
Abstract
Alzheimer’s disease (AD) is the most common progressive neurodegenerative disorder. A defining hallmark of the AD brain is the presence of intraneuronal neurofibrillary tangles (NFTs) which are made up of abnormally modified tau, with aberrant phosphorylation being the most studied posttranslational modification (PTM). Although the accumulation of tau as NFTs is an invariant feature of the AD brain, it has become evident that these insoluble aggregates are likely not the primary pathogenic form of tau, rather soluble forms of tau with abnormal PTMs are the mediators of toxicity. The most prevalent PTM on tau is phosphorylation, with the abnormal modification of specific residues on tau playing a key role in its toxicity. Even though it is widely accepted that tau with aberrant PTMs facilitates neurodegeneration, the precise cellular mechanisms remain unknown. Nonetheless, there is an evolving conceptual framework that an important contributing factor may be selective pathological tau species compromising mitochondrial biology. Understanding the mechanisms by which tau with site-specific PTM impacts mitochondria is crucial for understanding the role tau plays in AD. Here, we provide a brief introduction to tau and its phosphorylation and function in a physiological context, followed by a discussion of the impact of soluble phosphorylated tau species on neuronal processes in general and mitochondria more specifically. We also discuss how therapeutic strategies that attenuate pathological tau species in combination with treatments that improve mitochondrial biology could be a potential therapeutic avenue to mitigate disease progression in AD and other tauopathies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common degenerative brain disease in the aged population. It is characterized by the progressive decline of cognition and memory, as well as changes in behavior and personality [1]. A defining pathological hallmark of the AD brain is the presence of insoluble neurofibrillary tangles (NFTs), which are primarily composed of abnormally modified tau [2]. Tau isolated from AD brain tissues exhibits a number of posttranslational modifications (PTMs), with increases in phosphorylation being the most prevalent and well-studied, although there is an increasing awareness that tau is also abnormally acetylated and both these PTMs likely impair tau function [3, 4]. These PTMs were originally characterized in terms of their ability to influence tau aggregation. However, the concept of insoluble fibrillary structures in AD and other tauopathies being the principle mediators of neuronal toxicity has been gradually abandoned [5], and the mechanism by which specific tau PTMs contribute to the toxicity of soluble tau forms is increasingly the focus of ongoing investigations. Evolving evidences indicate that expression of human tau contributes to mitochondrial dysfunction in AD animal models [6]. In addition, accumulation of dysfunctional mitochondria can cause or magnify visible tau pathologies and are believed to play a central role in AD pathogenesis [7]. Thus, there is reciprocity between pathological tau species and impaired mitochondrial biology.
Mitochondria lie at the nexus of a broad spectrum of critical cellular functions, such as energy supply and intracellular communication [8], and mitochondrial dysfunction is characteristic of many neurodegenerative diseases including AD [9]. Recent studies have suggested that one possible mechanism by which pathological tau species may compromise neuronal function is by negatively impacting mitochondria [10]. Mitochondrial abnormalities that have been reported in response to the pathological accumulation of tau include imbalances in mitochondrial dynamics and impaired oxidative phosphorylation (OXPHOS), leading to increased production of reactive oxygen species (ROS) [11]. In addition, there are causative mechanistic links between tau with AD-relevant PTMs and impaired mitophagy and mitochondrial trafficking [12].
The focus of this review is on how pathogenic tau impairs neuronal function with an emphasis on mitochondria. Here, we will detail how current findings are beginning to shape our understanding of this relationship and highlight knowledge gaps that need to be addressed. We will also illustrate how model organism research including worms, flies, and mice has contributed to our appreciation of the impact of tau with AD-relevant PTMs on mitochondrial biology. Finally, we discuss how delineating these mechanisms may help us develop a combinatorial therapeutic approach for the treatment of AD patients.
Tau Structure and Function
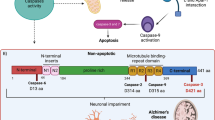
Tau is a neuronal protein and is central to the pathogenesis of AD and other neurodegenerative diseases termed tauopathies [13]. Human tau is encoded by the MAPT gene which has 16 exons and is located on chromosome 17q21.31. Alternate splicing of exons 2, 3, and 10 generates the six primary tau isoforms ranging from 352 to 441 amino acids in the adult human brain [14]. These six isoforms differ by the presence of zero, one, or two N-terminal inserts (resulting from the splicing in or out of exons 2 and 3) (0N, 1N, or 2N, respectively) and either three (3R) or four (4R) microtubule-binding repeats (resulting from the splicing in or out of exon 10) in the C-terminal half of tau [15, 16].
Decades of research have led to a model where tau maintains neuronal processes by regulating axonal microtubule structure and thus function [17,18,19]. The interaction between tau and neuronal microtubules is highly dynamic, as demonstrated by fast single-molecule tracking of tau in living neurons [20]. This interaction of tau with microtubules has been described as a “kiss and hop mechanism,” where tau dwells on a single microtubule in the range of milliseconds before it hops on to a neighboring one, residing only transiently on each of them [20]. Importantly, it explained why tau, although binding to microtubules, can interact with various proteins and fulfills diverse functions in different neuronal compartments [21]. Indeed, tau is defined as an intrinsically disordered protein (IDP), which are known to interact with many binding partners and are considered to modulate various signaling, regulation, and recognition processes [22, 23]. Intriguingly, recent findings have provided evidence that instead of stabilizing axonal microtubules, tau may enable axonal microtubules to have long labile domains, in part by outcompeting genuine stabilizer proteins, such as MAP6 [24, 25]. These are new findings and need to be further investigated; nonetheless, they indicate that perhaps the current dogma about the function of tau in mediating microtubule stability in neurons needs to be re-examined. Other functions of tau in normal neuronal physiology include but are not limited to maintenance of healthy synapses, neurogenesis, and neuronal polarity in development [26, 27] (Fig. 1). Clearly, tau plays a pivotal role in regulating various normal neuronal functions. However, in pathophysiological conditions, tau may induce neurotoxicity owing to its loss-of-function, toxic gain-of-function, or its mislocalization to the pre- or postsynaptic compartment leading to synaptic dysfunction or even a combination of these factors. PTMs alter tau oligomerization and can contribute to pathophysiological processes [28]. Although the mechanisms by which tau with AD-relevant PTMs causes neuronal toxicity are not fully understood, it is likely that this toxicity results from soluble tau species, and not the insoluble NFTs that characterize the AD brains [5]. Indeed, there is a growing body of evidence that soluble tau species that are modified at specific epitopes likely cause toxicity [29,30,31]. In this context, tau neurotoxicity is correlated with an increased phosphorylation (hyperphosphorylation) at various epitopes, which affects the strength and spectrum of its interaction with itself and many other cellular components [32]. While it is recognized that in addition to phosphorylation, pathological tau species from AD exhibit numerous PTMs including differential cleavage by calpains and caspases, which likely contribute to aggregation and toxicity [33,34,35,36,37,38,39,40], the cellular and functional implications of these other modifications of tau have not been as extensively studied and therefore, we will focus predominantly on phosphorylation.

Tau function and dysfunction. a In the physiological state, tau plays numerous roles including regulation of axonal microtubule assembly, contributing to neuronal polarity development and outgrowth. b In the pathological state, tau with abnormal PTMs exhibits decreased affinity for microtubules and can contribute to mitochondrial damage, as well as impair synaptic transmission culminating in neurodegenerative processes
Tau Phosphorylation
Phosphorylation is the most common tau PTM, with its dynamic regulation playing a critical role in modulating tau function [3, 26]. However, in AD and in other tauopathies, tau becomes disproportionally phosphorylated at key sites, which likely leads to its toxicity [41]. The longest tau isoform in human brain (2N4R) contains 85 potential phosphorylation sites (80 Ser or Thr, and 5 Tyr), with phosphorylation of serine 214 (S214), threonine 231 (T231), and serine 262 (S262), among others, playing key roles in regulating tau-microtubule interactions [16]. Studies to date have provided evidence that phosphorylation of tau at T231 occurs early in the evolution of tau pathology; for example, increased staining for this epitope is observed in “pre-tangle” neurons [42]. Further, increased phospho-T231 tau has been observed in neurons differentiated from iPSCs of sporadic AD cases [43]. In addition, phosphorylation of tau at T231 precedes the formation of tau oligomers [44] which likely contribute to tau toxicity [5]. Under pathophysiological conditions, it has been reported that tau phosphorylation at other sites such as S214 and S262 also decreases the ability of tau to bind microtubules, thus increasing the levels of “free” tau and enabling interaction with other cellular partners [45]. It has been proposed that “detached” tau gets redistributed from neuronal processes to the soma, undergoes self-aggregation, forming oligomers, leading to synaptic dysfunction [46] and ultimately neuronal cell death [47] (Fig. 1). Interestingly, another study demonstrated that the association of tau with microtubules was dramatically decreased when 10 key phosphorylation sites were pseudo-phosphorylated (S/T → E). This was due to a robust decrease in the association rate with no major change in the dissociation rate [48]. Thus, disease-associated phosphorylation is likely crucial in the pathogenesis of AD and therefore may represent a putative therapeutic target.
In addition to the aforementioned disease relevant sites, there exist unique sites where phosphorylation may actually make tau more protective and have beneficial effects in the context of Aβ toxicity. For example, at least at early stages of pathogenesis, site-specific phosphorylation of tau at threonine 205 (T205) inhibited Aβ toxicity in amyloid precursor protein (APP) transgenic mice. This specific tau phosphorylation was mediated by mitogen-activated protein kinase p38γ, which attenuated postsynaptic excitotoxic signaling in response to Aβ [49]. This example counters the idea that increased phosphorylation at all sites on tau are detrimental. A recent in vitro study also demonstrated that phosphorylation of a unique residue serine 305 (S305), which lies within the microtubule-binding domain, significantly inhibited tau aggregation. Furthermore, consistent with this data, phosphorylation of S305 was not detected in pathological tau inclusions in AD brain tissue [50]. This critical finding supports developing an in vivo model that will provide further mechanistic insight how select tau PTMs may be protective against aggregation. Moreover, evidence indicates the presence of other phosphorylated sites, which are not critical in terms of disease progression and are present at relatively low levels such as Serine 68 (S68), threonine 69 (T69) and threonine (T71). These residues are located in 1N and 2N tau isoforms, but not 0N [51]. Thus, it is clear that there is selectivity in terms of tau phosphorylation sites associated with dysfunction, where hyperphosphorylation of certain sites can be more pathogenic compared to others. This difference in pathogenicity can depend upon the location of the individual site in a specific tau isoform. For example, the 2N tau isoform is expressed at relatively low levels compared to other four brain tau isoforms [52]. To further understand how the different isoform’s toxicity might relate to the presence or absence of individual sites of PTM, in vivo models can be utilized which express all six, non-mutated, human tau isoforms, along with site-specific disease relevant phospho-mimetic mutant transgenic animals. Collectively, these studies will have important implications for potential kinase therapies as described in the later section.
Impact of Pathogenic Tau on Mitochondrial Biology
Mitochondria impact a broad spectrum of critical cellular processes and play a vital role in metabolic homeostasis, with implications for cell signaling, aging, and cell death. A substantial body of evidence suggests that mitochondrial dysfunction is associated with many neurodegenerative diseases, including AD [53, 54]. A well-recognized role of mitochondria is to supply ATP through OXPHOS to coordinate energy demand with expenditure [55]. Unsurprisingly, since neurons are highly energetic cells, maintenance of a healthy mitochondrial pool is essential for neuronal health. However, the physical and molecular processes that contribute to mitochondrial dysfunction in AD are still being explored. Consistent with their vital role in cell health, there are a variety of specific molecular mechanisms geared toward promoting mitochondrial quality control. Some of these mechanisms involve signaling between the mitochondria and the nucleus, leading to adaptive changes in gene expression [56], while other mechanisms involve physical processes that impact mitochondrial morphology, movement, and turnover [57]. Cellular processes involved in the later class include (i) mitochondrial fission and fusion, leading to morphologic dynamics; (ii) mitophagy, leading to the removal of dysfunctional mitochondria; and (iii) mitochondrial transport, leading to the dynamic movement of mitochondria via anterograde/retrograde trafficking along the neuron. In the following section, we focus on these processes and explore how pathogenic forms of tau can impact them and thus can be a parallel hit for the progression of the disease. As indicated above, tau is a soluble, natively unfolded multifunctional protein that has numerous interaction partners in addition to microtubules and shows a remarkable structural plasticity allowing for rapid changes in response to alterations in the cellular environment by in large through dynamic changes in PTMs, as thoroughly discussed in these excellent articles [23, 58]. Given the number of possible interactors, effects of pathogenic tau on mitochondria could be rather indirect. Nevertheless, it is crucial to understand the different mitochondrial mechanisms that are affected by pathogenic tau (directly or indirectly), which could serve as potential therapeutic targets for AD.
OXPHOS
Neurons heavily depend on mitochondria to meet the energy demanded by synaptic transmission. Mitochondria generate ATP through the OXPHOS system of the electron transport chain (ETC). The ETC comprises four biochemically linked multi-subunit complexes I, II, III, and IV, as well as two electron carriers, coenzyme Q and cytochrome C, localized at the inner mitochondrial membrane (IMM) (Fig. 2). By using the energy that is stored in nutritional sources, the respiratory chain generates a proton pump across the IMM to drive ATP synthesis via ATP synthase (complex V), while at the same time transferring electrons to oxygen and producing water [59]. Mitochondria are the major source of ROS and at the same time a target of ROS toxicity [60,61,62]. Damaged mitochondria lead to a reduction in cellular energy levels causing leakage of electrons. This promotes formation of ROS that can be detrimental for proteins, membrane lipids, and nucleic acids, ultimately hampering cellular homeostasis. Neurons have a high-energy demand, and hence, it comes as no surprise that abnormalities in mitochondrial bioenergetics have been associated with various neurodegenerative disorders, including tauopathies [63]. More surprising though is the degree to which individual select tau PTMs, as well as frontotemporal lobar dementia (FTLD) mutations, appear to be associated with specific bioenergetics changes.
Studies have demonstrated that phosphorylated tau can promote mitochondrial dysfunction in neurons of FTLD mutant tau (P301L) transgenic mice (pR5) by impairing complex I of ETC. Aged P301L tau mice that have significant accumulation of hyperphosphorylated tau showed impaired mitochondrial respiration, reduced complex V activity, and higher levels of ROS than wild-type controls [6]. This evidence indicated that tau pathology could lead to impaired mitochondrial respiration, ATP synthesis, and oxidative stress as in AD (Fig. 2). Expression of tau pseudo-phosphorylated at the AD-relevant sites S396/404 in aged cortical neurons resulted in mitochondrial depolarization and an increase in superoxide production [44]. It has been demonstrated that human tau overexpression in primary neuron cultures decreases both ATP levels and the ratio of ATP/ADP, as well as inhibits complex I activity [64]. Moreover, a recent study has reported that human-induced pluripotent stem cells (iPSC)–derived neurons carrying the FTLD 10 + 16 MAPT mutation present with hyperpolarized mitochondria, inhibition of complex I respiration, decreased ATP production, and oxidative stress [65]. Together, these findings support the concept that pathological tau species have a profound effect on mitochondrial functions causing inappropriate ROS production. This results in mitochondrial energy deprivation, oxidative stress at the synapses, and synaptic failure. Thus, mitigating ROS may provide novel therapeutic strategies for tauopathies

Mitochondrial bioenergetics is impaired by tau with pathological PTMs. a The OXPHOS machinery is located at the inner mitochondrial membrane (IMM). It consists of five complexes where complex I–IV are involved in electron transfer and proton export to maintain proton gradient. Finally, complex V generates ATP to support neuronal function and activity. In pathological conditions, phosphorylated tau interferes with the mitochondrial respiratory chain, affecting mitochondrial membrane potential (MMP), inducing oxidative stress, and stimulating higher production of reactive oxygen species (ROS). Moreover, hyperphosphorylated tau decreases the expression of complex I and V, with a concomitant reduction in complex I activity and ATP production
.
Mitochondria Fission and Fusion
Mitochondria are highly dynamic cellular organelles, with the ability to change size and shape. Two established coordinated processes regulate these changes: fission and fusion [66]. In a given cell, mitochondria are undergoing both fission and fusion simultaneously to maintain functionality and a healthy mitochondrial network. The key mitochondrial fission and fusion proteins are members of the dynamin family. Fusion proteins include the GTPases Mitofusin 1 (Mfn1) and Mitofusin 2 (Mfn2) located in the outer mitochondrial membrane (OMM) and optic atrophy 1 (Opa1), at the IMM. Mitochondrial fission involves dynamin-related protein 1 (Drp1), primarily a cytosolic protein that is dynamically recruited to mitochondria, and fission 1 (Fis1), which is anchored in the OMM [67, 68]. In addition, there are two negative regulators of Drp1-dependent fission, MIEF1/MiD51 and MiD49, which bind and inhibit Drp1. Fis1 promotes mitochondrial fission by its ability to sequester MIEF1/MiD51, preventing it from inhibiting mitochondrial fission [69]. The core proteins (Drp1, Fis1, Mfn1/2, and Opa1) are highly involved in mitochondrial dynamics (Fig. 3) and thus, any alterations in this machinery could have profound effects on mitochondrial function, energy, and redox homeostasis.

Mitochondrial dynamics and turnover. a A simplified overview of the processes through which mitochondria undergo morphological changes and recycling. Over time with stress, a mitochondrion accumulates damage, which undergoes fission through Drp1 and other proteins and helps in segregation of damaged mitochondria. A damaged mitochondrion is marked and recognized by the macroautophagic machinery, forming a mitophagosome. This then fuses with a lysosome to be degraded by mitophagy. After the fission event, a healthy daughter mitochondrion undergoes recovery and fusion with the help of Mfn1, Mfn2, Opa1 proteins. b Under normal physiological conditions, there is a balance between fission and fusion events and mitochondria maintain healthy life cycles. However, under pathological conditions, alteration in fission and fusion processes causes mitochondrial dynamics imbalance leading to altered mitochondrial morphology
It has been demonstrated that phosphorylated tau specifically interacts with Drp1 in AD brain [70]. Using cortical protein lysates from brain tissue from severe AD cases (Braak stages V and VI) and of age matched control subjects, phosphorylated tau was found to co-precipitate with Drp1. This study was further validated using brain tissue from APP/presenilin1 (PS1) mice, 3xTg-AD mice (13-month-old) and age-matched wild-type mice, with phosphorylated tau co-immunoprecipitating with Drp1 from lysates of APP/PS1 and 3xTg-AD mouse brain, but not wild type [70]. In addition, immunohistochemical analysis of AD brains demonstrated that the immunoreactivity of Drp1 colocalized with phosphorylated tau, further indicating Drp1 interacts with pathologically phosphorylated tau [70]. These effects were accompanied by significantly increased levels of GTPase activity in the cerebral cortex tissues from the APP/PS1 and 3xTg-AD mice, relative to age-matched wild-type controls. This indicates that increased physical interaction between phosphorylated tau and Drp1 is associated with enhanced GTPase activity, which may be the possible mechanism behind enhanced mitochondrial fragmentation in AD neurons [70]. In addition, in a later study, to reduce the level of Drp1, Drp1+/− mice were crossed with P301L transgenic mice (JNPL3 line) to create double mutant (TauXDrp1+/−) mice. Reducing Drp1 potentially attenuated synaptic damage in 6-month-old P301L mice. TauXDrp1+/− mice also displayed reduced mitochondrial dysfunction and enhanced mitochondrial biogenesis as evidenced by increased RNA transcript levels of PGC1α, Nrf1, Nrf2, and TFAM [71]. Further, the levels of pS202/T205 were significantly reduced in TauXDrp1+/− mice compared to the P301L mice, indicating that partial reduction of Drp1 reduced the levels of tau phosphorylated at this epitope. These studies suggest that toxic pathogenic forms of tau may dysregulate Drp1 function resulting in abnormal mitochondrial dynamics [71]. An elegant review recently published by Oliver and Reddy details these findings more thoroughly [72].
In another recent study, neuronal mitochondrial morphology in postmortem brain samples from age and sex matched AD patients and healthy controls was examined [73]. The mitochondrial morphology was found to be abnormal in hippocampal neurons in the AD samples as evidenced by reduced size and excessive mitochondrial damage compared to the controls [73]. Overall, these data indicate that in AD brain mitochondrial morphology in neurons is abnormal, which may in part be due to phospho-tau mediated Drp1 dysregulation. Interestingly prior to recruitment of Drp1 to mitochondria, the endoplasmic reticulum (ER) wraps around the mitochondria [74]. These ER-mitochondria contacts have been suggested to facilitate Drp1 assembly on the mitochondria and fission, followed by Drp1 disassembly and dissociation from ER-mitochondria contacts for further rounds of fission [74]. Furthermore, an increase in the number of contacts between ER and mitochondria was observed in the motor neurons of JNPL3 (P301L tau) mice, as compared to wild-type mice [74]. Many human diseases including AD are associated with excessive mitochondrial fragmentation, raising the possibility that these diseases could involve an alteration of the ER-mitochondrial contacts possibly mediated by selective PTMs of tau. Interestingly findings have suggested that ER-mitochondria contacts were affected by the caspase 3-cleaved form truncated at the C-terminus (2N4R∆C20) of tau altering mitochondrial Ca2+ homeostasis. This defective ER-mitochondria communication might be an important pathological event in tau-related dysfunction and contributing to neurodegeneration [75]. Using a heterologous yeast system, it has also been reported that tau interacts with the Ca2+-regulated plasma membrane protein annexin A2 (AnxA2), as well as annexin A6 (AnxA6) via a domain encoded by exon 1 [23, 76]. These interactions of tau with the membrane components are affected by phosphorylation or familial AD tau mutations (R406W), which may contribute to the redistribution of tau from the axon to the somatodendritic compartment in AD [77, 78].
Another study placed phosphorylation of tau upstream of altered mitochondrial morphology/dynamics; however, in this case, expression of pathological modified human tau (hTau) lead to an increase in elongated mitochondria. In this study, a Drosophila model expressing a pseudo hyperphosphorylated form of tau (tauE14) exhibited longer mitochondria in neurons as compared to what was observed with the expression of R406W tau (a FTLD mutation) or wild-type tau [79]. The fact that tauE14 indirectly resulted in elongated mitochondria could be due to tau binding and stabilizing the actin cytoskeletal, disrupting physical association of mitochondria and Drp1, and thus preventing Drp1 protein from reaching the organelles in tauE14 transgenic flies. The mechanism could be sabotaging mitochondria, possibly leading to decreased fission and increased fusion. As mentioned, a delicate balance of fusion and fission maintain mitochondria at just the right length, and if the balance goes awry, there can be serious consequences for mitochondrial health. To determine whether abnormal mitochondrial morphology correlated with increased oxidative stress, ROS was measured in whole mount brains of the flies using fluorescent probe, dihydroethidium (DHE). In agreement with the previous data, brains of tauE14 animals showed a significant increase in superoxide production compared with tau controls, which could be rescued in vivo by genetically restoring the proper balance of mitochondrial fission and fusion [79]. Moreover, in neurons from tauE14 transgenic flies, Drp1 foci were infrequent and did not colocalize with mitochondria, whereas in neurons from R406W or wild-type tau expressing flies, Drp1 signals were discrete and exclusively localized to mitochondria. This event seems to be related to Drp1 mislocalization due to phosphorylated tau expression causing it to accumulate on F-actin and failing to translocate to mitochondria, leading to abnormal mitochondrial dynamics [79]. In the same study, the authors further demonstrated that MARF (the fly homolog of mammalian MFN) knockdown rescued tau-induced mitochondrial elongation and toxic effects in tauE14 transgenic flies, thus indicating a connection between pathogenic tau with abnormal mitochondrial morphology. Importantly, normalization of mitochondrial length in tauE14 transgenic flies by MARF knockdown was followed by significant rescue of neurotoxicity, as monitored with TUNEL staining to identify dying neurons [79]. In addition, in a prior study, suppression of Drp1 levels and abnormal mitochondrial distribution characterized by elongated mitochondria was observed in fibroblasts from AD patients [80], suggesting a similar chain of events may unfold in people with AD. However, it was unclear whether phospho-tau was a potential pathogenic factor as the baseline level of tau in fibroblasts is low.
Collectively, these studies support an effect of phospho-tau on mitochondrial morphology, though the precise mechanisms remain to be elucidated, and clearly indicate that perturbation of mitochondrial dynamics by pathogenic tau species can be detrimental. It is still under current investigation whether mitochondria are fragmented or elongated in human disease, and it is even possible that this may vary from case to case in AD [81]. Nonetheless, it is clear that a disruption of mitochondrial fission and fusion processes results in morphologically abnormal and dysfunctional mitochondria that accumulate in the soma with a subsequent shortage of healthy mitochondria in the distal processes, which is detrimental to neuronal function and survival [82, 83].
Mitophagy
Accumulation of damaged mitochondria due to inefficient turnover is characteristic of various diseases, including neurodegenerative diseases such as AD [6]. Mitophagy represents a form of selective macroautophagy that removes dysfunctional or damaged mitochondria, which otherwise could generate toxic ROS and/or trigger cell death [84]. During mitophagy, damaged mitochondria are recognized by an isolation membrane (phagophore) and subsequently sequestered into double membrane structures called mitophagosomes which fuse with lysosomes resulting in damaged mitochondrial degradation [9, 36]. This process limits the potentially detrimental effects of defective mitochondria and maintains the integrity of the mitochondrial pool. Mitophagy is initiated when the mitochondrial membrane potential dissipates because of functional impairment. This depolarization activates the PTEN-induced putative kinase 1/parkin RBR E3 ubiquitin protein ligase (PINK1/PARKIN) system, whereby the kinase PINK1 accumulates on the OMM, flagging the damaged mitochondrion for removal. PINK1 then phosphorylates ubiquitin on the OMM, which then binds to and activates the ubiquitin ligase Parkin, causing cytosolic Parkin to translocate to the surface of the targeted mitochondrion. The ubiquitin chains can interact with autophagy adaptors, recruiting LC3BII-positive autophagosomal membranes around the dysfunctional mitochondrion, which is then trafficked to the lysosome for degradation [85, 86]. The primary receptors for PINK1/Parkin-mediated autophagy are NDP52 and optineurin [87]. PINK1 directly activates mitophagy through recruitment of ubiquitin-binding proteins, NDP52 and optineurin. PINK1-generated phospho-ubiquitin acts as mitophagy signal, and ubiquitin binding by NDP52 is essential for mitophagy [88]. In addition to the classical PINK1/Parkin-related mitophagy, other ubiquitin-independent mitophagy receptors include AMBRA1, FUNDC1, and Nix/BNIP3L in mammals [89]. These mitophagy receptors bind via their LC3-interacting region (LIR) motifs to LC3 and GABARAP family proteins that are covalently bound to the phagophore membrane lipid. Engulfing the mitochondrion is completed by the formation of the protein bridges between the OMM and the phagophore membrane resulting in elongation (mediated by LC3 proteins) and closure mediated by GABARAP proteins. The final stage of mitophagy involves the fusion of the mitophagosomes with a lysosome, mediated by the phagophore LC3-binding proteins, and the lysosome membrane-associated protein Rab7. Lysosomal hydrolases then degrade the damaged mitochondrion [90].
Studies in living AD patients and postmortem brain tissue have provided evidence that in affected brain regions, there is impaired mitochondrial function and an accumulation of unrepaired damaged mitochondria [57, 89]. There are data indicating that pathological tau species may impair mitophagy; however, the precise mechanism of tau-induced mitophagy impairment remains to be delineated. It has been demonstrated that both human wild-type tau (hTau) and FTLD tau (P301L) inhibited mitophagy in neuroblastoma cells, by reducing mitochondrial translocation of Parkin [10]. Tau specifically impaired Parkin recruitment to defective mitochondria by sequestering it in the cytosol. This sequestration was mediated by aberrant interactions of Parkin with the projection domain of Tau [10]. Recently, in our lab, we have used Caenorhabditis elegans as a genetic model to express human tau in a defined set of mechanosensory neurons via single-copy gene integration as a means of interrogating the role of tau PTM on mitochondria [91]. Toward this end, CRISPR-Cas9 gene editing was then used to introduce AD-associated phosphorylation mimicking (T → E) or a nonphosphorylatable (T → A) mutation at the T231 position of the wild-type tau isoform. Surprisingly, we found that wild-type tau had no effect on baseline mitophagy, and that the AD-relevant tau mutant exerted only a slight effect on its own. However, the same mutant completely suppressed mitophagy that was induced in response to low doses of a mitochondrial toxin paraquat, supporting the idea that pathological modifications of tau may sensitize neurons to stress, and that the molecular mechanism may include disrupting stress-induced mitochondrial turnover [92].
Mitochondrial Axonal Transport
Mitochondrial fission, fusion, mitophagy, and transport all combine to maintain mitochondrial integrity, and overall cellular homeostasis. Mitochondria are actively trafficked throughout the cytosol, mainly along the microtubules to the sites of greatest energy demand [57]. Neurons are rich in mitochondria with a high demand for energy. Mitochondria are produced within the neuronal cell body, but must be transported to the synaptic endings to energize neurotransmission. Mitochondrial movement is not only critical for energy supply, but also control the clearance and replenishing of mitochondria in the distal tips of the neurons. Defects in mitochondrial trafficking can lead to the accumulation of dysfunctional mitochondria, which could contribute to AD pathology [93].
Mitochondrial transport is regulated by a series of adaptor proteins that mediate the attachment of mitochondria to molecular motors. For example, syntabulin, mitochondrial Rho small GTPases (Miro-1 and Miro-2), and Milton (TRAK1/2) bind to mitochondria and are associated with motor proteins of the kinesin-1 and kinesin-3 family to transport mitochondria toward the (+) end of microtubules [94, 95]. Milton protein is associated with mitochondria primarily in axons and synapses [94] and is required for kinesin-mediated transport of mitochondria to nerve terminals. In Drosophila, photoreceptors mutant for Milton show aberrant synaptic transmission despite normal photo transduction. The synaptic terminals and axons of the mutant fly lack mitochondria, although they are abundant in the neuronal cell bodies [94]. Similarly, in the Drosophila mutant for Miro (dMiro), orthologous to human Miro, abnormal mitochondrial distribution was observed in neurons [95]. In the dMiro mutant fly, mitochondria accumulate in parallel rows in neuronal soma rather than being transported in to axons and dendrites. Importantly, neuronal expression of dMiro was able to restore viability and transport of mitochondria to neuromuscular junctions (NMJs) [95]. Together, these findings suggest that mitochondrial transport is impaired in the absence of Milton or Miro, as synaptic terminals and axons lack mitochondria, leading to severe neurotransmission impairment and synaptic degeneration [96].
In the context of AD, there are data indicating that hyperphosphorylated forms of tau disrupt axonal transport, and cause synaptic damage [68, 97]. For example, it was shown that tau with sites that encompass the AT8 epitope (S199, S202, and T205) pseudo-phosphorylated inhibits mitochondrial transport to a greater degree than wild-type tau. It was speculated that this may be due to phosphorylation of tau at these sites causing extension of the projection domain away from the surface of the microtubule, increasing repulsive forces between microtubules which would result in a stronger resistance against mitochondria moving inside the microtubule bundles. These results suggest that phosphorylation of sites of the AT8 epitope may contribute to axonal degeneration by disrupting mitochondrial transport in AD [98]. Using transgenic Drosophila expressing human 0N4R tau, it was demonstrated that RNAi-mediated knockdown of Milton or Miro enhanced human tau–induced neurodegeneration. Further analysis indicated that phosphorylation of tau at S262 was increased with the knockdown of Milton or Miro. Partitioning defective-1 (PAR-1), the Drosophila homolog of mammalian microtubule affinity-regulating kinase (MARK), mediated this increase of tau phosphorylation [99]. Blocking tau phosphorylation at S262 site by PAR-1 or mutating the tau S262 site to nonphosphorylatable alanine resulted in decrease of neurodegeneration caused by Milton knockdown. This study suggests that the restriction of mitochondria to the cell body leading to depletion of axonal mitochondria trigger a change in the phosphorylation state of tau, causing increased neurodegeneration and toxicity [99]. In another study using C. elegans as a model system, perturbation of axonal transport of mitochondria was reported when pro-aggregant tau (∆K280 FTLD mutation) was expressed in neurons [100].
That tau can impair axonal transport of mitochondria in the absence of hyperphosphorylation has been shown in SH-SY5Y cell line stably overexpressing either human wild-type tau or P301L tau. Expression of P301L tau significantly decreased mitochondrial movement, causing a destabilization of the microtubule network, leading to perinuclear localization of the mitochondria [101]. In transgenic mice that robustly overexpress P301L tau (rTg4510 line) [102], aberrant mitochondrial distribution is observed in neurites along with perinuclear mitochondrial clumping as a function age [103]. A more recent study showed that cortical neurons from the knockin (KI) mice expressing physiological levels of murine tau with a mutation equivalent to human P301L displayed an age-dependent reduction in the number of axonal mitochondria (50% reduction in the number of mitochondria compared to wild-type neurons) as well as mitochondrial transport defects [104].
To date, a substantial body of evidence demonstrates an association between various forms of soluble pathogenic tau and impaired mitochondrial axonal transport, coupled with downregulation of OXPHOS complex activity that could contribute to the pathogenesis of neurodegenerative diseases including AD [12, 93, 98, 105]. Studies utilizing transgenic animals have made important contributions to the field, but overexpression of tau can potentially lead to synthetic toxic or gain-of-function phenotypes, and this caveat must always be kept in mind when extrapolating results to the human disease [12, 106, 107]. Therefore, much remains to be learned about the biology of soluble forms of tau with abnormal modification at disease relevant sites resulting in changes to OXPHOS cycle and mitochondrial trafficking. To facilitate achieving these latter ends, the use of single-copy, transgenic C. elegans strains expressing tau with site-specific PTMs will be helpful in identifying how they impact specific mitochondrial health parameters. In addition, there is need to devise novel approaches that will prevent or reduce the negative impact of tau PTM-related damage, which we describe in the following section.
Therapeutic Strategies
There is a growing body of evidence linking tau PTMs, especially hyperphosphorylation with mitochondrial dysfunction, and the mechanisms by which they influence disease progression and genetic networks involved are gradually being elucidated. An important caveat to keep in mind, however, is that for the most part monotherapeutic approaches have thus far been deemed unsuccessful for the treatment of AD. Nonetheless, it has been suggested that combinatorial treatment regimens using parallel strategies could be more effective, particularly if initiated at an early stage [108, 109]. Hence, it is worth considering individual approaches with such a goal in mind. Here, we focus on studies that target tau phosphorylation and mitochondria, and provide rationale for these approaches.
Reduction of Tau Hyperphosphorylation
Numerous kinases phosphorylate tau, including proline-directed serine/threonine protein kinases, such as glycogen synthase kinase-3β (GSK-3β), cyclin-dependent kinase 5 (CDK5) and mitogen-activated protein kinases (MAPKs), non-proline-directed serine/threonine protein kinases, such as MARK, and tyrosine protein kinases (for review, see [110]). The most widely studied as potential targets for reducing tau hyperphosphorylation are GSK-3β and CDK5 as both phosphorylate tau at multiple sites and their expression levels and activity in the brain are high [109, 110].
Over the past several years, there has been increasing interest in re-purposing lithium, a GSK-3β inhibitor, for diseases involving neurodegeneration like AD [109]. Lithium is commonly used for the treatment of bipolar disorder and seems to be well tolerated in humans [111]. In a mouse model expressing triple FTLD mutations (G272V, P301L, and R406W), chronic administration of lithium prevented tau hyperphosphorylation and NFT formation [112]. Recently, the McGill-R-Thy1-APP transgenic rat model of AD was treated with a novel micro dose lithium formulation (NP03) at early pre-plaque stages [113]. The therapeutic formulation of NP03 consists of sub clinical dose of lithium encapsulated in water-in-oil microemulsion [114, 115]. The compound was able to rescue functional deficiencies in object recognition, reduced loss of cholinergic boutons in the hippocampus, reduced soluble and insoluble cortical amyloid-β42 (Aβ42) levels, and reduced hippocampal Aβ plaque number [113]. A human clinical trial (NCT01055392) in amnestic mild cognitive impairment (MCI) patients suggested that lithium treatment was associated with a decrease in phospho-tau in the CSF and better cognitive performance [116]. However, no benefits were observed from short-term treatment (6 weeks) with lithium and results from other lithium trials have been mixed (NCT00088387, NCT02129348, and NCT00870311). Currently, there are two ongoing trials with lithium in AD patients with a much larger sample size and long-term treatment with lithium (2 years), NCT02129348 (phase II) and NCT03185208 (phase IV).
A problem with using protein kinase inhibitors to decrease tau phosphorylation is that they invariably have off-target effects. Therefore, identifying molecules that selectively inhibit tau phosphorylation at specific disease relevant epitopes may be an approach that has therapeutic potential. Recently, Reddy and his group used an in silico approach and tau pharmacophore modeling to identify selective, site-specific serine protein kinase–based tau inhibitors. [117]. Thus, given the evidence implicating phosphorylated tau in AD, developing selective inhibitors of phosphorylated tau represents a reasonable approach to the treatment of AD in addition to tau clearance, as described in a later section.
Activation of Tau Phosphatases
Another approach to reverse tau hyperphosphorylation is upregulation of tau phosphatases [118]. Protein phosphatase 2A (PP2A), the major tau phosphatase is responsible for over 70% of tau dephosphorylation [119]. It is also involved in dephosphorylation and inactivation of protein kinases involved in tau phosphorylation, which are over activated in the AD brain, whereas PP2A levels and activity are downregulated [119]. Thus, correcting PP2A levels and enhancing PP2A activity may be a feasible alternative to kinase inhibition for reducing tau phosphorylation in AD [120]. Multiple compounds have been identified that reverse PP2A inhibition, among which memantine is an FDA approved drug for the treatment of AD. It is a low-affinity voltage-dependent uncompetitive antagonist of glutamatergic N-methyl-d-aspartate receptors (NMDARs) and has been shown to restore PP2A activity in AD models [121]. Moreover, a year treatment with memantine resulted in a significant decrease in phosphorylated tau in the CSF of AD patients [122]. Currently, there are several clinical trials ongoing to examine the effects of memantine on brain function and the symptoms of AD (NCT03703856 (phase IV), NCT04117178 (phase IV), NCT02288000 (phase I), and NCT03959124). Sodium selenate is a specific agonist for PP2A able to stabilize PP2A-tau complexes, causing reduced phosphorylation of tau and completely abrogating NFT formation both in vitro and in vivo. Oral administration of sodium selenite in two independent tau transgenic AD mouse models with NFT pathology, P301L mutant pR5 and K369I mutant K3, mitigated tau pathology by reducing tau hyperphosphorylation and rescued behavioral impairment in terms of memory and motor functions, as well as prevented neuronal loss [123, 124]. Taken together, although sodium selenite mitigated tau pathology in several pre-clinical AD models, its efficacy needs to be tested further in clinical trials for tau-targeted treatment of AD and related dementias.
Tau Clearance Approaches
Initially, pharmacological manipulation using tau-directed therapeutics such as tau phosphatase activators or tau kinase inhibitors although seemed promising; however, none of these approaches has translated into successful clinical applications with disease-modifying outcomes (for review, see [125]). They have potential limitations and pose critical challenges due to low affinity and specificity as well as strong immune responses.
More recently, scientists from Arvinas have developed an alternative approach to reduce or eliminate the levels of pathological tau species. This is a new therapeutic strategy for tauopathies where highly potent brain penetrant PROTAC (PROteolysis TArgeting Chimeras) “degrader” molecules are being developed that target pathological tau protein species [126]. In a pre-clinical tauopathy mouse model, a tau-targeted PROTAC protein degrader effectively crossed the blood brain barrier (BBB) and decreased more than 95% of pathologic tau species [127]. The fact that these molecules specifically facilitate the removal of pathologic tau has created significant interest in developing this approach for the treatment of AD and other tauopathies [127]. PROTAC protein degraders offers numerous potential therapeutic advantages as compounds for the treatment of AD, including broad tissue distribution, routes of administration such as oral delivery, and simpler manufacturing than other new modalities, like cell-based therapies [128].
Another potential therapeutic avenue for treating AD is tau immunotherapy which has shown great promise in available animal models. In a recent study, 3xTg-AD mice at mild to moderate stages of the disease (12 months old) were treated with two doses of mouse monoclonal tau antibodies 43D (against tau amino acids 6–18) and 77E9 (against tau amino acids 184–195) [129]. Although both the antibodies in this paradigm were able to reduce levels of total tau, they had no significant impact on the levels of hyperphosphorylated tau. However, six doses of 43D lowered levels of total tau and tau hyperphosphorylated at S262/356 and S396/404 epitopes in the hippocampus. The tau lowering capacities of antibodies were followed by rescue of tau-induced spatial and short-term memory impairments in 3xTg-AD mice. In addition, the beneficial effect of the antibodies on cognitive performance was sustained up to 3 months after discontinuation if immunization [129]. A phase II study of the first tau immunotherapy target active immunization against tau (amino acids 294–305) in AD patients has been completed (NCT02579252). Among active vaccines in clinical trial currently at phase Ib/2a trial, ACI-35 (AC immune AG) is promising, targeting tau amino acids 393–408. It is a liposomal-based 16 amino acid phosphotau peptide including phosphorylated S396 and S404 epitopes. The vaccine elicited a rapid immune response against the immunogen in wild-type and P301L mice, resulting in a mild reduction of hyperphosphorylated pathological tau and improved motor functions of vaccinated mice [130].
Improving Mitochondrial Health in AD
Due to the complex pathophysiology of AD, clinical efficacy will most likely be achieved using combinatorial approaches, e.g., by reducing pathological soluble tau oligomeric species concurrent with improving mitochondrial health. Given the evidence that pathological tau-induced loss of mitochondrial health plays an important role in AD pathogenesis [6], therapeutic efforts aimed at the removal of free radicals, prevention of mitochondrial fragmentation, increase ATP production, and promote mitochondrial transport in combination with reducing pathological tau species seem promising [98]. To that end, there has been a considerable effort to correlate mitochondrial health with tau pathologies in a variety of model organisms ranging from invertebrates to vertebrates in order to gain mechanistic insights [10, 12, 33]. These studies also highlight the importance of using model organisms in designing and delineating putative therapeutic targets that could otherwise been extremely challenging to determine. Here, we summarize therapeutic strategies specifically related to mitochondria in AD.
A beneficial effect is observed when mitochondrial fission is inhibited by using a selective peptide inhibitor of Drp1, such as P110 [131]. SH-SY5Y cells were treated with mitochondrial complex I inhibitor MPP+ and the mitochondrial uncoupler CCCP, which resulted in excessive mitochondrial fragmentation. P110 reduced the mitochondrial fragmentation to a level similar to Drp1 siRNA treatment under analogous conditions. More importantly, P110 did not affect mitochondrial network integrity under basal conditions [131]. In addition, pretreatment of pryramidal neurons from CRND8 APP transgenic mice with mitochondrial division inhibitor-1 (mdivi-1), an inhibitor of Drp1, rescued mitochondrial fragmentation and distribution deficits as well as improved mitochondrial function [132]. More importantly, amelioration of mitochondrial dynamics deficits by mdivi-1 treatment markedly decreased extracellular amyloid deposition and improved cognitive deficits and synaptic parameters in CRND8 mice [132]. Growing evidence suggests that the inhibitor enhances mitochondrial fusion, inhibits fission by blocking Drp-1 self-assembly, and increases mitochondrial biogenesis and levels of synaptic protein [133, 134]. Both in vivo and in vitro studies reveal development of molecules that reduce mitochondrial fission while maintaining normal mitochondrial fusion [135]. Treatment with P110 and mdivi-1 has been widely successful in various pre-clinical models of neurodegenerative diseases such as Parkinson’s disease, Huntington’s disease, and AD and is currently being considered for clinical trials [72].
The role of the natural dietary, microflora-derived metabolite Urolithin A (UA) has recently been investigated for improving mitochondrial health in humans [136]. In a study with animal models, it has been shown that restoration of neuronal mitophagy by UA abolished AD-related tau hyperphosphorylation in human neuronal cells and reverses memory impairment in transgenic tau mice [73]. UA also inhibited phosphorylation of several sites on tau such as T181, S202/T205, T231, and S262 in human SH-SY5Y neuroblastoma cells overexpressing 2N4R, 1N4R, or 2N3R tau [73]. To test the conserved beneficial role of mitophagy induction in the presence of phospho-tau, 3xTgAD mice were treated with UA. The treatment improved performance in both object recognition and Y maze tests in 3xTg-AD mice to that of wild-type vehicle-treated mice. Moreover, UA treatment showed strong inhibition of the same phosphorylation sites on tau, similar to in the results in the tau overexpressing human SH-SY5Y cells [73]. Currently, a clinical trial is ongoing with UA (NCT04160312), if results are beneficial, it can used as a potential therapeutic option to ramp up mitophagy in AD patients.
Approaches to improve mitochondrial transport defects in AD have also been explored as an AD therapeutic. Treatment with tubacin, a specific inhibitor of HDAC6, significantly enhanced mitochondrial movement in cultured hippocampal neurons. This evidence suggests that decreasing HDAC6 is preventive against mitochondrial transport defects [137, 138]. Treatment with the mitochondrial-targeted antioxidant peptide, 6′-di-methyltyrosine-Lys-Phe-NH2 (SS-31) prevented mitochondrial transport decreases and reversed excess occurrence of mitochondrial fission in primary neurons from Tg2576 mice [139]. SS-31 binds selectively to cardiolipin, a phospholipid that is localized specifically to the IMM, and promotes the efficiency of mitochondrial OXPHOS machinery. Due to multi-efficacies of SS-31 (improves mitochondrial trafficking and decreases fission and oxidative damage) and the fact that it appears to be safe in adults, it is currently being considered for human clinical trials for mitochondrial myopathy [140,141,142].
Mitochondrial biogenesis in brain is regulated in part by the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), a basic leucine zipper (bZIP) protein that also regulates expression of other mitochondrial proteins involved in ETC and antioxidant enzymes that prevent excessive ROS production [143]. AD brain samples compared with age-matched controls showed significant reduction in the expression of Nrf2 [144]. Furthermore, the Nrf2 pathway is inhibited in APP/PS1 mice [137]. The Nrf2 pathway is activated by various natural compounds such as curcumin and pyrrolidine dithiocarbamate, which result in the upregulation of antioxidant response element (ARE) responsive cytoprotective genes, and has been shown to result in an amelioration of cognitive defects in an AD transgenic mouse model [144,145,146]. In neurons, activation of Nrf2 pathway reduces the levels of phosphorylated tau by induction of an autophagy adapter protein NDP52 in neurons. The expression of NDP52 is strongly induced by Nrf2 and its overexpression facilitates clearance of phosphorylated tau in the presence of an autophagy stimulator [147]. Deletion of Nrf2 in APP/PS1 mice significantly impaired spatial learning and memory. Increased Aβ and tau phosphorylated at S404 was also observed in the hippocampus of these APP/PS1 mice with Nrf2 deletion, along with increased astroglial and microglial activation and upregulation of the proinflammatory cytokines IL-1β, IL-6, and TNF-α [148]. SKN-1 is the C. elegans ortholog of Nrf2 and is a key determinant in promoting healthy aging and increased lifespan [149]. It will be interesting to observe whether SKN-1/Nrf2 activation in our single-copy tau PTM mimetic strains ameliorates neurodegeneration and neuronal behavior [92]. Thus, given the evidence for Nrf2 in neuroprotective effects, it represents a target for drug development for the treatment of AD.
It is worth mentioning that antioxidant therapies were once considered as popular therapeutic routes to bolster mitochondrial health. Research indicated that treatment of cortical neurons with antioxidants like N-acetylcysteine (NAC), vitamins E and C, improved axonal mitochondrial transport in neurons and are now emerging as treatment options for vascular and nonvascular neurological disorders [150]. However, additional investigations into their molecular targets are needed using model organisms, especially if they are considered for clinical trials, to ensure safety and efficacy of the application in humans. Dysfunctional mitochondria generate excessive amount of ROS, which damages cellular homeostasis [57, 89]. Novel approaches which include antioxidants such as MitoQ (mitoquinone mesylate: 10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4 cyclohexadienyl) decyltriphenyl-phosphonium methane sulfonate) which is specifically targeted to mitochondria and concentrates there have the capability of reducing ROS generated by dysfunctional mitochondria [151]. Oral administration of MitoQ produced encouraging results diminishing oxidative stress in multiple animal models as well as in healthy older adults [152, 153]. Currently, patients who are suffering from mild cognitive impairment (MCI) are being recruited for a clinical study (NCT03514875). However, using different genetic model organisms, it has been demonstrated that ROS at normal physiological levels plays a beneficial role in supporting neuron health and function [154], promoting longevity, and slowing aging [155]. There is also data to suggest that ROS regulates neuronal plasticity and maintains synaptic transmission at neuromuscular junctions [156, 157]. Thus, although ROS has been extensively classified as damaging agents associated with aging and neurodegenerative diseases, abolishing ROS from the system or disturbing ROS balance could have detrimental side effects with respect to therapeutic targeting.
Mitochondrial dysfunction likely contributes to the neurodegeneration in AD by affecting energy supply and reducing antioxidant defenses and thus mitochondrial injury could participate in the establishment of tauopathy [8, 73]. Thus, the evolution of AD pathology can be considered as multi-hit process, as toxic tau oligomers alone are not fully responsible for AD. Decreasing soluble pathological tau species and improving mitochondrial health concurrently are like to provide beneficial outcomes in AD patients. In this scenario, we can use model organisms where pathological tau species are not over expressed and which are amenable to high throughput drug screens, notably worms and flies to determine efficacy of the drug combinations. They can be treated with novel drug combinations to improve mitochondrial health and attenuate toxic, soluble tau species followed by appropriate analyses. Translating these treatment strategies into humans will likely require the identification of individuals at high risk for developing AD as they are likely to be most successful in initiated prior to advance tau pathology. As we understand better the mechanisms by which soluble tau species with specific AD-relevant PTMs impair mitochondrial health, this strategy can be fine-tuned and thus its effectiveness increased as studies are translated into AD vertebrate models. One of the goals in our lab is to develop a pipeline where single-copy gene insertions in invertebrate model systems—circumventing the confounds of tau overexpression—are used to gain mechanistic insights into the toxicity of pathological tau species, primarily through genetic approaches, as well as for initial screens of combination drug studies (targeting both tau and mitochondria). Results from these simpler models will provide rigorous premise for moving into pre-clinical trials in vertebrate AD models. We anticipate that this pipeline will expedite the process of developing and/or validating effective treatments for AD.
Concluding Remarks
Numerous different model systems have provided compelling data indicating that soluble tau species with disease relevant PTMs likely contribute to mitochondrial dependent disease pathologies. These models, especially those in which pathological tau is not overexpressed, are helping us to gain a mechanistic understanding of how soluble oligomeric tau species can cause dysfunctional mitochondria. In particular, the use of animal models with shorter lifespans (e.g., worms and flies) has allowed the monitoring of the progression of pathology as function of age and its link with mitochondrial abnormalities. The studies presented in this review highlight how various forms of soluble pathogenic tau affect three critical aspects of mitochondrial function: (1) mitochondrial dynamics (inducing fragmentation), (2) bioenergetics (ATP and ROS production), and (3) mitochondrial movement (synaptic function) (Fig. 4). This knowledge of cellular mechanistic insights may allow for the development of combinatorial therapeutic strategies that target tau modifications to improve outcomes in AD and simultaneously energize mitochondria through the improvement of mitochondrial dynamics, bioenergetics, and transport.

Soluble tau species with specific AD-relevant tau PTMs impair mitochondrial health by causing fragmentation, reducing mitophagy and trafficking of dysfunctional mitochondria, depleting ATP production, and increasing mitochondrial ROS production. All of these collective failures disturb cellular homeostasis and lead to neuronal cell death aiding to the progression of AD
References
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766. https://doi.org/10.1152/physrev.2001.81.2.741
Avila J, Lucas JJ, Perez M, Hernandez F (2004) Role of tau protein in both physiological and pathological conditions. Physiol Rev 84(2):361–384. https://doi.org/10.1152/physrev.00024.2003
Neddens J, Temmel M, Flunkert S, Kerschbaumer B, Hoeller C, Loeffler T, Niederkofler V, Daum G et al (2018) Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun 6(1):52. https://doi.org/10.1186/s40478-018-0557-6
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VMY, Trojanowski JQ (2012) Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135(3):807–818. https://doi.org/10.1093/brain/aws013
Mroczko B, Groblewska M, Litman-Zawadzka A (2019) The role of protein misfolding and tau oligomers (TauOs) in Alzheimer’s disease (AD). Int J Mol Sci 20(19). https://doi.org/10.3390/ijms20194661
David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Drose S et al (2005) Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem 280(25):23802–23814. https://doi.org/10.1074/jbc.M500356200
Oliver DMA, Reddy PH (2019) Molecular basis of Alzheimer’s disease: focus on mitochondria. Journal of Alzheimer’s Disease. https://doi.org/10.3233/JAD-190048
Swerdlow RH (2018) Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis 62(3):1403–1416. https://doi.org/10.3233/JAD-170585
Hu H, Tan CC, Tan L, Yu JT (2017) A mitocentric view of Alzheimer’s disease. Mol Neurobiol 54(8):6046–6060. https://doi.org/10.1007/s12035-016-0117-7
Cummins N, Tweedie A, Zuryn S, Bertran-Gonzalez J, Götz J (2019) Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J 38(3):e99360. https://doi.org/10.15252/embj.201899360
Perez MJ, Jara C, Quintanilla RA (2018) Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front Neurosci 12(441). https://doi.org/10.3389/fnins.2018.00441
Butler VJ, Salazar DA, Soriano-Castell D, Alves-Ferreira M, Dennissen FJA, Vohra M, Oses-Prieto JA, Li KH et al (2019) Tau/MAPT disease-associated variant A152T alters tau function and toxicity via impaired retrograde axonal transport. Hum Mol Genet 28(9):1498–1514. https://doi.org/10.1093/hmg/ddy442
Chong FP, Ng KY, Koh RY, Chye SM (2018) Tau proteins and tauopathies in Alzheimer’s disease. Cell Mol Neurobiol 38(5):965–980. https://doi.org/10.1007/s10571-017-0574-1
Liu F, Gong CX (2008) Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener 3:8. https://doi.org/10.1186/1750-1326-3-8
Wu X-L, Piña-Crespo J, Zhang Y-W, Chen X-C, Xu H-X (2017) Tau-mediated neurodegeneration and potential implications in diagnosis and treatment of Alzheimer’s disease. Chin Med J 130(24):2978–2990. https://doi.org/10.4103/0366-6999.220313
LaFerla FM, Oddo S (2005) Alzheimer’s disease: Abeta, tau and synaptic dysfunction. Trends Mol Med 11(4):170–176. https://doi.org/10.1016/j.molmed.2005.02.009
Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol 133(5):665–704. https://doi.org/10.1007/s00401-017-1707-9
Feinstein SC, Wilson L (2005) Inability of tau to properly regulate neuronal microtubule dynamics: a loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim Biophys Acta 1739(2–3):268–279. https://doi.org/10.1016/j.bbadis.2004.07.002
Mandelkow EM, Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2(7):a006247. https://doi.org/10.1101/cshperspect.a006247
Janning D, Igaev M, Sündermann F, Brühmann J, Beutel O, Heinisch JJ, Bakota L, Piehler J et al (2014) Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol Biol Cell 25(22):3541–3551. https://doi.org/10.1091/mbc.E14-06-1099
Morris M, Maeda S, Vossel K, Mucke L (2011) The many faces of tau. Neuron 70(3):410–426. https://doi.org/10.1016/j.neuron.2011.04.009
Brandt R, Leschik J (2004) Functional interactions of tau and their relevance for Alzheimer’s disease. Curr Alzheimer Res 1(4):255–269. https://doi.org/10.2174/1567205043332054
Trushina NI, Bakota L, Mulkidjanian AY, Brandt R (2019) The evolution of tau phosphorylation and interactions. Front Aging Neurosci 11(256). https://doi.org/10.3389/fnagi.2019.00256
Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, Yu W, Baas PW (2018) Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr Biol 28(13):2181–2189 e2184. https://doi.org/10.1016/j.cub.2018.05.045
Baas PW, Qiang L (2019) Tau: it’s not what you think. Trends Cell Biol 29(6):452–461. https://doi.org/10.1016/j.tcb.2019.02.007
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17(1):5–21. https://doi.org/10.1038/nrn.2015.1
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156(6):1051–1063. https://doi.org/10.1083/jcb.200108057
Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 103(1):26–35. https://doi.org/10.1007/s004010100423
Fox LM, William CM, Adamowicz DH, Pitstick R, Carlson GA, Spires-Jones TL, Hyman BT (2011) Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. J Neuropathol Exp Neurol 70(7):588–595. https://doi.org/10.1097/NEN.0b013e318220a658
Ghag G, Bhatt N, Cantu DV, Guerrero-Munoz MJ, Ellsworth A, Sengupta U, Kayed R (2018) Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci 27(11):1901–1909. https://doi.org/10.1002/pro.3499
Liu P, Smith BR, Montonye ML, Kemper LJ, Leinonen-Wright K, Nelson KM, Higgins L, Guerrero CR et al (2020) A soluble truncated tau species related to cognitive dysfunction is elevated in the brain of cognitively impaired human individuals. Sci Rep 10(1):3869. https://doi.org/10.1038/s41598-020-60777-x
Heinisch JJ, Brandt R (2016) Signaling pathways and posttranslational modifications of tau in Alzheimer’s disease: the humanization of yeast cells. Microb Cell 3(4):135–146. https://doi.org/10.15698/mic2016.04.489
Min S-W, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS et al (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21(10):1154–1162. https://doi.org/10.1038/nm.3951
Tracy TE, Gan L (2017) Acetylated tau in Alzheimer’s disease: an instigator of synaptic dysfunction underlying memory loss: increased levels of acetylated tau blocks the postsynaptic signaling required for plasticity and promotes memory deficits associated with tauopathy. Bioessays 39(4). https://doi.org/10.1002/bies.201600224
Wang JZ, Grundke-Iqbal I, Iqbal K (1996) Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer’s disease. Nat Med 2(8):871–875. https://doi.org/10.1038/nm0896-871
Cao J, Zhong MB, Toro CA, Zhang L, Cai D (2019) Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis. Neurosci Lett 703:68–78. https://doi.org/10.1016/j.neulet.2019.03.016
Luo HB, Xia YY, Shu XJ, Liu ZC, Feng Y, Liu XH, Yu G, Yin G et al (2014) SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci U S A 111(46):16586–16591. https://doi.org/10.1073/pnas.1417548111
Kontaxi C, Piccardo P, Gill AC (2017) Lysine-directed post-translational modifications of tau protein in Alzheimer’s disease and related tauopathies. Front Mol Biosci 4(56). https://doi.org/10.3389/fmolb.2017.00056
Park SY, Tournell C, Sinjoanu RC, Ferreira A (2007) Caspase-3- and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons. Neuroscience 144(1):119–127. https://doi.org/10.1016/j.neuroscience.2006.09.012
Ferreira A, Bigio EH (2011) Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med 17(7–8):676–685. https://doi.org/10.2119/molmed.2010.00220
Šimić G, Babić Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milošević N, Bažadona D, Buée L et al (2016) Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6(1):6. https://doi.org/10.3390/biom6010006
Mi K, Johnson GV (2006) The role of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 3(5):449–463. https://doi.org/10.2174/156720506779025279
Ochalek A, Mihalik B, Avci HX, Chandrasekaran A, Teglasi A, Bock I, Giudice ML, Tancos Z et al (2017) Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res Ther 9(1):90. https://doi.org/10.1186/s13195-017-0317-z
Quintanilla RA, von Bernhardi R, Godoy JA, Inestrosa NC, Johnson GV (2014) Phosphorylated tau potentiates Abeta-induced mitochondrial damage in mature neurons. Neurobiol Dis 71:260–269. https://doi.org/10.1016/j.nbd.2014.08.016
Chen Q, Zhou Z, Zhang L, Xu S, Chen C, Yu Z (2014) The cellular distribution and Ser262 phosphorylation of tau protein are regulated by BDNF in vitro. PLoS One 9(3):e91793. https://doi.org/10.1371/journal.pone.0091793
Zempel H, Dennissen FJA, Kumar Y, Luedtke J, Biernat J, Mandelkow EM, Mandelkow E (2017) Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem 292(29):12192–12207. https://doi.org/10.1074/jbc.M117.784702
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA et al (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68(6):1067–1081. https://doi.org/10.1016/j.neuron.2010.11.030
Niewidok B, Igaev M, Sündermann F, Janning D, Bakota L, Brandt R (2016) Presence of a carboxy-terminal pseudorepeat and disease-like pseudohyperphosphorylation critically influence tau's interaction with microtubules in axon-like processes. Mol Biol Cell 27(22):3537–3549. https://doi.org/10.1091/mbc.E16-06-0402
Ittner A, Chua SW, Bertz J, Volkerling A, van der Hoven J, Gladbach A, Przybyla M, Bi M et al (2016) Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 354(6314):904–908. https://doi.org/10.1126/science.aah6205
Strang KH, Sorrentino ZA, Riffe CJ, Gorion KM, Vijayaraghavan N, Golde TE, Giasson BI (2019) Phosphorylation of serine 305 in tau inhibits aggregation. Neurosci Lett 692:187–192. https://doi.org/10.1016/j.neulet.2018.11.011
Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, Seereeram A, Reynolds CH, Ward MA et al (2007) Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem 282(32):23645–23654. https://doi.org/10.1074/jbc.M703269200
Espinoza M, de Silva R, Dickson DW, Davies P (2008) Differential incorporation of tau isoforms in Alzheimer’s disease. J Alzheimers Dis 14(1):1–16. https://doi.org/10.3233/jad-2008-14101
Reddy PH, Oliver DM (2019) Amyloid beta and phosphorylated Tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 8(5). https://doi.org/10.3390/cells8050488
Albensi BC (2019) Dysfunction of mitochondria: implications for Alzheimer’s disease. Int Rev Neurobiol 145:13–27. https://doi.org/10.1016/bs.irn.2019.03.001
Roger AJ, Munoz-Gomez SA, Kamikawa R (2017) The origin and diversification of mitochondria. Curr Biol 27(21):R1177–R1192. https://doi.org/10.1016/j.cub.2017.09.015
Lin Y-F, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM (2016) Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533(7603):416–419. https://doi.org/10.1038/nature17989
Flannery PJ, Trushina E (2019) Mitochondrial dynamics and transport in Alzheimer’s disease. Mol Cell Neurosci 98:109–120. https://doi.org/10.1016/j.mcn.2019.06.009
Bakota L, Ussif A, Jeserich G, Brandt R (2017) Systemic and network functions of the microtubule-associated protein tau: implications for tau-based therapies. Mol Cell Neurosci 84:132–141. https://doi.org/10.1016/j.mcn.2017.03.003
Onukwufor JO, Berry BJ, Wojtovich AP (2019) Physiologic implications of reactive oxygen species production by mitochondrial complex I reverse electron transport. Antioxidants (Basel) 8(8). https://doi.org/10.3390/antiox8080285
Chan SHH, Chan JYH (2017) Mitochondria and reactive oxygen species contribute to neurogenic hypertension. Physiology (Bethesda) 32(4):308–321. https://doi.org/10.1152/physiol.00006.2017
Dan Dunn J, Alvarez LA, Zhang X, Soldati T (2015) Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol 6:472–485. https://doi.org/10.1016/j.redox.2015.09.005
Lambert AJ, Brand MD (2009) Reactive oxygen species production by mitochondria. Methods Mol Biol 554:165–181. https://doi.org/10.1007/978-1-59745-521-3_11
Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60(5):748–766. https://doi.org/10.1016/j.neuron.2008.10.010
Li XC, Hu Y, Wang ZH, Luo Y, Zhang Y, Liu XP, Feng Q, Wang Q et al (2016) Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep 6:24756. https://doi.org/10.1038/srep24756
Esteras N, Rohrer JD, Hardy J, Wray S, Abramov AY (2017) Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Biol 12:410–422. https://doi.org/10.1016/j.redox.2017.03.008
Scott I, Youle RJ (2010) Mitochondrial fission and fusion. Essays Biochem 47:85–98. https://doi.org/10.1042/bse0470085
van der Bliek AM, Shen Q, Kawajiri S (2013) Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol 5(6). https://doi.org/10.1101/cshperspect.a011072
Beharry C, Cohen LS, Di J, Ibrahim K, Briffa-Mirabella S, Alonso Adel C (2014) Tau-induced neurodegeneration: mechanisms and targets. Neurosci Bull 30(2):346–358. https://doi.org/10.1007/s12264-013-1414-z
Dikov D, Reichert AS (2011) How to split up: lessons from mitochondria. EMBO J 30(14):2751–2753. https://doi.org/10.1038/emboj.2011.219
Manczak M, Reddy PH (2012) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21(11):2538–2547. https://doi.org/10.1093/hmg/dds072
Kandimalla R, Manczak M, Fry D, Suneetha Y, Sesaki H, Reddy PH (2016) Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet 25(22):4881–4897. https://doi.org/10.1093/hmg/ddw312
Oliver D, Reddy PH (2019) Dynamics of dynamin-related protein 1 in Alzheimer’s disease and other neurodegenerative diseases. Cells 8(9). https://doi.org/10.3390/cells8090961
Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM et al (2019) Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22(3):401–412. https://doi.org/10.1038/s41593-018-0332-9
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334(6054):358–362. https://doi.org/10.1126/science.1207385
Cieri D, Vicario M, Vallese F, D'Orsi B, Berto P, Grinzato A, Catoni C, De Stefani D et al (2018) Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca(2+) handling. Biochim Biophys Acta Mol basis Dis 1864(10):3247–3256. https://doi.org/10.1016/j.bbadis.2018.07.011
Gauthier-Kemper A, Suárez Alonso M, Sündermann F, Niewidok B, Fernandez MP, Bakota L, Heinisch JJ, Brandt R (2018) Annexins A2 and A6 interact with the extreme N terminus of tau and thereby contribute to tau's axonal localization. J Biol Chem 293(21):8065–8076. https://doi.org/10.1074/jbc.RA117.000490
Gauthier-Kemper A, Weissmann C, Golovyashkina N, Sebö-Lemke Z, Drewes G, Gerke V, Heinisch JJ, Brandt R (2011) The frontotemporal dementia mutation R406W blocks tau's interaction with the membrane in an annexin A2-dependent manner. J Cell Biol 192(4):647–661. https://doi.org/10.1083/jcb.201007161
Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol 12(6):609–622. https://doi.org/10.1016/s1474-4422(13)70090-5
DuBoff B, Gotz J, Feany MB (2012) Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75(4):618–632. https://doi.org/10.1016/j.neuron.2012.06.026
Wang X, Su B, Fujioka H, Zhu X (2008) Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol 173(2):470–482. https://doi.org/10.2353/ajpath.2008.071208
DuBoff B, Feany M, Gotz J (2013) Why size matters - balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci 36(6):325–335. https://doi.org/10.1016/j.tins.2013.03.002
Flippo KH, Strack S (2017) Mitochondrial dynamics in neuronal injury, development and plasticity. J Cell Sci 130(4):671–681. https://doi.org/10.1242/jcs.171017
Byrne JJ, Soh MS, Chandhok G, Vijayaraghavan T, Teoh JS, Crawford S, Cobham AE, Yapa NMB et al (2019) Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell Mol Life Sci 76(10):1967–1985. https://doi.org/10.1007/s00018-019-03024-5
Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20(9):1013–1022. https://doi.org/10.1038/s41556-018-0176-2
Wei H, Liu L, Chen Q (2015) Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta 1853(10 Pt B):2784–2790. https://doi.org/10.1016/j.bbamcr.2015.03.013
Audano M, Schneider A, Mitro N (2018) Mitochondria, lysosomes, and dysfunction: their meaning in neurodegeneration. J Neurochem 147(3):291–309. https://doi.org/10.1111/jnc.14471
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524(7565):309–314. https://doi.org/10.1038/nature14893
Yamaguchi O, Murakawa T, Nishida K, Otsu K (2016) Receptor-mediated mitophagy. J Mol Cell Cardiol 95:50–56. https://doi.org/10.1016/j.yjmcc.2016.03.010
Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF (2017) Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci 40(3):151–166. https://doi.org/10.1016/j.tins.2017.01.002
Liu L, Sakakibara K, Chen Q, Okamoto K (2014) Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res 24(7):787–795. https://doi.org/10.1038/cr.2014.75
Suzuki H, Kerr R, Bianchi L, Frokjaer-Jensen C, Slone D, Xue J, Gerstbrein B, Driscoll M et al (2003) In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron 39(6):1005–1017. https://doi.org/10.1016/j.neuron.2003.08.015
Guha S, Fischer S, Johnson GV, Nehrke K (2020) Alzheimer’s disease-relevant tau modifications selectively impact neurodegeneration and mitophagy in a novel <em>C. elegans</em> single-copy transgenic model. bioRxiv:2020.2002.2012.946004. https://doi.org/10.1101/2020.02.12.946004
Schwarz TL (2013) Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5(6). https://doi.org/10.1101/cshperspect.a011304
Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL (2002) Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36(6):1063–1077. https://doi.org/10.1016/s0896-6273(02)01094-2
Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP et al (2005) The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47(3):379–393. https://doi.org/10.1016/j.neuron.2005.06.027
Fransson S, Ruusala A, Aspenstrom P (2006) The atypical rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun 344(2):500–510. https://doi.org/10.1016/j.bbrc.2006.03.163
Gendron TF, Petrucelli L (2009) The role of tau in neurodegeneration. Mol Neurodegener 4:13. https://doi.org/10.1186/1750-1326-4-13
Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M et al (2012) Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J Neurosci 32(7):2430–2441. https://doi.org/10.1523/JNEUROSCI.5927-11.2012
Iijima-Ando K, Sekiya M, Maruko-Otake A, Ohtake Y, Suzuki E, Lu B, Iijima KM (2012) Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer’s disease-related tau phosphorylation via PAR-1. PLoS Genet 8(8):e1002918. https://doi.org/10.1371/journal.pgen.1002918
Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow EM, Schmidt E, Baumeister R (2012) Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum Mol Genet 21(16):3587–3603. https://doi.org/10.1093/hmg/dds190
Schulz KL, Eckert A, Rhein V, Mai S, Haase W, Reichert AS, Jendrach M, Muller WE et al (2012) A new link to mitochondrial impairment in tauopathies. Mol Neurobiol 46(1):205–216. https://doi.org/10.1007/s12035-012-8308-3
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M et al (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309(5733):476–481. https://doi.org/10.1126/science.1113694
Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP et al (2011) Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol 179(4):2071–2082. https://doi.org/10.1016/j.ajpath.2011.07.004
Rodriguez-Martin T, Pooler AM, Lau DHW, Morotz GM, De Vos KJ, Gilley J, Coleman MP, Hanger DP (2016) Reduced number of axonal mitochondria and tau hypophosphorylation in mouse P301L tau knockin neurons. Neurobiol Dis 85:1–10. https://doi.org/10.1016/j.nbd.2015.10.007
Eckert A, Nisbet R, Grimm A, Gotz J (2014) March separate, strike together--role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 1842(8):1258–1266. https://doi.org/10.1016/j.bbadis.2013.08.013
Kosmidis S, Grammenoudi S, Papanikolopoulou K, Skoulakis EMC (2010) Differential effects of tau on the integrity and function of neurons essential for learning in Drosophila. J Neurosci 30(2):464–477. https://doi.org/10.1523/jneurosci.1490-09.2010
Maeda S, Djukic B, Taneja P, Yu G-Q, Lo I, Davis A, Craft R, Guo W et al (2016) Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep 17(4):530–551. https://doi.org/10.15252/embr.201541438
Cabezas-Opazo FA, Vergara-Pulgar K, Perez MJ, Jara C, Osorio-Fuentealba C, Quintanilla RA (2015) Mitochondrial dysfunction contributes to the pathogenesis of Alzheimer’s disease. Oxidative Med Cell Longev 2015(509654). https://doi.org/10.1155/2015/509654
Coughlin D, Irwin DJ (2017) Emerging diagnostic and therapeutic strategies for Tauopathies. Curr Neurol Neurosci Rep 17(9):72. https://doi.org/10.1007/s11910-017-0779-1
Yoshiyama Y, Lee VM, Trojanowski JQ (2013) Therapeutic strategies for tau mediated neurodegeneration. J Neurol Neurosurg Psychiatry 84(7):784–795. https://doi.org/10.1136/jnnp-2012-303144
Paul P, Iyer S, Nadella RK, Nayak R, Chellappa AS, Ambardar S, Sud R, Sukumaran SK et al (2020) Lithium response in bipolar disorder correlates with improved cell viability of patient derived cell lines. Sci Rep 10(1):7428. https://doi.org/10.1038/s41598-020-64202-1
Engel T, Goni-Oliver P, Lucas JJ, Avila J, Hernandez F (2006) Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J Neurochem 99(6):1445–1455. https://doi.org/10.1111/j.1471-4159.2006.04139.x
Wilson EN, Do Carmo S, Welikovitch LA, Hall H, Aguilar LF, Foret MK, Iulita MF, Jia DT et al (2020) NP03, a microdose lithium formulation, blunts early amyloid post-plaque neuropathology in McGill-R-Thy1-APP Alzheimer-like transgenic rats. J Alzheimers Dis 73(2):723–739. https://doi.org/10.3233/JAD-190862
Pouladi MA, Brillaud E, Xie Y, Conforti P, Graham RK, Ehrnhoefer DE, Franciosi S, Zhang W et al (2012) NP03, a novel low-dose lithium formulation, is neuroprotective in the YAC128 mouse model of Huntington disease. Neurobiol Dis 48(3):282–289. https://doi.org/10.1016/j.nbd.2012.06.026
Mouri A, Legrand P, El Ghzaoui A, Dorandeu C, Maurel JC, Devoisselle JM (2016) Formulation, physicochemical characterization and stability study of lithium-loaded microemulsion system. Int J Pharm 502(1–2):117–124. https://doi.org/10.1016/j.ijpharm.2016.01.072
Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF (2011) Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry 198(5):351–356. https://doi.org/10.1192/bjp.bp.110.080044
Pradeepkiran JA, Reddy PH (2019) Structure based design and molecular docking studies for phosphorylated tau inhibitors in Alzheimer’s disease. Cells 8(3). https://doi.org/10.3390/cells8030260
Gong CX, Iqbal K (2008) Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem 15(23):2321–2328. https://doi.org/10.2174/092986708785909111
Voronkov M, Braithwaite SP, Stock JB (2011) Phosphoprotein phosphatase 2A: a novel druggable target for Alzheimer’s disease. Future Med Chem 3(7):821–833. https://doi.org/10.4155/fmc.11.47
Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, Yu JW, Strack S et al (2006) Structure of the protein phosphatase 2A holoenzyme. Cell 127(6):1239–1251. https://doi.org/10.1016/j.cell.2006.11.033
Kishi T, Matsunaga S, Oya K, Nomura I, Ikuta T, Iwata N (2017) Memantine for Alzheimer’s disease: an updated systematic review and meta-analysis. J Alzheimers Dis 60(2):401–425. https://doi.org/10.3233/jad-170424
Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K (2004) Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett 566(1–3):261–269. https://doi.org/10.1016/j.febslet.2004.04.047
Corcoran NM, Martin D, Hutter-Paier B, Windisch M, Nguyen T, Nheu L, Sundstrom LE, Costello AJ et al (2010) Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J Clin Neurosci 17(8):1025–1033. https://doi.org/10.1016/j.jocn.2010.04.020
van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Gotz J, Ittner LM (2010) Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci U S A 107(31):13888–13893. https://doi.org/10.1073/pnas.1009038107
Iqbal K, Liu F, Gong CX (2016) Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12(1):15–27. https://doi.org/10.1038/nrneurol.2015.225
Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, Tong Y, Rao Y (2019) PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther 4:64. https://doi.org/10.1038/s41392-019-0101-6
Silva MC, Ferguson FM, Cai Q, Donovan KA, Nandi G, Patnaik D, Zhang T, Huang HT et al (2019) Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 8. https://doi.org/10.7554/eLife.45457
Kargbo RB (2019) Treatment of Alzheimer’s by PROTAC-tau protein degradation. ACS Med Chem Lett 10(5):699–700. https://doi.org/10.1021/acsmedchemlett.9b00083
Dai CL, Tung YC, Liu F, Gong CX, Iqbal K (2017) Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res Ther 9(1):1. https://doi.org/10.1186/s13195-016-0227-5
Panza F, Solfrizzi V, Seripa D, Imbimbo BP, Lozupone M, Santamato A, Zecca C, Barulli MR et al (2016) Tau-centric targets and drugs in clinical development for the treatment of Alzheimer’s disease. Biomed Res Int 2016(3245935). https://doi.org/10.1155/2016/3245935
Qi X, Qvit N, Su YC, Mochly-Rosen D (2013) A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci 126(Pt 3):789–802. https://doi.org/10.1242/jcs.114439
Wang W, Yin J, Ma X, Zhao F, Siedlak SL, Wang Z, Torres S, Fujioka H et al (2017) Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum Mol Genet 26(21):4118–4131. https://doi.org/10.1093/hmg/ddx299
Manczak M, Kandimalla R, Yin X, Reddy PH (2019) Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum Mol Genet 28(2):177–199. https://doi.org/10.1093/hmg/ddy335
Wu Q, Xia SX, Li QQ, Gao Y, Shen X, Ma L, Zhang MY, Wang T et al (2016) Mitochondrial division inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing cell death and improving functional outcome in a mouse model of traumatic brain injury. Brain Res 1630:134–143. https://doi.org/10.1016/j.brainres.2015.11.016
Smith G, Gallo G (2017) To mdivi-1 or not to mdivi-1: Is that the question? Dev Neurobiol 77(11):1260–1268. https://doi.org/10.1002/dneu.22519
Lin J, Zhuge J, Zheng X, Wu Y, Zhang Z, Xu T, Meftah Z, Xu H et al (2020) Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway. Free Radic Biol Med 150:109–119. https://doi.org/10.1016/j.freeradbiomed.2020.02.024
Govindarajan N, Rao P, Burkhardt S, Sananbenesi F, Schluter OM, Bradke F, Lu J, Fischer A (2013) Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol Med 5(1):52–63. https://doi.org/10.1002/emmm.201201923
Chen S, Owens GC, Makarenkova H, Edelman DB (2010) HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One 5(5):e10848. https://doi.org/10.1371/journal.pone.0010848
Calkins MJ, Manczak M, Reddy PH (2012) Mitochondria-targeted antioxidant SS31 prevents amyloid beta-induced mitochondrial abnormalities and synaptic degeneration in Alzheimer’s disease. Pharmaceuticals (Basel) 5(10):1103–1119. https://doi.org/10.3390/ph5101103
Feniouk BA, Skulachev VP (2017) Cellular and molecular mechanisms of action of mitochondria-targeted antioxidants. Curr Aging Sci 10(1):41–48. https://doi.org/10.2174/1874609809666160921113706
Machiraju P, Wang X, Sabouny R, Huang J, Zhao T, Iqbal F, King M, Prasher D et al (2019) SS-31 peptide reverses the mitochondrial fragmentation present in fibroblasts from patients with DCMA, a mitochondrial cardiomyopathy. Front Cardiovasc Med 6:167. https://doi.org/10.3389/fcvm.2019.00167
Chavez JD, Tang X, Campbell MD, Reyes G, Kramer PA, Stuppard R, Keller A, Marcinek DJ et al (2019) Mitochondrial protein interaction landscape of SS-31. bioRxiv:739128. https://doi.org/10.1101/739128
Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem 120(3):419–429. https://doi.org/10.1111/j.1471-4159.2011.07581.x
Kanninen K, Malm TM, Jyrkkanen HK, Goldsteins G, Keksa-Goldsteine V, Tanila H, Yamamoto M, Yla-Herttuala S et al (2008) Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci 39(3):302–313. https://doi.org/10.1016/j.mcn.2008.07.010
Kanninen K, Heikkinen R, Malm T, Rolova T, Kuhmonen S, Leinonen H, Ylä-Herttuala S, Tanila H et al (2009) Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106(38):16505–16510. https://doi.org/10.1073/pnas.0908397106
Vomhof-Dekrey EE, Picklo MJ Sr (2012) The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J Nutr Biochem 23(10):1201–1206. https://doi.org/10.1016/j.jnutbio.2012.03.005
Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV (2014) Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun 5:3496. https://doi.org/10.1038/ncomms4496
Ren P, Chen J, Li B, Zhang M, Yang B, Guo X, Chen Z, Cheng H et al (2020) Nrf2 ablation promotes Alzheimer’s disease-like pathology in APP/PS1 transgenic mice: the role of neuroinflammation and oxidative stress. Oxidative Med Cell Longev 2020:3050971. https://doi.org/10.1155/2020/3050971
Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, Bose N, Gugliucci A, Kapahi P (2018) The role of advanced glycation end products in aging and metabolic diseases: bridging association and causality. Cell Metab 28(3):337–352. https://doi.org/10.1016/j.cmet.2018.08.014
Bavarsad Shahripour R, Harrigan MR, Alexandrov AV (2014) N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav 4(2):108–122. https://doi.org/10.1002/brb3.208
Murphy MP, Hartley RC (2018) Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov 17(12):865–886. https://doi.org/10.1038/nrd.2018.174
Young ML, Franklin JL (2019) The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol Cell Neurosci 101:103409. https://doi.org/10.1016/j.mcn.2019.103409
Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, Woodward KA, Chonchol M et al (2018) Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 71(6):1056–1063. https://doi.org/10.1161/HYPERTENSIONAHA.117.10787
Li G, Gong J, Lei H, Liu J, Xu XZ (2016) Promotion of behavior and neuronal function by reactive oxygen species in C. elegans. Nat Commun 7:13234. https://doi.org/10.1038/ncomms13234
Wei Y, Kenyon C (2016) Roles for ROS and hydrogen sulfide in the longevity response to germline loss in Caenorhabditis elegans. Proc Natl Acad Sci U S A 113(20):E2832–E2841. https://doi.org/10.1073/pnas.1524727113
Oswald MC, Brooks PS, Zwart MF, Mukherjee A, West RJ, Giachello CN, Morarach K, Baines RA et al (2018) Reactive oxygen species regulate activity-dependent neuronal plasticity in Drosophila. Elife 7. https://doi.org/10.7554/eLife.39393
Oswald MCW, Garnham N, Sweeney ST, Landgraf M (2018) Regulation of neuronal development and function by ROS. FEBS Lett 592(5):679–691. https://doi.org/10.1002/1873-3468.12972
Acknowledgments
We thank the members of the Mitochondrial Research and Interest Group at the University of Rochester Medical Center for their valuable suggestions and helpful discussions. The authors would like to acknowledge Bio-render for providing an online paid subscription platform (BioRender.com) to create all the figures.
Funding
This work was supported by NIH (R21AG060627 and R01AG067617) (GJ and KN).
Author information
Authors and Affiliations
Contributions
SG wrote the paper, made the figures, and reviewed all the cited articles. GJ and KN helped in writing the paper and critically revised the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guha, S., Johnson, G.V.W. & Nehrke, K. The Crosstalk Between Pathological Tau Phosphorylation and Mitochondrial Dysfunction as a Key to Understanding and Treating Alzheimer’s Disease. Mol Neurobiol 57, 5103–5120 (2020). https://doi.org/10.1007/s12035-020-02084-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02084-0