.17.811
Fernández DJ, Lamkanfi M (2015) Inflammatory caspases: key regulators of inflammation and cell death. Biol Chem 396:193–203. https://doi.org/10.1515/hsz-2014-0253
Cotman CW, Poon WW, Rissman RA, Blurton-Jones M (2001) The role of caspase cleavage of tau in Alzheimer disease neuropathology. pdf. J Neuropathol Exp Neurol. https://doi.org/10.1093/jnen/64.2.104
Dhage PA, Sharbidre AA, Magdum SM (2023) Interlacing the relevance of caspase activation in the onset and progression of Alzheimer’s disease. Brain Res Bull 192:83–92. https://doi.org/10.1016/j.brainresbull.2022.11.008
Thornberry NA (1997) The caspase family of cysteine proteases. Brit Med Bull 53:478–490. https://doi.org/10.1093/oxfordjournals.bmb.a011625
McIlwain DR, Berger T, Mak TW (2013) Caspase functions in cell death and disease. Csh Perspect Biol 5:a008656. https://doi.org/10.1101/cshperspect.a008656
Troy CM, Akpan N, Jean YY (2011) Chapter 7 regulation of caspases in the nervous system implications for functions in health and disease. Prog Mol Biol Transl 99:265–305. https://doi.org/10.1016/b978-0-12-385504-6.00007-5
Tait SWG, Green DR (2013) Mitochondrial regulation of cell death. Csh Perspect Biol 5:a008706. https://doi.org/10.1101/cshperspect.a008706
Yeh W-C, Itie A, Elia AJ et al (2000) Requirement for Casper (c-FLIP) in regulation of death receptor–induced apoptosis and embryonic development. Immunity 12:633–642. https://doi.org/10.1016/s1074-7613(00)80214-9
Annunziato L, Amoroso S, Pannaccione A et al (2003) Apoptosis induced in neuronal cells by oxidative stress: role played by caspases and intracellular calcium ions. Toxicol Lett 139:125–133. https://doi.org/10.1016/s0378-4274(02)00427-7
Keramaris E, Stefanis L, MacLaurin J et al (2000) Involvement of caspase 3 in apoptotic death of cortical neurons evoked by DNA damage. Mol Cell Neurosci 15:368–379. https://doi.org/10.1006/mcne.2000.0838
Doyle KM, Kennedy D, Gorman AM et al (2011) Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J Cell Mol Med 15:2025–2039. https://doi.org/10.1111/j.1582-4934.2011.01374.x
Ho FY, Tsang WP, Kong SK, Kwok TT (2006) The critical role of caspases activation in hypoxia/reoxygenation induced apoptosis. Biochem Bioph Res Co 345:1131–1137. https://doi.org/10.1016/j.bbrc.2006.04.178
Jiang X, Wang X (2000) Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1*. J Biol Chem 275:31199–31203. https://doi.org/10.1074/jbc.c000405200
Lamkanfi M, Kanneganti T-D (2010) Caspase-7: a protease involved in apoptosis and inflammation. Int J Biochem Cell Biology 42:21–24. https://doi.org/10.1016/j.biocel.2009.09.013
Oakley DH, Klickstein N, Commins C et al (2021) Continuous monitoring of tau-induced neurotoxicity in patient-derived iPSC-neurons. J Neurosci 41:4335–4348. https://doi.org/10.1523/jneurosci.2590-20.2021
D’Amelio M, Cavallucci V, Cecconi F (2010) Neuronal caspase-3 signaling: not only cell death. Cell Death Differ 17:1104–1114. https://doi.org/10.1038/cdd.2009.180
Wang J-Y, Luo Z-G (2014) Non-apoptotic role of caspase-3 in synapse refinement. Neurosci Bull 30:667–670. https://doi.org/10.1007/s12264-014-1454-4
Li Z, Sheng M (2012) Caspases in synaptic plasticity. Mol. Brain 5:15. https://doi.org/10.1186/1756-6606-5-15
Villavicencio-Tejo F, Olesen MA, Aránguiz A, Quintanilla RA (2022) Activation of the Nrf2 pathway prevents mitochondrial dysfunction induced by caspase-3 cleaved tau: implications for Alzheimer’s disease. Antioxidants 11:515. https://doi.org/10.3390/antiox11030515
Zheng J, Akbari M, Schirmer C et al (2020) Hippocampal tau oligomerization early in tau pathology coincides with a transient alteration of mitochondrial homeostasis and DNA repair in a mouse model of tauopathy. Acta Neuropathologica Commun 8:25. https://doi.org/10.1186/s40478-020-00896-8
Bravarenko NI, Onufriev MV, Stepanichev MY et al (2006) Caspase-like activity is essential for long-term synaptic plasticity in the terrestrial snail Helix. Eur J Neurosci 23:129–140. https://doi.org/10.1111/j.1460-9568.2005.04549.x
Huesmann GR, Clayton DF (2006) Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron 52:1061–1072. https://doi.org/10.1016/j.neuron.2006.10.033
Zhang A, Lorke DE, Wu S-X, Yew DT (2006) Caspase-3 immunoreactivity in different cortical areas of young and aging macaque (Macaca mulatta) monkeys. Neurosignals 15:64–73. https://doi.org/10.1159/000094602
Seaman JE, Julien O, Lee PS et al (2016) Cacidases: caspases can cleave after aspartate, glutamate and phosphoserine residues. Cell Death Differ 23:1717–1726. https://doi.org/10.1038/cdd.2016.62
Zhao Y, Tseng I-C, Heyser CJ et al (2015) Appoptosin-mediated caspase cleavage of tau contributes to progressive supranuclear palsy pathogenesis. Neuron 87:963–975. https://doi.org/10.1016/j.neuron.2015.08.020
Friedrich MG, Skora A, Hancock SE et al (2021) Tau is truncated in five regions of the normal adult human brain. Int J Mol Sci 22:3521. https://doi.org/10.3390/ijms22073521
Gu J, Xu W, Jin N et al (2020) Truncation of Tau selectively facilitates its pathological activities. J Biol Chem 295:13812–13828. https://doi.org/10.1074/jbc.ra120.012587
Nunez WA, Combs B, Gamblin TC, Ackley BD (2022) Age-dependent accumulation of tau aggregation in Caenorhabditis elegans. Frontiers Aging 3:928574. https://doi.org/10.3389/fragi.2022.928574
García-Sierra F, Mondragón-Rodríguez S, Basurto-Islas G (2008) Truncation of tau protein and its pathological significance in Alzheimer’s disease. J Alzheimers Dis 14:401–409. https://doi.org/10.3233/jad-2008-14407
Fasulo L, Ugolini G, Visintin M et al (2000) The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem 75:624–633. https://doi.org/10.1046/j.1471-4159.2000.0750624.x
Chung C-W, Song Y-H, Kim I-K et al (2001) Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis 8:162–172. https://doi.org/10.1006/nbdi.2000.0335
Yogev S, Cooper R, Fetter R et al (2016) Microtubule organization determines axonal transport dynamics. Neuron 92:449–460. https://doi.org/10.1016/j.neuron.2016.09.036
Venkatramani A, Panda D (2019) Regulation of neuronal microtubule dynamics by tau: implications for tauopathies. Int J Biol Macromol 133:473–483. https://doi.org/10.1016/j.ijbiomac.2019.04.120
Ding H, Matthews TA, Johnson GVW (2006) Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation*. J Biol Chem 281:19107–19114. https://doi.org/10.1074/jbc.m511697200
Kandel ER, Dudai Y, Mayford MR (2014) The molecular and systems biology of memory. Cell 157:163–186. https://doi.org/10.1016/j.cell.2014.03.001
Di J, Cohen LS, Corbo CP et al (2016) Abnormal tau induces cognitive impairment through two different mechanisms: synaptic dysfunction and neuronal loss. Sci Rep-uk 6:20833. https://doi.org/10.1038/srep20833
Usenovic M, Niroomand S, Drolet RE et al (2015) Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci 35:14234–14250. https://doi.org/10.1523/jneurosci.1523-15.2015
Cowan CM, Quraishe S, Hands S et al (2015) Rescue from tau-induced neuronal dysfunction produces insoluble tau oligomers. Sci Rep-uk 5:17191. https://doi.org/10.1038/srep17191
Hoover BR, Reed MN, Su J et al (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68:1067–1081. https://doi.org/10.1016/j.neuron.2010.11.030
Zhou L, McInnes J, Wierda K et al (2017) Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun 8:15295. https://doi.org/10.1038/ncomms15295
Teravskis PJ, Ashe KH, Liao D (2020) The accumulation of tau in postsynaptic structures: a common feature in multiple neurodegenerative diseases? Neurosci 26:503–520. https://doi.org/10.1177/1073858420916696
Jadhav S, Katina S, Kovac A et al (2015) Truncated tau deregulates synaptic markers in rat model for human tauopathy. Front Cell Neurosci 9:24. https://doi.org/10.3389/fncel.2015.00024
Loon A, Zamudio F, Sanneh A et al (2022) Accumulation of C-terminal cleaved tau is distinctly associated with cognitive deficits, synaptic plasticity impairment, and neurodegeneration in aged mice. Geroscience 44:173–194. https://doi.org/10.1007/s11357-021-00408-z
Kim Y, Choi H, Lee W et al (2016) Caspase-cleaved tau exhibits rapid memory impairment associated with tau oligomers in a transgenic mouse model. Neurobiol Dis 87:19–28. https://doi.org/10.1016/j.nbd.2015.12.006
Tapia-Monsalves C, Olesen MA, Villavicencio-Tejo F, Quintanilla RA (2023) Cyclosporine A (CsA) prevents synaptic impairment caused by truncated tau by caspase-3. Mol Cell Neurosci 125:103861. https://doi.org/10.1016/j.mcn.2023.103861
LeBlanc A, Liu H, Goodyer C et al (1999) Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease*. J Biol Chem 274:23426–23436. https://doi.org/10.1074/jbc.274.33.23426
Louneva N, Cohen JW, Han L-Y et al (2008) Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am J Pathology 173:1488–1495. https://doi.org/10.2353/ajpath.2008.080434
Kudryashov IE, Yakovlev AA, Kudryashova IV, Gulyaeva NV (2004) Inhibition of caspase-3 blocks long-term potentiation in hippocampal slices. Neurosci Behav Physiol 34:877–880. https://doi.org/10.1023/b:neab.0000042571.86110.28
Conze C, Rierola M, Trushina NI et al (2022) Caspase-cleaved tau is senescence-associated and induces a toxic gain of function by putting a brake on axonal transport. Mol Psychiatr 27:3010–3023. https://doi.org/10.1038/s41380-022-01538-2
Noël A, Foveau B, LeBlanc AC (2021) Caspase-6-cleaved tau fails to induce tau hyperphosphorylation and aggregation, neurodegeneration, glial inflammation, and cognitive deficits. Cell Death Dis 12:227. https://doi.org/10.1038/s41419-021-03506-0
Guo H, Albrecht S, Bourdeau M et al (2004) Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease.pdf. Am J Pathol. 165:523–531. https://doi.org/10.1016/s0002-9440(10)63317-2
Theofilas P, Ambrose AJ, Butler D, Wang C, Morales DO, Petersen C, Chin B, Yang et al (2021) Caspase inhibition mitigates tau cleavage and neurotoxicity in iPSC-induced neurons with the V337M MAPT mutation. Alzheimer's Dement 17: e051471. https://doi.org/10.1002/alz.051471
Ramcharitar J, Afonso VM, Albrecht S et al (2013) Caspase-6 activity predicts lower episodic memory ability in aged individuals. Neurobiol Aging 34:1815–1824. https://doi.org/10.1016/j.neurobiolaging.2013.01.007
Ramcharitar J, Albrecht S, Afonso VM et al (2013) Cerebrospinal fluid tau cleaved by caspase-6 reflects brain levels and cognition in aging and Alzheimer disease.pdf. J Neuropathol Exp Neurol 72:824–832. https://doi.org/10.1097/nen.0b013e3182a0a39f
Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol 133:665–704. https://doi.org/10.1007/s00401-017-1707-9
Zhao X, Kotilinek LA, Smith B et al (2016) Caspase-2 cleavage of tau reversibly impairs memory. Nat Med 22:1268–1276. https://doi.org/10.1038/nm.4199
Tamayev R, Akpan N, Arancio O et al (2012) Caspase-9 mediates synaptic plasticity and memory deficits of Danish dementia knock-in mice: caspase-9 inhibition provides therapeutic protection. Mol Neurodegener 7:60. https://doi.org/10.1186/1750-1326-7-60
Bekris LM, Yu C-E, Bird TD, Tsuang DW (2010) Review Article: Genetics of Alzheimer disease. J Geriatr Psych Neur 23:213–227. https://doi.org/10.1177/0891988710383571
DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457–464. https://doi.org/10.1002/ana.410270502
Tönnies E, Trushina E (2017) Oxidative stress, synaptic dysfunction, and Alzheimer’s disease.pdf. Journal of Alzheimer’s Disease 57:1105–1121. https://doi.org/10.3233/jad-161088
Gomez-Nicola D, Boche D (2015) Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimer’s Res Ther 7:42. https://doi.org/10.1186/s13195-015-0126-1
Medeiros R, Baglietto-Vargas D, LaFerla FM (2011) The role of tau in Alzheimer’s disease and related disorders. Cns Neurosci Ther 17:514–524. https://doi.org/10.1111/j.1755-5949.2010.00177.x
Matthews K (2006) Tau protein abnormalities correlate with the severity of dementia in Alzheimer’s disease. Nat Clin Pract Neuro 2:178–178. https://doi.org/10.1038/ncpneuro0139
Moloney CM, Lowe VJ, Murray ME (2021) Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: a clinicopathologic perspective for biomarker research. Alzheimer’s Dementia 17:1554–1574. https://doi.org/10.1002/alz.12321
Brion J-P (2006) Immunological demonstration of tau protein in neurofibrillary tangles of Alzheimer’s disease. J Alzheimer’s Dis 9:177–185. https://doi.org/10.3233/jad-2006-9s321
Polydoro M, Dzhala VI, Pooler AM et al (2014) Soluble pathological tau in the entorhinal cortex leads to presynaptic deficits in an early Alzheimer’s disease model. Acta Neuropathol 127:257–270. https://doi.org/10.1007/s00401-013-1215-5
Rissman RA, Poon WW, Blurton-Jones M et al (2004) Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114:121–130. https://doi.org/10.1172/jci20640
Basurto-Islas G, Luna-Muñoz J, Guillozet-Bongaarts AL et al (2008) Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol 67:470–483. https://doi.org/10.1097/nen.0b013e31817275c7
Albrecht S, Bourdeau M, Bennett D et al (2007) Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol 170:1200–1209. https://doi.org/10.2353/ajpath.2007.060974
Klaiman G, Petzke TL, Hammond J, LeBlanc AC (2008) Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol Cell Proteomics 7:1541–1555. https://doi.org/10.1074/mcp.m800007-mcp200
Theofilas P, Piergies AMH, Oh I et al (2022) Caspase-6-cleaved tau is relevant in Alzheimer’s disease and marginal in four-repeat tauopathies: diagnostic and therapeutic implications. Neuropath Appl Neuro 48:e12819. https://doi.org/10.1111/nan.12819
LeBlanc AC, Ramcharitar J, Afonso V et al (2014) Caspase-6 activity in the CA1 region of the hippocampus induces age-dependent memory impairment. Cell Death Differ 21:696–706. https://doi.org/10.1038/cdd.2013.194
Shimohama S, Tanino H, Fujimoto S (1999) Changes in caspase expression in Alzheimer’s disease: comparison with development and aging. Biochem Bioph Res Co 256:381–384. https://doi.org/10.1006/bbrc.1999.0344
Hlynialuk C, Kemper L, Leinonen-Wright K et al (2022) Caspase-2 mRNA levels are not elevated in mild cognitive impairment, Alzheimer’s disease, Huntington’s disease, or Lewy Body dementia.pdf. Plos One 17:e0274784. https://doi.org/10.1371/journal.pone.0274784
Sheng Z-H (2014) Mitochondrial trafficking and anchoring in neurons: new insight and implications. J Cell Biol 204:1087–1098. https://doi.org/10.1083/jcb.201312123
Wang L, Liu M, Gao J et al (2021) Mitochondrial fusion suppresses tau pathology-induced neurodegeneration and cognitive decline. J Alzheimer’s Dis 84:1057–1069. https://doi.org/10.3233/jad-215175
Chang DTW, Honick AS, Reynolds IJ (2006) Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci Official J Soc Neurosci 26:7035–7045. https://doi.org/10.1523/jneurosci.1012-06.2006
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metabolism 21:1133–1145. https://doi.org/10.1097/00004647-200110000-00001
Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75:762–777. https://doi.org/10.1016/j.neuron.2012.08.019
Kühlbrandt W (2015) Structure and function of mitochondrial membrane protein complexes. Bmc Biol 13:89. https://doi.org/10.1186/s12915-015-0201-x
van der Bliek AM, Shen Q, Kawajiri S (2013) Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol 5:a011072. https://doi.org/10.1101/cshperspect.a011072
Castora FJ, Kerns KA, Pflanzer HK et al (2022) Expression changes in mitochondrial genes affecting mitochondrial morphology, transmembrane potential, fragmentation, amyloidosis, and neuronal cell death found in brains of Alzheimer’s disease patients. J Alzheimer’s Dis 90:119–137. https://doi.org/10.3233/jad-220161
Wang X, Su B, Lee HG et al (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090–9103. https://doi.org/10.1523/jneurosci.1357-09.2009
Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20:2495–2509. https://doi.org/10.1093/hmg/ddr139
Calkins MJ, Manczak M, Mao P et al (2011) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet 20:4515–4529. https://doi.org/10.1093/hmg/ddr381
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48:158–167. https://doi.org/10.1016/j.molcel.2012.09.025
Yin Z, Burger N, Kula-Alwar D et al (2021) Structural basis for a complex I mutation that blocks pathological ROS production. Nat Commun 12:707. https://doi.org/10.1038/s41467-021-20942-w
Muller FL, Liu Y, Remmen HV (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane*. J Biol Chem 279:49064–49073. https://doi.org/10.1074/jbc.m407715200
Marchi ED, Bonora M, Giorgi C, Pinton P (2014) The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 56:1–13. https://doi.org/10.1016/j.ceca.2014.03.004
Stefani DD, Patron M, Rizzuto R (2015) Structure and function of the mitochondrial calcium uniporter complex. Biochimica Et Biophysica Acta Bba - Mol Cell Res 1853:2006–2011. https://doi.org/10.1016/j.bbamcr.2015.04.008
Luongo TS, Lambert JP, Gross P et al (2017) The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545:93–97. https://doi.org/10.1038/nature22082
Natarajan GK, Glait L, Mishra J et al (2020) Total matrix Ca2+ modulates Ca2+ efflux via the Ca2+/H+ exchanger in cardiac mitochondria. Front Physiol 11:510600. https://doi.org/10.3389/fphys.2020.510600
Vos M, Lauwers E, Verstreken P (2010) Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci 2:139. https://doi.org/10.3389/fnsyn.2010.00139
Sanganahalli BG, Herman P, Hyder F, Kannurpatti SS (2013) Mitochondrial calcium uptake capacity modulates neocortical excitability. J Cereb Blood Flow Metabolism 33:1115–1126. https://doi.org/10.1038/jcbfm.2013.61
Reiss AB, Ahmed S, Dayaramani C et al (2022) The role of mitochondrial dysfunction in Alzheimer’s disease: a potential pathway to treatment. Exp Gerontol 164:111828. https://doi.org/10.1016/j.exger.2022.111828
Sharma C, Kim S, Nam Y et al (2021) Mitochondrial dysfunction as a driver of cognitive impairment in Alzheimer’s disease. Int J Mol Sci 22:4850. https://doi.org/10.3390/ijms22094850
Hirai K, Aliev G, Nunomura A et al (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21:3017–3023. https://doi.org/10.1523/jneurosci.21-09-03017.2001
Yao J, Irwin RW, Zhao L et al (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc National Acad Sci 106:14670–14675. https://doi.org/10.1073/pnas.0903563106
Blass JP, Sheu RK, Gibson GE (2000) Inherent abnormalities in energy metabolism in Alzheimer disease: interaction with cerebrovascular compromise. Ann Ny Acad Sci 903:204–221. https://doi.org/10.1111/j.1749-6632.2000.tb06370.x
Ishii K, Sasaki M, Kitagaki H et al (1997) Reduction of cerebellar glucose metabolism in advanced Alzheimer’s disease. J Nucl Med Off Publ Soc Nucl Med 38:925–928
David DC, Hauptmann S, Scherping I et al (2005) Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice*. J Biol Chem 280:23802–23814. https://doi.org/10.1074/jbc.m500356200
Brooks WM, Lynch PJ, Ingle CC et al (2007) Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res 1127:127–135. https://doi.org/10.1016/j.brainres.2006.09.106
Xie H, Guan J, Borrelli LA et al (2013) Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J Neurosci 33:17042–17051. https://doi.org/10.1523/jneurosci.1836-13.2013
Dragicevic N, Mamcarz M, Zhu Y et al (2010) Mitochondrial amyloid-β levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J Alzheimer’s Dis 20:S535–S550. https://doi.org/10.3233/jad-2010-100342
Manczak M, Park BS, Jung Y, Reddy PH (2004) Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromol Med 5:147–162. https://doi.org/10.1385/nmm:5:2:147
Jadiya P, Kolmetzky DW, Tomar D et al (2019) Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat Commun 10:3885. https://doi.org/10.1038/s41467-019-11813-6
Mattson MP, Barger SW, Cheng B et al (1993) β-Amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer’s disease. Trends Neurosci 16:409–414. https://doi.org/10.1016/0166-2236(93)90009-b
Baumgartner HK, Gerasimenko JV, Thorne C et al (2009) Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening*. J Biol Chem 284:20796–20803. https://doi.org/10.1074/jbc.m109.025353
Schinder AF, Olson EC, Spitzer NC, Montal M (1996) Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci 16:6125–6133. https://doi.org/10.1523/jneurosci.16-19-06125.1996
Moreira PI, Santos MS, Moreno A et al (2002) Effect of amyloid ?-peptide on permeability transition pore: a comparative study. J Neurosci Res 69:257–267. https://doi.org/10.1002/jnr.10282
Du H, Guo L, Fang F et al (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14:1097–1105. https://doi.org/10.1038/nm.1868
Jara C, Cerpa W, Tapia-Rojas C, Quintanilla RA (2021) Tau deletion prevents cognitive impairment and mitochondrial dysfunction age associated by a mechanism dependent on cyclophilin-D. Front Neurosci-switz 14:586710. https://doi.org/10.3389/fnins.2020.586710
Lopes S, Teplytska L, Vaz-Silva J et al (2016) Tau deletion prevents stress-induced dendritic atrophy in prefrontal cortex: role of synaptic mitochondria. Cereb Cortex 27:2580–2591. https://doi.org/10.1093/cercor/bhw057
Bertholet AM, Delerue T, Millet AM et al (2016) Mitochondrial fusion:fission dynamics in neurodegeneration and neuronal plasticity.pdf. Neurobiol Dis 90:3–19. https://doi.org/10.1016/j.nbd.2015.10.011
de la Cueva M, Antequera D, Ordoñez-Gutierrez L et al (2022) Amyloid-β impairs mitochondrial dynamics and autophagy in Alzheimer’s disease experimental models. Sci Rep-uk 12:10092. https://doi.org/10.1038/s41598-022-13683-3
Szabo L, Eckert A, Grimm A (2020) Insights into disease-associated tau impact on mitochondria. Int J Mol Sci 21:6344. https://doi.org/10.3390/ijms21176344
Kandimalla R, Manczak M, Pradeepkiran JA et al (2021) A partial reduction of Drp1 improves cognitive behavior and enhances mitophagy, autophagy and dendritic spines in a transgenic Tau mouse model of Alzheimer disease. Hum Mol Genet 31:1788–1805. https://doi.org/10.1093/hmg/ddab360
Bevan RJ, Williams PA, Waters CT et al (2020) OPA1 deficiency accelerates hippocampal synaptic remodelling and age-related deficits in learning and memory. Brain Commun 2:fcaa101. https://doi.org/10.1093/braincomms/fcaa101
Quintanilla RA, Dolan PJ, Jin YN, Johnson GVW (2012) Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol Aging 33:619.e25-619.e35. https://doi.org/10.1016/j.neurobiolaging.2011.02.007
Pathak D, Shields LY, Mendelsohn BA et al (2015) The role of mitochondrially derived ATP in synaptic vesicle recycling* ♦. J Biol Chem 290:22325–22336. https://doi.org/10.1074/jbc.m115.656405
Marland JRK, Hasel P, Bonnycastle K, Cousin MA (2016) Mitochondrial calcium uptake modulates synaptic vesicle endocytosis in central nerve terminals*. J Biol Chem 291:2080–2086. https://doi.org/10.1074/jbc.m115.686956
Li Z, Okamoto K-I, Hayashi Y, Sheng M (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119:873–887. https://doi.org/10.1016/j.cell.2004.11.003
Cheng A, Hou Y, Mattson MP (2010) Mitochondria and neuroplasticity. Asn Neuro 2:e00045. https://doi.org/10.1042/an20100019
Pilling AD, Horiuchi D, Lively CM, Saxton WM (2006) Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell 17:2057–2068. https://doi.org/10.1091/mbc.e05-06-0526
Fenton AR, Jongens TA, Holzbaur ELF (2021) Mitochondrial adaptor TRAK2 activates and functionally links opposing kinesin and dynein motors. Nat Commun 12:4578. https://doi.org/10.1038/s41467-021-24862-7
Cai Q, Davis ML, Sheng Z-H (2011) Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci Res 70:9–15. https://doi.org/10.1016/j.neures.2011.02.005
Fransson Å, Ruusala A, Aspenström P (2003) Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis*. J Biol Chem 278:6495–6502. https://doi.org/10.1074/jbc.m208609200
Shahpasand K, Uemura I, Saito T et al (2012) Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J Neurosci Off J Soc Neurosci 32:2430–2441. https://doi.org/10.1523/jneurosci.5927-11.2012
Kopeikina KJ, Carlson GA, Pitstick R et al (2011) Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol 179:2071–2082. https://doi.org/10.1016/j.ajpath.2011.07.004
Ebneth A, Godemann R, Stamer K et al (1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol 143:777–794. https://doi.org/10.1083/jcb.143.3.777
Quintanilla RA, von Bernhardi R, Godoy JA et al (2014) Phosphorylated tau potentiates Aβ-induced mitochondrial damage in mature neurons. Neurobiol Dis 71:260–269. https://doi.org/10.1016/j.nbd.2014.08.016
van Spronsen M, Mikhaylova M, Lipka J et al (2013) TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77:485–502. https://doi.org/10.1016/j.neuron.2012.11.027
Jash K, Gondaliya P, Kirave P et al (2020) Cognitive dysfunction: a growing link between diabetes and Alzheimer’s disease. Drug Dev Res 81:144–164. https://doi.org/10.1002/ddr.21579
Dolan C, Glynn R, Griffin S et al (2018) Brain complications of diabetes mellitus: a cross-sectional study of awareness among individuals with diabetes and the general population in Ireland. Diabet Med 35:871–879. https://doi.org/10.1111/dme.13639
Gaspar JM, Baptista FI, Macedo MP, Ambrósio AF (2016) Inside the Diabetic Brain: Role of Different Players Involved in Cognitive Decline. Acs Chem Neurosci 7:131–142. https://doi.org/10.1021/acschemneuro.5b00240
Jayaraj RL, Azimullah S, Beiram R (2020) Diabetes as a risk factor for Alzheimer’s disease in the Middle East and its shared pathological mediators. Saudi J Biol Sci 27:736–750. https://doi.org/10.1016/j.sjbs.2019.12.028
Macauley SL, Stanley M, Caesar EE et al (2015) Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J Clin Invest 125:2463–2467. https://doi.org/10.1172/jci79742
Silzer TK, Phillips NR (2018) Etiology of type 2 diabetes and Alzheimer’s disease: exploring the mitochondria. Mitochondrion 43:16–24. https://doi.org/10.1016/j.mito.2018.04.004
Hobday AL, Parmar MS (2021) The link between diabetes mellitus and tau hyperphosphorylation: implications for risk of Alzheimer’s disease. Cureus 13:e18362. https://doi.org/10.7759/cureus.18362
Clodfelder-Miller BJ, Zmijewska AA, Johnson GVW, Jope RS (2006) Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 55:3320–3325. https://doi.org/10.2337/db06-0485
Reagan LP, Magariños AM, Yee DK et al (2000) Oxidative stress and HNE conjugation of GLUT3 are increased in the hippocampus of diabetic rats subjected to stress. Brain Res 862:292–300. https://doi.org/10.1016/s0006-8993(00)02212-5
Moreira PI, Santos MS, Seiça R, Oliveira CR (2007) Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J Neurol Sci 257:206–214. https://doi.org/10.1016/j.jns.2007.01.017
Kim B, Backus C, Oh S, Feldman EL (2013) Hyperglycemia-induced tau cleavage in vitro and in vivo: a possible link between diabetes and Alzheimer’s disease. J Alzheimer’s Dis 34:727–739. https://doi.org/10.3233/jad-121669
Kim B, Backus C, Oh S et al (2009) Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 150:5294–5301. https://doi.org/10.1210/en.2009-0695
Latina V, Giacovazzo G, Calissano P et al (2021) Tau cleavage contributes to cognitive dysfunction in strepto-zotocin-induced sporadic Alzheimer’s disease (sAD) mouse model. Int J Mol Sci 22:12158. https://doi.org/10.3390/ijms222212158
Funding
This work was supported by ANID, GRANT FONDECYT # 1200178, Santiago, Chile. MAO thanked Universidad Autónoma de Chile (PhD fellowship).
Author information
Authors and Affiliations
Contributions
MAO made the figures and conceived, wrote, and edited manuscript; RAQ conceived, edited, and financed this research.
Corresponding author
Ethics declarations
Ethics Approval
N/A.
Consent to Participate
N/A
Consent for Publication
N/A
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Olesen, M.A., Quintanilla, R.A. Pathological Impact of Tau Proteolytical Process on Neuronal and Mitochondrial Function: a Crucial Role in Alzheimer’s Disease. Mol Neurobiol 60, 5691–5707 (2023). https://doi.org/10.1007/s12035-023-03434-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03434-4