
Abstract
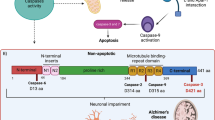
Alzheimer’s disease (AD) is characterized by the presence of aggregates of tau protein. Tau truncated by caspase-3 (D421) or tau hyperphosphorylated at Ser396/S404 might play a role in the pathogenesis of AD. Mitochondria are dynamic organelles that modify their size and function through mitochondrial dynamics. Recent studies have shown that alterations of mitochondrial dynamics affect synaptic communication. Therefore, we studied the effects of pathological forms of tau on the regulation of mitochondrial dynamics. We used primary cortical neurons from tau(−/−) knockout mice and immortalized cortical neurons (CN1.4) that were transfected with plasmids containing green fluorescent protein (GFP) or GFP with different tau forms: full-length (GFP-T4), truncated (GFP-T4C3), pseudophosphorylated (GFP-T42EC), or both truncated and pseudophosphorylated modifications of tau (GFP-T4C3-2EC). Cells expressing truncated tau showed fragmented mitochondria compared to cells that expressed full-length tau. These findings were corroborated using primary neurons from tau(−/−) knockout mice that expressed the truncated and both truncated and pseudophosphorylated forms of tau. Interestingly, mitochondrial fragmentation was accompanied by a significant reduction in levels of optic atrophy protein 1 (Opa1) in cells expressing the truncated form of tau. In addition, treatment with low concentrations of amyloid-beta (Aβ) significantly reduced mitochondrial membrane potential, cell viability, and mitochondrial length in cortical cells and primary neurons from tau(−/−) mice that express truncated tau. These results indicate that the presence of tau pathology impairs mitochondrial dynamics by reducing Opa1 levels, an event that could lead to mitochondrial impairment observed in AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common form of dementia in the elderly [1]. AD is characterized by the accumulation of extracellular senile plaques formed by deposits of amyloid-beta (Aβ) peptide and the presence of intracellular aggregates known as neurofibrillary tangles (NFTs), which are formed by pathological forms of the tau protein [1]. These protein aggregates are neurotoxic and are responsible for synaptic dysfunction, mitochondrial damage, oxidative stress, and ultimately death of significant numbers of neurons in the hippocampus and cerebral cortex [2–5]. Tau is a microtubule-associated protein which is mainly expressed in axons [6–8], where it regulates vesicular and mitochondrial transport [9]. Post-translational modifications such as abnormal phosphorylation and cleavage of tau are considered hallmarks of AD [10, 11]. Modifications such as tau hyperphosphorylation at serine 396 (S396) and serine 404 (S404) and tau truncation or cleavage by caspase-3 (D421) have been detected in AD brains [12]. Interestingly, previous studies from our group and others indicate an association between tau pathology and mitochondrial impairment in AD [13–16].
Mitochondrial dysfunction is a contributing factor in AD and is considered an early event in the development of this pathology [15, 17]. Studies in cellular and animal models of AD have shown a significant compromise of mitochondrial bioenergetics indicated for a reduction in mitochondrial respiration and membrane potential [17–19]. Further studies have shown that impairment of mitochondrial transport, resulting in reduced numbers of mitochondria at the synaptic terminal, is an event that contributes to the synaptic dysfunction seen in AD [20]. Furthermore, defects in regulation of the mitochondrial cycle of fission/fusion (dynamics) have been described in different models of AD [13–16]. In AD, control of mitochondrial dynamics is affected by alterations in the expression of proteins involved in mitochondrial fission, such as dynamin-related protein 1 (Drp1), and mitochondrial fusion: mitofusins 1 and 2 (Mfn1/2) and optic atrophy protein 1 (Opa1), among other proteins [16, 20, 21].
Previous studies by our group have shown that pathological forms of tau affect mitochondrial bioenergetics, transport, and morphology [11, 13, 21]. In addition, treatment with Aβ oligomers and the presence of tau pathology decrease the expression of complexes I and IV of the electron transport chain (ETC) and affect mitochondrial respiration [22–24]. Furthermore, it has been reported that truncated tau affects mitochondrial morphology and induces mitochondrial fragmentation [11, 21]; however, the underlying mechanism by which pathological forms of tau affect mitochondrial dynamics and function remains unknown.
Here, we show that expression of truncated tau induces mitochondrial fragmentation, accompanied by a significant reduction in levels of Opa1 in cortical neurons. In addition, treatment with low concentrations of Aβ significantly reduces mitochondrial membrane potential, cell viability, and mitochondrial length in immortalized cells and primary neurons from tau(−/−) knockout mice. These results suggest that the presence of tau pathology, specially truncated form of tau, impairs the mitochondrial fission/fusion cycle through the downregulation of Opa1. These novel observations suggest an important role for caspase-cleaved tau in the pathogenesis of AD through the impairment of mitochondrial dynamics.
Experimental Procedures
Materials
All materials for cell culture and transfection were obtained from Sigma-Aldrich (St. Louis, MO, USA), Invitrogen (Carlsbad, CA, 119 USA), and Mediatech Inc. (Corning, NY, USA). Tetramethylrhodamine methyl ester (TMRM) was obtained from Molecular Probes (OR, USA). Amyloid peptide1–40 (Aβ1–40) was obtained from Calbiochem (MED Millipore, MA, USA).
Tau Constructs
Full-length (T4) and mutant tau constructs [T4-2EC (pseudophosphorylated, S396/S404E), T4C3 (truncated at D421), and T4-C3-2EC (pseudophosphorylated and truncated)] were generated by mutagenesis according to Ding et al. [25]. Constructs of full-length tau tagged with GFP [GFP-T4 (full length), GFP-T42EC (pseudophosphorylated tau), and GFP-T42EC-C3 (pseudophosphorylated and truncated tau)] were obtained by sub-cloning, as previously described by Matthews-Roberson et al. [26]. For mitochondrial length studies, Mito-mCherry was developed as we previously described in Quintanilla et al. [21].
Aβ Fibrils
Aβ1–40 fibrils were prepared as previously described in Quintanilla et al. [21]. Briefly, stock solutions were prepared by dissolving aliquots of Aβ1–40 in dimethyl sulfoxide (DMSO) to 1 M, and this solution was further diluted with 0.1 M Tris–HCl (pH 7.4) to a final concentration of 100 μM Aβ. The solutions were stirred continuously (210 rpm) at 22 °C or 48 h.
Cell Culture and Transfection
CN1.4 cells were cultured in 1× Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech Inc., Corning, NY, USA) supplemented with 5% inactivated fetal bovine serum (Mediatech Inc., Corning, NY, USA) and 1% penicillin/streptomycin (Mediatech Inc., Corning, NY, USA), and cells were incubated at 33 °C and 5% CO2. Cells were transiently transfected 48 h post-incubation with plasmids containing GFP, GFP full-length tau, and GFP link to the pathological tau constructs. Co-transfections were made using Mito-mCherry to detect mitochondrial morphology. The media was changed 24 h post-transfection, and studies with Aβ were carried out 48 h post-transfection.
Cortical neuronal cultures were prepared from the cortices of tau(−/−) mice (B6.129 × 1-Mapttm1Hnd/J, 7251) obtained from Jackson Laboratories (USA). Cortices were dissected from embryos, and primary cortical neurons were prepared as previously described [21]. All procedures were performed in agreement with the Institutional Bioethics Committee of Universidad Autonoma de Chile.
Tau(−/−) cortical neurons were transiently transfected with 0.8 μg of the different plasmids using Lipofectamine 2000 (Invitrogen) diluted in OptiMEM (Gibco, MA, USA) after 4 days in vitro (DIV) [21]. The transfection medium was replaced with Neurobasal medium (NBM) supplemented with B27 and 0.4 mM glutamine after 24 h. Neurons were subsequently maintained on a cell incubator for at least 7 days.
Measurement of Mitochondrial Length
An estimate of mitochondrial length was obtained by measuring the lengths of individual mitochondria in untreated and Aβ-treated cells (immortalized and primary tau(−/−) neurons) double-transfected with GFP/Mito-mCherry, GFP-T4/Mito-mCherry, GFP-T42EC/Mito-mCherry, GFP-T4C3/Mito-mCherry, and GFP-T42EC-C3/Mito-mCherry. Fluorescence images were obtained using high-resolution fluorescence microscopy (Leica, Germany) using a 63× oil objective. We analyzed the mitochondrial population of 30–40 cells, measuring 25 images for each experiment using ImageJ software for analysis.
Western Blot Analysis
Transfected cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (AMRESCO, OH, USA) that contained protease and phosphatase inhibitor cocktails (Roche, IN, USA). Total protein extracts (30 μg) were separated on a sodium dodecyl sulfate (SDS)-polyacrylamide gel and subsequently transfered onto nitrocellulose membranes. After blocking in 2% non-fat milk and 0.1% Tween-20 in phosphate-buffered saline (PBS), the membranes were incubated with mouse monoclonal anti-GFP (1:1000, Roche, IN, USA), rabbit polyclonal anti-Opa1 (1:1000; Thermo Fisher, MA, USA), rabbit polyclonal anti-Drp1 (1:1000; Thermo Fisher, MA, USA), mouse monoclonal anti-pDrp1 (1:1000; Cell Signaling, MA, USA), rabbit polyclonal anti-Mfn1 (1:1000; Santa Cruz, TX, USA), and rabbit polyclonal anti-Mfn2 (1:1000; Santa Cruz, TX, USA) antibodies. To confirm equal loading and transfer, membranes were subsequently re-probed with anti-actin (1:5000; Santa Cruz, TX, USA) antibody as indicated. After treatment with horseradish peroxidase (HRP)-linked goat anti-mouse or anti-rabbit secondary antibodies (1:2000; Thermo Fisher, MA, USA), immunoreactive proteins were detected using enhanced chemiluminescence (ECL, Thermo Fisher, MA, USA).
Determination of Mitochondrial Membrane Potential in Live Cells
The mitochondrial membrane potential was evaluated using the mitochondrial dye TMRM in a non-quenching mode [11, 13, 21]. Prior to treatment with Aβ, transfected cells were preloaded with TMRM (100 nM) in Krebs-Ringer-Henseleit (KRH) buffer (136 mM NaCl, 20 mM HEPES, 4.7 mM KCl, 1.5 mM MgSO4, 1.25 mM CaCl2, 5 mM glucose; pH = 7.4) for 45 min, and changes in fluorescence intensity were captured using a high-resolution fluorescence microscope LX6000 (Leica, Germany) under a 63× oil objective. We analyzed 30–40 cells, measuring at least 25 images for each experiment. Results are presented as the pseudo ratio (ΔF/F0) [11, 13, 21].
Cell Viability
CN1.4 cells were growing in 96-well plates (around 5000 cells per well) (Corning® Nunclon Delta Surface, Corning, NY, USA), and 48 h post-transfection, cells were treated with Aβ fibrils for 24 h. Cell viability was determined by incubating plates for 1 h at 37 °C and 5% CO2 with AlamarBlue™ (Thermo Fisher, MA, USA) 10% (v/v) in KRH buffer. Cell viability was determined according to the manufacturer’s instructions.
Statistical Analysis
Results are expressed as mean ± standard error (SE), and they were analyzed using one-way ANOVA tests. “n” represents the number of experiments carried out. Differences were considered significant at p < 0.05.
Results
Truncated Tau Induces Mitochondrial Fragmentation in Immortalized Cortical Cells
To study the effects of pathological forms of tau on mitochondrial morphology, CN1.4 cells were co-transfected with GFP, full-length (GFP-T4), and pathological forms of tau [truncated (GFP-T4C3), pseudophosphorylated (GFP-T42EC), and truncated and pseudophosphorylated (GFP-T4C3-2EC)] along with Mito-mCherry (m-Ch), which carries an outer mitochondrial membrane-specific destination sequence [11, 21]. High-resolution fluorescence images were captured from these cells 48 h post-transfection using an LSM 6000× fluorescence microscope (Leica, Germany). For mitochondrial length measurements, ImageJ software was calibrated using the scale determined by the Leica Application Suite software (Leica, Germany) when images were captured [21]. Cells that expressed GFP and Mito-mCherry exhibited long and reticular form of mitochondria (Fig. 1a, b) with an average length of 2.008 ± 0.03531 μm, which value was similar to those cells that expressed full-length tau (GFP-T4) and Mito-mCherry, whose mitochondrial length was 2.094 ± 0.04107 μm (Fig. 1a, b). However, cells co-expressing Mito-mCherry and truncated tau (GFP-T4C3) or pseudophosphorylated plus truncated tau (GFP-T4C3-2EC) showed fragmented and “drop-shaped” mitochondria (Fig. 1a, b), with lengths of 1.279 ± 0.02710 and 1.363 ± 0.1073 μm, respectively (Fig. 1a, b). In contrast, mitochondria from cells expressing GFP-T42EC showed similar mitochondrial morphology to those expressing GFP and full-length tau (Fig. 1a, b).

Expression of caspase-cleaved tau reduces mitochondrial length in neuronal cells. a Representative images from immortalized cortical neurons (CN1.4) transfected with green fluorescent protein (GFP) and tau constructs [GFP full-length tau (GFP-T4), GFP pseudophosphorylated tau (GFP-T42EC), GFP caspase-cleaved tau (GFP-T4C3), and caspase-cleaved psedophosphorylated tau (GFP-T4CE-2EC)] plus Mito-mCherry (m-Ch) to evaluate mitochondrial length. b Images of m-Ch fluorescence channels taken from insert displayed in a that show mitochondrial morphology in cells transfected with GFP and GFP-tau constructs. Truncated tau expression reduced mitochondrial length compared with full-length tau expression. c Quantitative analysis of mitochondrial length from four independent experiments. Data are expressed as mean ± SE, n = 4. Scale bars in a 10 μm and b 5 μm. *p < 0.05
Caspase-Cleaved Tau Expression Affects the Balance of the Mitochondrial Fission/Fusion Cycle in Neuronal Cells
To determine if pathological forms of tau affects the expression of proteins that regulate mitochondrial dynamics, CN1.4 cells were transfected with constructs containing GFP and pathological forms of tau coupled with GFP, to obtain total extracts of these cells 48 h post-transfection (Fig. 2). Similar expression levels of GFP, full-length, and pathological forms of tau were observed in total extracts from CN1.4 cells transfected with the constructs mentioned before (Fig. 2a, b). In western blot analysis, a band was observed for GFP at ∼30 kDa, while a band for GFP linked with full-length and pathological forms of tau was observed at ∼100 kDa. This corroborates the link between GFP and tau [25]. Untransfected CN1.4 cells did not produce any signals for GFP or tau proteins (Fig. 2a). Simultaneously, the relative expression levels of mitochondrial fission/fusion proteins Drp1 (Fig. 2c, d), Mfn1 (Fig. S1B), Mfn 2 (Fig. 3a, b), and Opa1 (Fig. 3c, d) were examined in extracts from CN1.4 cells transfected with the same tau constructs. Analysis of the expression levels of these proteins showed that Drp1 showed a signal with a prominent band at ∼50 kDa and a double band between 75 and 50 kDa (Fig. 2b), Mfn2 appeared as a double band between ∼50 and 75 kDa (Fig. 3a), and Opa1 produced a band at ∼120 kDa (Fig. 3c), where the heaviest band corresponded to the dimer and the other to monomeric forms (Fig. 3b). We found similar levels of Drp1 in untransfected cells and those cells that express GFP, full-length (T4), and pseudophosphorylated tau (T42EC) (Fig. 2c, d). Complementary to these studies, we evaluated pDrp1 levels in CN1.4 cells transfected with same GFP and tau constructs (Fig. S1A). Expression of truncated tau did not affect pDrp1 levels compared to cells that expressed GFP and full-length tau (Fig. S1A). Interestingly, cells expressing truncated tau (T4C3) and truncated and phosphorylated tau (T4C3-2EC) showed a partial reduction in total Drp1 levels compared with untransfected and GFP full-length tau transfected cells (Fig. 2c). However, this decrease was not significant (Fig. 2d; p < 0.05 for both conditions). We also analyzed levels of Mfn2 in extracts obtained from cells transfected with GFP and the GFP-tau constructs. Mfn2 levels were similar in all representative samples obtained from cells that express all GFP-tau forms (Fig. 3a, b). In addition, Mfn1 levels were not affected in CN1.4 cells transfected with GFP and GFP-tau forms (Fig. S1B). Opa1 levels showed a similar pattern of expression in untransfected cells compared to those expressing GFP, T4, and T42EC (Fig. 3c, d). More importantly, in extracts from cells expressing T4C3 and T4C32EC, the relative expression of Opa1 was significantly reduced compared to untransfected and GFP-positive cells (Fig. 3d). These results suggest that expression of tau cleaved by caspase-3 (T4C3 and T4C3-2EC) induces a decrease in the expression of Opa1, an event that could explain the mitochondrial fragmentation observed in the cells that expressed truncated tau.

Effects of caspase-cleaved tau expression on the mitochondrial fusion protein Drp1 in immortalized cortical neurons. a CN1.4 cells were transfected with GFP and tau constructs [GFP-T4, GFP-T42EC, GFP-T4C3, and GFP-T4C3-2EC] to determine the expression of the different forms of tau using GFP by western blot. Whole cell extracts were collected, and GFP levels were evaluated 48 h after transfection. Representative images show that GFP expression levels are similar in all GFP-tau constructs, indicating equal expression levels of the different forms of tau. Images also show the levels of β-actin as a loading control. b Semi-quantitative analysis of four independent experiments assessing GFP levels normalized against β-actin levels. Data are expressed as mean ± SE, n = 4. c Total Drp1 levels were determined in whole cell extracts from CN1.4 cells transfected with GFP and GFP-tau constructs. Expression of truncated tau decreased Drp1 levels compared to cells expressing full-length tau or GFP. β-actin protein levels were evaluated as a loading control. d Quantitative analysis of four independent experiments representing Drp1 levels in cells transfected with GFP or GFP-tau constructs showed no significant differences in total Drp1 levels under all compared conditions. Data are expressed as mean ± SE, n = 4

Expression of caspase-cleaved tau affects Opa1 expression. a CN1.4 cells were transfected with GFP, full-length tau (GFP-T4), phosphorylated tau (GFP-T42EC), truncated tau (GFP-T4C3), and truncated-peudophosphorylated tau (GFP-T4C3-2EC), and levels of Mfn2 were evaluated by western blot. Expression of GFP or GFP-tau constructs did not change total Mfn2 levels. b Quantitative analysis of three experiments evaluating Mfn2 levels. Data are expressed as mean ± SE, n = 4. c Total Opa1 levels were evaluated in CN1.4 cells transfected under the conditions indicated above. Truncated and truncated-pseudophosphorylated tau reduced Opa1 expression when compared to cells expressing GFP and full-length tau. d Quantitative analysis of three experiments showing significant differences in Opa1 levels in cells expressing truncated and truncated-pseudophosphorylated tau. Data are expressed as mean ± SE, n = 3. *p < 0.05
Expression of Caspase-Cleaved Tau Does Not Affect Cell Viability in Neurons
To investigate the effects of pathological forms of tau on mitochondrial function, CN1.4 cells were transfected with GFP, normal tau, and other GFP-tau constructs, and the mitochondrial membrane potential was tested with TMRM, as a measure of mitochondrial bioenergetics [21]. Fluorescence images of these cells were obtained, and mitochondrial potential levels were evaluated (Fig. 4a). Relative fluorescence intensity was quantified using the Leica Application Suite® and ImageJ software, normalizing each image to its background fluorescence (Fig. 4a, b) [21]. Analysis of mitochondrial potential showed similar levels in untransfected cells compared to those transfected with GFP and all GFP-tau constructs (Fig. 4b). Furthermore, we analyzed the effects of pathological forms of tau on cell viability in CN1.4 cells. Cell viability was examined 48 h post-transfection with AlamarBlue™. Cell viability percentages were determined by subtracting the background measurement of all samples, and the rate of the reaction reduced by untransfected controls was considered as 100% cell viability (Fig. 4c). The viability of cells expressing GFP and full-length tau was not altered compared to untransfected cells (Fig. 4c). In addition, cell viability in cells expressing pathological forms of tau [T4C3, T42EC, and T4C3-2EC] was not significantly reduced compared to control cells (Fig. 4c). Altogether, these results suggest that the expression of pathological forms of tau does not affect cell viability under basal conditions.

Expression of truncated tau did not affect mitochondrial function or cell viability. a Representative fluorescence images from CN1.4 cells transfected with green fluorescent protein (GFP), full-length tau (GFP-T4), phosphorylated tau (GFP-T42EC), truncated tau (GFP-T4C3), and truncated-psedophosphorylated tau (GFP-T4C3-2EC), loaded with TMRM for mitochondrial membrane potential determinations. b Quantitative analysis of TMRM fluorescence intensity of three experiments. Data are expressed as mean ± SE, n = 3. Scale bar, 10 μm. c Cells were transfected with GFP and GFP-tau forms as indicated above, and cell viability was measured using AlamarBlue™ assay. Graph represents the quantitative analysis of four experiments. Data are expressed as mean ± SE, n = 4
β-Amyloid Treatment Enhances Mitochondrial Fragmentation Induced by Tau Pathology
Several studies have shown that Aβ affects mitochondrial function in neuronal cells (reviewed in [20, 27]). For instance, expression of amyloid precursor protein (APP) in neuronal cells and mice (APP mice) induces mitochondrial fragmentation and bioenergetics failure, and later deterioration of memory and cognitive function [18, 19]. Therefore, it is important to study the effects of Aβ in neuronal cells in where truncated tau is present. Immortalized cortical neurons were transfected with GFP and all GFP-tau constructs plus Mito-mCherry (m-Ch) to examine effects on mitochondrial morphology (Fig. 5). Cells were treated during 24 h with 0.5 μM Aβ after transfection, and they were subsequently mounted in a microscope chamber for obtaining representative images (see “Experimental Procedures” section) from cells transfected under different conditions. Cells transfected with the vector (GFP), GFP full-length tau, and GFP-T42EC and treated with 0.5 μM Aβ showed a tubular-like mitochondrial morphology (Fig. 5a, b), with no apparent differences in mitochondrial length after quantification (Fig. 5c). In addition, cells transfected with truncated tau and subsequently treated with 0.5 μM Aβ did not present any significant differences in mitochondrial length after comparison with T4C3 untreated cells (Fig. 5c). However, Aβ treatment significantly reduced mitochondrial length in cells expressing truncated plus the pseudophosphorylated tau form (GFP-T4C3-2EC) compared with untreated cells (Fig. 5c).

Aβ treatment enhances mitochondrial fragmentation induced by caspase-cleaved tau in neuronal cells. a Representative images from CN1.4 cells transfected with green fluorescent protein (GFP) and GFP-tau constructs [GFP-T4, GFP-T42EC, GFP-T4C3, GFP-T4C3-2EC], in addition to Mito-mCherry (m-Ch) that were treated with 0.5 μM Aβ for 24 h. b Images of m-Ch fluorescence channel taken from the insert displayed in a that shows mitochondrial morphology in cells transfected with GFP and GFP-tau constructs treated with Aβ. Treatment with 0.5 μM Aβ reduced mitochondrial length in cortical cells expressing caspase-cleaved plus pseudophosphorylated tau (GFP-T4C3-2EC) compared to untreated cells. c Quantitative analysis of mitochondrial length from four independent experiments. Data are expressed as mean ± SE, n = 4. Scale bars in a 10 μm and b 5 μm. *p < 0.05
Effects of Aβ and Tau Pathology Expression on Mitochondrial Dynamics
As we have shown previously, Aβ treatment enhanced the mitochondrial fragmentation induced by the expression of truncated and pseudophosphorylated tau (GFP-T4C3-2EC) in immortalized cortical neurons (Fig. 5c). Mitochondrial morphology is regulated by proteins such as Drp1, Fis1 (mitochondrial fusion), Mnf1/2, and Opa1 (mitochondrial fission) [24]. Next, we analyzed the expression levels of Drp1 and Opa1 in cortical neurons transfected with tau forms and subsequently treated with 0.5 μM Aβ for 24 h (Fig. 6). Our previous observations showed that levels of both proteins (Drp1 and Opa1) were altered in immortalized cortical neurons transfected with truncated and truncated plus pseudophosphorylated tau (Figs. 2 and 3). However, treatment with Aβ did not significantly affect Drp1 (Fig. 6a, b) or Opa1 (Fig. 6c, d) levels in cells expressing GFP, GFP full-length (GFP-T4), GFP pseudophosphorylated (GFP-T42EC), and GFP truncated tau (GFP-T4C3). However, cells that expressed the truncated plus the pseudophosphorylated tau form showed a significant increase in Opa1 levels after treatment with Aβ (Fig. 6c, d).

Effect of Aβ treatment on Drp1 and Opa1 levels in neuronal cells that express pathological forms of tau. a CN1.4 cells were transfected with green fluorescent protein (GFP) and GFP-tau constructs [GFP-T4, GFP-T42EC, GFP-T4C3, GFP-T4C3-2EC] and treated with 0.5 μM Aβ for 24 h to determine the expression of Drp1 and Opa1 in whole cell extracts. Treatment with Aβ did not alter Drp1 expression levels in cells transfected with GFP and GFP-tau constructs. Levels of β-actin were evaluated as a loading control. b Quantitative analysis of three independent experiments representing Drp1 levels in cells transfected with GFP or GFP-tau constructs and treated with Aβ shows no significant differences in total Drp1 levels in all conditions. Data are expressed as mean ± SE, n = 3. c Western blot analysis of CN1.4 cells transfected with GFP and GFP-tau constructs previously indicated and treated with 0.5 μM Aβ for 24 h. Treatment with Aβ significantly increased Opa1 levels in cortical cells expressing caspase-cleaved plus pseudohyperphosphorylated tau. d Quantitative analysis of three experiments shows significant differences in Opa1 levels in cells expressing truncated plus pseudophosphorylated tau (GFP-T4C3-2EC) and treated with Aβ. Data are expressed as mean ± SE, n = 3. *p < 0.05
β-Amyloid Reduces Mitochondrial Membrane Potential and Cell Viability in Immortalized Neurons that Express Truncated Tau
Aβ is an important contributing factor to the pathogenesis of AD, and its toxic effects against neurons are mediated by the presence of tau [28]. Repressing tau expression in a knockout mouse model prevents the loss of cell viability, the mitochondrial calcium defects, and the impairment of axonal transport induced by Aβ [29–31]. Therefore, we analyzed cell viability and mitochondrial function in cortical neurons expressing truncated and pseudophosphorylated tau in immortalized cortical neurons (Fig. 7). To determine cell viability, cells expressing pathological forms of tau were treated with Aβ (0.5 and 2.5 μM) for 24 h, and then, they were incubated with AlamarBlue™ (Fig. 7a). Treatment with low doses of Aβ fibrils (0.5 μM) did not affect cell viability in any of the cells transfected with the tau constructs (Fig. 7a). Interestingly, exposure to 2.5 μM Aβ significantly decreased cell viability in cortical neurons transfected with truncated and truncated plus pseudophosphorylated forms of tau (Fig. 7a). However, these changes were significant only in cells expressing both pathological forms of tau, i.e., both truncated and pseudophosphorylated forms (Fig. 7a). To complement these findings, we evaluated the changes in mitochondrial membrane potential induced by acute Aβ treatment in cells expressing all tau constructs using TMRM and cell imaging (Fig. 7b) [17]. Cells transfected with different tau constructs were incubated with TMRM and transferred to a microscope chamber in order to determine changes in mitochondrial potential during Aβ exposure (Fig. 7b). Cells expressing truncated and truncated plus pseudophosphorylated forms of tau showed a significant reduction in mitochondrial membrane potential levels after Aβ treatment (Fig. 7b). Interestingly, cells expressing pseudophosphorylated tau (GFP-T42EC) showed a slight decrease in mitochondrial membrane potential compared to those expressing GFP or full-length tau (Fig. 7b). However, the effects observed in GFP-T42EC cells were not significant compared to control cells (Fig. 7b).

Aβ treatment induces mitochondrial dysfunction and cell viability loss in cells expressing caspase-cleaved tau. a Cells were transfected with GFP and GFP-tau constructs as indicated previously, and they were treated with 0.5 or 2.5 μM Aβ fibrils for 24 h. Treatment with 2.5 μM Aβ significantly reduced cell viability in cortical cells with truncated plus pseudophosphorylated tau (GFP-T4C3-2EC). Quantitative analysis of three experiments shows significant differences in the viability of cells expressing truncated plus pseudophosphorylated tau (GFP-T4C3-2EC) treated with Aβ. Data are expressed as mean ± SE, n = 3. *p < 0.05. b Mitochondrial membrane potential levels from immortalized cortical neurons transfected with GFP and GFP-tau constructs and treated with 0.5 μM Aβ fibrils as indicated. The line arrow indicates the time point at which Aβ was added. Changes in mitochondrial membrane potential levels were subsequently recorded for 1 h. Aβ treatment significantly reduced the mitochondrial membrane potential in cells expressing caspase-cleaved (GFP-T4C3) (*p < 0.05) and caspase-cleaved plus pseudohyperphosphorylated tau (GFP-T4C3-2EC) (**p < 0.05). Data are expressed as mean ± SE, n = 4
Expression of Truncated Tau Induces Mitochondrial Fragmentation and Enhances Aβ-Induced Mitochondrial Injury in Primary Cortical Neurons from Tau(−/−) Knockout Mice
Tau pathology is a key player in the pathogenesis of AD. Tau expression is necessary to mediate Aβ-induced toxicity in neurons [28]. Expression of truncated tau enhances mitochondrial injury in Aβ-treated cells, suggesting a synergic action between these two factors (Fig. 5). However, to rule out the possibility that endogenous tau might participate in Aβ-induced mitochondrial impairment, we expressed tau constructs in primary cortical neurons isolated from tau(−/−) knockout mice [29–31]. First, we double-transfected cortical neurons from tau(−/−) knockout mice with GFP-tau constructs and Mito-mCherry, and we subsequently examined mitochondrial length in vivo (Fig. 8). Cortical neurons expressing GFP, full-length, and pseudophosphorylated tau forms showed elongated mitochondrial morphology with a similar length (Fig. 8a, b). Interestingly, cortical neurons transfected with truncated and truncated plus pseudophosphorylated tau showed a significant reduction of mitochondrial length in axons and neuronal bodies (Fig. 8a, b, c). To complement these studies, we studied mitochondrial membrane potential in cortical neurons from tau(−/−) knockout mice expressing GFP-tau constructs followed by the acute administration of 0.5 μM Aβ in vivo (Fig. 8d). Corroborating our previous observations in immortalized cortical neurons (Fig. 7), the expression of GFP and full-length tau did not significantly affect mitochondrial membrane potential in response to Aβ treatment (Fig. 8d). In addition, expression of pseudophosphorylated tau reduced mitochondrial membrane potential in neurons treated with Aβ (Fig. 8d). However, this decrease in mitochondrial potential was not significant compared to untreated neurons (Fig. 8d). Both truncated tau forms (GFP-T4C3 and GFP-T4C3-2EC) enhanced the mitochondrial membrane potential loss induced by Aβ in cortical neurons corroborating our previous findings in CN1.4 cells (Fig. 7a).

Expression of truncated tau induces mitochondrial fragmentation and enhances the mitochondrial injury induced by Aβ in cortical neurons from tau(−/−) knockout mice. a Representative images from tau(−/−) knockout mouse cortical neurons transfected with GFP and tau constructs [GFP-T4, GFP-T42EC, GFP-T4C3, and GFP-T4C3-2EC], plus Mito-mCherry (m-Ch) for evaluation of mitochondrial length. b Fluorescence images of m-Ch channel from the insert displayed in a show mitochondrial morphology in neurons transfected with GFP and GFP-tau constructs. Caspase-cleaved tau (GFP-T4C3 or GFP-T4C3-2EC) reduced mitochondrial length compared to full-length tau (GFP-T4). c Quantitative analysis of mitochondrial length from four independent experiments. Data are expressed as mean ± SE, n = 4. Scale bars in a 10 μm and b 5 μm. *, **, ***p < 0.05. d Quantitative analysis of mitochondrial membrane potential levels from cortical neurons obtained from tau(−/−) knockout mice transfected with GFP and GFP-tau constructs and treated with 0.5 μM Aβ fibrils for 1 h as indicated. Treatment with Aβ significantly reduced the mitochondrial membrane potential in neurons expressing caspase-cleaved (GFP-T4C3) (*p < 0.05), pseudophosphorylated (GFP-T42EC) (**p < 0.05), and caspase-cleaved plus pseudophosphorylated tau (GFP-T4C3-2EC) (***p < 0.05). Data are expressed as mean ± SE, n = 3
Overall, these data indicate that truncated tau affects neuronal and mitochondrial function by the impairing of mitochondrial dynamics. Mitochondrial fragmentation induced by truncated tau may occur because of the reduction in Opa1 levels observed in neurons expressing this tau form. These results suggest an important role for truncated tau in the pathogenesis of AD, through the modification of mitochondrial function in the brain, which has been indicated as an early contributor factor to the pathogenesis of AD.
Discussion
AD is characterized by progressive loss of cognitive capabilities and memory due to the loss of synaptic function [32]. Mitochondrial function is an important player in the development, maintenance, and function of synapses, which are highly dependent on adenosine triphosphate (ATP) production by mitochondria. Mitochondria also contribute to neurotransmitter release and the handling of cytosolic Ca2+, which is taken up into neurons after a neuronal impulse [33–38]. Therefore, mitochondrial size and morphology must be precisely regulated to support the production of energy and antioxidant defense in neurons [20]. In this study, we show that the presence of truncated tau impairs mitochondrial dynamics by reducing Opa1 levels in neuronal cells. This is the first study that shows a negative effect of tau pathology against mitochondrial dynamics through downregulation of Opa1. These effects are likely to affect mitochondrial bioenergetics and neuronal function, since the presence of truncated tau enhances mitochondrial damage and cell viability loss induced by Aβ (Figs. 7 and 8). These are important observations because Aβ and pathological forms of tau participate in the genesis and progression of AD [20, 32]. Complementary our data, it has been shown that alterations in the balance of the mitochondrial fission/fusion cycle impair bioenergetics and synaptogenesis [39–43]. Regarding to Opa1, interesting studies have shown the importance of this protein in mitochondrial function and synaptic regulation. For instance, Bertholet et al. showed that ablation of Opa1 in the Opa1 knockout mouse produces mitochondrial fragmentation, a decrease in the mitochondrial population in dendrites and axons, and defects in mitochondrial bioenergetics [41]. Interestingly, these events were associated with a reduced expression of both presynaptic and postsynaptic proteins and the number of synapses [41]. In addition, synaptic defects were also observed in vivo in retinal ganglion cells (RGCs) from a dominant optic atrophy (DOA) mouse model [42, 43]. Here, Kushnareva et al. showed that decreases in Opa1 expression results in mitochondrial fragmentation, deficiencies in oxidative phosphorylation, decreased ATP production, decreased mitochondrial Ca2+ retention capacity, and sensitization to apoptotic insults in RGC neurons [43]. More importantly, this Opa1 deficiency decreases buffering of cytosolic Ca2+ and sensitizes RGCs to excitotoxic injury due to glutamate exposure, indicating a potential role for Opa1 in synaptic transmission [43]. These studies show that both the presynaptic and postsynaptic compartments could be affected by inactivation of Opa1. Thus, dysregulation of Opa1 could play an important role in the pathological mechanisms involved in neurodegenerative diseases such as AD [20, 41].
Along with the clinical manifestations of memory impairment and aggregates of Aβ, the presence of aberrantly modified tau in the NFTs is a prominent feature in the brain of individuals with AD and has been implicated in the pathogenesis of this disease [44–46]. Tau that is hyperphosphorylated at residues S396 and S404 shows an increased tendency to self-associate, and it has been found in the NFTs of the AD brain [44]. However, based on immunohistochemical analyses, increased phosphorylation of tau at S396/404 appears to be a relatively late event in AD [47]. Interestingly, cleavage of tau by caspase-3 at D421 (truncated tau) and its co-localization with NFTs appear to occur at an early stage in the brain of individuals with AD [47]. More importantly, recent studies have provided evidence suggesting that truncated tau might contribute to the cognitive impairment in AD [48–50]. For instance, Means et al. studied the role of truncated tau in age-related cognitive impairment [48]. Levels of activated caspases were increased in aged mice, which correlated specifically with an increase in truncated tau and the formation of NFTs [48]. In addition, they demonstrated that aged mice model showed significant cognitive impairment, which correlated with high levels of caspase-3 activity and truncated tau [48]. In a complementary study, Kim et al. investigated the role of truncated tau in memory and synaptic impairment [49]. They generated a transgenic mice line in where the expression of truncated tau was regulated by a specific neuronal promoter [49]. These mice exhibited a significant deficit in learning, spatial memory loss, and synaptic dysfunction at young age (2 months) [49]. Finally, Ozcelik et al. studied the contribution of truncated tau in neurodegeneration by co-expressing full-length and truncated tau to generate an inducible mouse model [50]. Co-expression of these two forms of tau induced tau aggregation, missorting of synaptic proteins, axonal transport failure, and mitochondrial aggregation [50]. Interestingly, this study and our previous work showed that the expression of truncated tau induces mitochondrial fragmentation, bioenergetics defects, and impairs axonal transport [11, 21]. Overall, this indicates that truncated tau plays an important role in the neurodegeneration seen in AD by impairing mitochondrial function in the brain.
The work presented here provides significant evidence linking tau pathology with defects in mitochondrial dynamics, which, in turn, could induce synaptic impairment. We found that expression of caspase-cleaved tau (T4C3) induces mitochondrial fragmentation and a significant decrease in mitochondrial length, which is in agreement with previous studies [21, 51, 52] showing that mitochondria are fragmented in AD and that caspase-cleaved tau induces mitochondrial fragmentation [11], respectively. Interestingly, we found that the expression of truncated tau reduced the relative expression of Opa1 and Drp1 (Figs. 3 and 4) with a significant effect on Opa1 levels. Opa1 expression determines the integrity of mitochondrial cristae maintaining the ETC function and cell survival [53, 54]. However, under basal conditions, the expression of truncated tau did not affect mitochondrial function or cell viability in neuronal cells. Interestingly, treatment with Aβ fibrils significantly decreased mitochondrial membrane potential levels and increase cell viability loss in neuronal cells expressing truncated tau. More importantly, these observations were verified using primary neuronal cultures from tau(−/−) knockout mice in where truncated tau was expressed to physiological levels (Fig. 8). These are very important observations because they rule out the possibility that truncated tau overexpression could be a causal factor in the impairment of mitochondrial dynamics observed in truncated tau-expressing cells. Together, our results suggest that the presence of truncated tau forms could sensitize mitochondria to external stimuli, i.e., Aβ peptide, an event that might be important in the neurotoxicity found in AD [29, 30].
Additionally, it has been shown that caspase activation precedes and promotes NFTs and that formation of caspase cleavage of tau occurs prior to the formation of NFTs [55]. This implies that the effects of caspase-cleaved tau on mitochondrial dynamics showed in our study could be considered an early event in the pathogenesis of AD and that would contribute to memory impairment through synaptic failure [56]. Since the mechanism by which caspase-cleaved tau decreases Opa1 is still unknown, further studies should elucidate how caspase-cleaved tau affects the expression of Opa1 and to determine if this Opa1 downregulation has direct consequences for mitochondrial integrity in AD.
References
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362(4):329–344. doi:10.1056/NEJMra0909142
Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT et al (2004) Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114(1):121–130. doi:10.1172/JCI20640
Cho JH, Johnson GV (2004) Glycogen synthase kinase 3 beta induces caspase-cleaved tau aggregation in situ. J Biol Chem 279(52):54716–54723. doi:10.1074/jbc.M403364200
Lee S, Shea TB (2012) Caspase-mediated truncation of tau potentiates aggregation. Int J Alzheimers Dis 2012:731063. doi:10.1155/2012/731063
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P et al (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307(5713):1282–1288. doi:10.1126/science.1105681
Cowan CM, Bossing T, Page A, Shepherd D, Mudher A (2010) Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol 120(5):593–604. doi:10.1007/s00401-010-0716-8
Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N (2012) Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One 7(5):e36873. doi:10.1371/journal.pone.0036873
Zhang Y, Chen L, Shen G, Zhao Q, Shangguan L, He M (2014) GRK5 dysfunction accelerates tau hyperphosphorylation in APP (swe) mice through impaired cholinergic activity. Neuroreport 25(7):542–547. doi:10.1097/WNR.0000000000000142
Dixit R, Ross JL, Goldman YE, Holzbaur EL (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319(5866):1086–1089. doi:10.1126/science.1152993
Dolan PJ, Johnson GV (2010) The role of tau kinases in Alzheimer’s disease. Curr Opin Drug Discov Dev 13(5):595–603
Quintanilla RA, Dolan PJ, Jin YN, Johnson GV (2012) Truncated tau and Abeta cooperatively impair mitochondria in primary neurons. Neurobiol Aging 33(3):e625–e635. doi:10.1016/j.neurobiolaging.2011.02.007 619
Zhang Q, Zhang X, Sun A (2009) Truncated tau at D421 is associated with neurodegeneration and tangle formation in the brain of Alzheimer transgenic models. Acta Neuropathol 117(6):687–697. doi:10.1007/s00401-009-0491-6
Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV (2009) Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem 284(28):18754–18766. doi:10.1074/jbc.M808908200
Kandimalla R, Reddy PH (2016) Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim Biophys Acta 1862(4):814–828. doi:10.1016/j.bbadis.2015.12.018
Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P (2011) Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev 67(1–2):103–118. doi:10.1016/j.brainresrev.2010.11.004
R Kandimalla, Manczak M, Fry D, Suneetha Y, Sesaki H, Reddy PH (2016) Reduced dynamin-related protein 1 protects against phosphorylated tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Human Molecular Genetics
Hauptmann S, Scherping DS, Brandt U, Schulz KL, Jendrach M, Leuner K, Eckert A, Müller WE (2009) Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging 30(10):1574–1586. doi:10.1016/j.neurobiolaging.2007.12.005
Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H et al (2009) Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci U S A 106(47):20057–20062. doi:10.1073/pnas.0905529106
Trushina E, Nemutlu E, Zhang S, Christensen T, Camp J, Mesa J, Siddiqui A, Tamura Y et al (2012) Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS One 7(2):e32737. doi:10.1371/journal.pone.0032737
Cabezas-Opazo FA, Vergara-Pulgar K, Perez MJ, Jara C, Osorio-Fuentealba C, Quintanilla RA (2015) Mitochondrial dysfunction contributes to the pathogenesis of Alzheimer’s disease. Oxidative Med Cell Longev 2015:509654. doi:10.1155/2015/509654
Quintanilla RA, von Bernhardi R, Godoy JA, Inestrosa NC, Johnson GV (2014) Phosphorylated tau potentiates Abeta-induced mitochondrial damage in mature neurons. Neurobiol Dis 71:260–269. doi:10.1016/j.nbd.2014.08.016
Takuma H, Tomiyama T, Kuida K, Mori H (2004) Amyloid beta peptide-induced cerebral neuronal loss is mediated by caspase-3 in vivo. J Neuropathol Exp Neurol 63(3):255–261
David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Drose S et al (2005) Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem 280(25):23802–23814. doi:10.1074/jbc.M500356200
Lasagna-Reeves CA, Glabe CG, Kayed R (2011) Amyloid-beta annular protofibrils evade fibrillar fate in Alzheimer disease brain. J Biol Chem 286(25):22122–22130. doi:10.1074/jbc.M111.236257
Ding H, Matthews TA, Johnson GV (2006) Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J Biol Chem 281(28):19107–19114. doi:10.1074/jbc.M511697200
Matthews-Roberson TA, Quintanilla RA, Ding H, Johnson GV (2008) Immortalized cortical neurons expressing caspase-cleaved tau are sensitized to endoplasmic reticulum stress induced cell death. Brain Res 1234:206–212. doi:10.1016/j.brainres.2008.07.111
Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9(7):505–518. doi:10.1038/nrn2417
Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 99(9):6364–6369. doi:10.1073/pnas.092136199
Pallo SP, Johnson GV (2015) Tau facilitates Abeta-induced loss of mitochondrial membrane potential independent of cytosolic calcium fluxes in mouse cortical neurons. Neurosci Lett 597:32–37. doi:10.1016/j.neulet.2015.04.021
Pallo SP, DiMaio J, Cook A, Nilsson B, Johnson GV (2016) Mechanisms of tau and Abeta-induced excitotoxicity. Brain Res 1634:119–131. doi:10.1016/j.brainres.2015.12.048
Vossel KA, Xu JC, Fomenko V, Miyamoto T, Suberbielle E, Knox JA, Ho K, Kim DH et al (2015) Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J Cell Biol 209(3):419–433. doi:10.1083/jcb.201407065
Selkoe DJ (2008) Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 192(1):106–113. doi:10.1016/j.bbr.2008.02.016
Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ (2005) Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47(3):365–378. doi:10.1016/j.neuron.2005.06.018
Ly CV, Verstreken P (2006) Mitochondria at the synapse. Neuroscientist 12(4):291–299. doi:10.1177/1073858406287661
Ivannikov MV, Macleod GT (2013) Mitochondrial free Ca(2)(+) levels and their effects on energy metabolism in Drosophila motor nerve terminals. Biophys J 104(11):2353–2361. doi:10.1016/j.bpj.2013.03.064
Rangaraju V, Calloway N, Ryan TA (2014) Activity-driven local ATP synthesis is required for synaptic function. Cell 156(4):825–835. doi:10.1016/j.cell.2013.12.042
Reyes RC, Parpura V (2008) Mitochondria modulate Ca2+-dependent glutamate release from rat cortical astrocytes. J Neurosci 28(39):9682–9691. doi:10.1523/JNEUROSCI.3484-08.2008
Li Z, Okamoto K, Hayashi Y, Sheng M (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119(6):873–887. doi:10.1016/j.cell.2004.11.003
Chen H, McCaffery JM, Chan DC (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130(3):548–562. doi:10.1016/j.cell.2007.06.026
Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH et al (2012) Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol 197(4):535–551. doi:10.1083/jcb.201110034
Bertholet AM, Millet AM, Guillermin O, Daloyau M, Davezac N, Miquel MC, Belenguer P (2013) OPA1 loss of function affects in vitro neuronal maturation. Brain 136(Pt 5):1518–1533. doi:10.1093/brain/awt060
Williams PA, Piechota M, von Ruhland C, Taylor E, Morgan JE, Votruba M (2012) Opa1 is essential for retinal ganglion cell synaptic architecture and connectivity. Brain 135(Pt 2):493–505. doi:10.1093/brain/awr330
Kushnareva YE, Gerencser AA, Bossy B, Ju WK, White AD, Waggoner J, Ellisman MH, Perkins G et al (2013) Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ 20(2):353–365. doi:10.1038/cdd.2012.128
Pritchard SM, Dolan PJ, Vitkus A, Johnson GV (2011) The toxicity of tau in Alzheimer disease: turnover, targets and potential therapeutics. J Cell Mol Med 15(8):1621–1635. doi:10.1111/j.1582-4934.2011.01273.x
Kosik KS, Joachim CL, Selkoe DJ (1986) Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 83(11):4044–4048
Basurto-Islas G, Luna-Munoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, Garcia-Sierra F (2008) Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol 67(5):470–483. doi:10.1097/NEN.0b013e31817275c7
Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, Cahill ME, Bigio EH et al (2005) Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging 26(7):1015–1022. doi:10.1016/j.neurobiolaging.2004.09.019
Means JC, Gerdes BC, Kaja S, Sumien N, Payne AJ, Stark DA, Borden PK, Price JL et al (2016) Caspase-3-dependent proteolytic cleavage of tau causes neurofibrillary tangles and results in cognitive impairment during normal aging. Neurochem Res 41(9):2278–2288. doi:10.1007/s11064-016-1942-9
Kim Y, Choi H, Lee W, Park H, Kam TI, Hong SH, Nah J, Jung S et al (2016) Caspase-cleaved tau exhibits rapid memory impairment associated with tau oligomers in a transgenic mouse model. Neurobiol Dis 87:19–28. doi:10.1016/j.nbd.2015.12.006
Ozcelik S, Sprenger F, Skachokova Z, Fraser G, Abramowski D, Clavaguera F, Probst A, Frank S et al (2016) Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol Psychiatry. doi:10.1038/mp.2015.228
Wang X, Su B, Fujioka H, Zhu X (2008) Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol 173(2):470–482. doi:10.2353/ajpath.2008.071208
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29(28):9090–9103. doi:10.1523/JNEUROSCI.1357-09.2009
Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS et al (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126(1):177–189. doi:10.1016/j.cell.2006.06.025
Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, Menabo R, Costa V et al (2015) The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 21(6):834–844. doi:10.1016/j.cmet.2015.05.007
de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464(7292):1201–1204. doi:10.1038/nature08890
Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova NN, Yang L, Starkov AA et al (2011) Behavioral deficit, oxidative stress, and mitochondrial dysfunction precede tau pathology in P301S transgenic mice. FASEB J 25(11):4063–4072. doi:10.1096/fj.11-186650
Acknowledgements
This work was supported by FONDECYT #1140968 and Anillo ACT1411 (RAQ).
Author information
Authors and Affiliations
Corresponding author
Additional information
María José Pérez and Katiana Vergara-Pulgar contributed equally to this work.
Electronic supplementary material
Figure supplementary 1
Caspase-cleaved tau expression did not affect pDrp1 and Mfn1 regulation in immortalized cortical neurons. (A) CN1.4 cells were transfected with GFP and tau constructs [GFP-T4, GFP-T42EC, GFP-T4C3, and GFP-T4C3-2EC] to determine the expression of pDrp1 using anti-pDrp1 antibody that detects Drp1 phosphorylation at S616. Representative western blot analysis from 3 independent experiments that show no significant differences in Drp1 activation by the presence of tau pathology. (B) Representative western blot image from 3 experiments of Mfn1 expression in CN1.4 cells transfected with pathological forms of tau. As well as, Mfn2, Mfn1 levels were not affected by the presence of tau pathology in these cells. In both western blot analysis the levels of β-actin were evaluated as a loading control. (GIF 857 kb)
Figure supplementary 2
Expression levels of Drp1 and Mfn2 in CN 1.4 cells transfected with pathological forms of tau. (A), (B) are representative western blots images from CN 1.4 cells transfected with GFP and GFP-tau forms in where the levels of total Drp1 and Mfn2 were evaluated. Images includes molecular weight standard to corroborate Drp1 and Mnf2 expression in CN 1.4 cells. β-actin levels were evaluated as a loading control. Images are representative of 4 independent experiments. (GIF 918 kb)
Rights and permissions
About this article

Cite this article
Pérez, M.J., Vergara-Pulgar, K., Jara, C. et al. Caspase-Cleaved Tau Impairs Mitochondrial Dynamics in Alzheimer’s Disease. Mol Neurobiol 55, 1004–1018 (2018). https://doi.org/10.1007/s12035-017-0385-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0385-x