
Abstract
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disorder characterized by hyperglycemic conditions. A higher risk of developing Parkinson’s disease (PD) in patients with T2DM has become evident in recent years. However, the molecular mechanisms underlying the interplay between T2DM and PD pathogenesis remain unknown. Nevertheless, emerging epidemiological studies have demonstrated many common molecular pathways that play an essential role in regulating normal cellular functioning are independently implicated in the progression and etiopathogenesis of T2DM and PD. This review summarizes some common shared pathophysiological mechanisms, including insulin resistance, inflammation, mitochondrial dysfunction, endoplasmic reticulum stress (ER stress), autophagy, and the ubiquitin–proteasome system (UPS) that independently mediate the onset and etiopathogenesis of T2DM and PD. In this review, we summarize the studies that have reported the relationship between T2DM and PD. This review will provide insights into the common involvement of molecular pathways that may provide alternative treatment strategies for both T2DM and PD.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Studies have observed that individuals with diabetes mellitus (T2DM) are at a greater risk of developing Parkinson’s disease (PD) [1]. T2DM is a metabolic disease characterized by the aggregation of amylin protein in the β-cells of pancreatic islets, insulin resistance, and chronic hyperglycemia [2]. PD is a neurodegenerative disorder characterized by the loss of dopaminergic (DA-ergic) neurons in the basal ganglia, substantia nigra pars compacta (SNpc) regions of the brain [3]. Previous clinical studies have reported an increased prevalence of T2DM in patients with PD [4]. These studies reported higher insulin resistance in PD patients compared to healthy individuals, suggesting a potential link may exist between T2DM and PD [1]. Preclinical studies have also demonstrated the crucial role of insulin in regulating brain DA-ergic activity and its dysregulation in developing PD-like symptoms [1]. Previous reports suggest that mitochondrial dysfunction, endoplasmic reticulum (ER) stress, dysfunction in the UPS, and alterations in autophagy may be common underlying mechanisms in the course of T2DM and PD [5]. Another commonality is the formation of amylin protein-mediated amyloids fiber which plays a central role in the pathogenesis of T2DM and PD [6]. This suggests that the most potent drugs for T2DM could be used as a potential therapeutic approach against amyloidogenic neurodegenerative disorders [7]. For example, exenatide, glucagon-like peptide–l (GLP-1) agonists, and antidiabetic agents have been shown to exhibit neuroprotective and neurotrophic effects in the treatment of PD [8]. These observations raise the intriguing question of how T2DM could contribute to the pathogenesis of PD and whether, from a therapeutic perspective, the development of strategies targeting T2DM becomes a valuable approach to prevent PD. In this review article, we have focused on the common signaling pathways between T2DM and PD and the molecular mechanisms. Targeting these common signaling pathways could provide alternative and efficient treatment strategies for T2DM and PD.
T2DM and PD Comorbidity
An association between T2DM and PD was reported a few decades ago and ever since, many epidemiological and molecular studies have investigated a correlation between T2DM and PD development, summarized in Table 1 [9]. Most of these epidemiological studies suggest that T2DM is associated with an increased risk of PD in diverse populations. Reports indicate that T2DM may predispose to PD-like symptoms and may even exhibit a more aggressive phenotype when showing comorbidity with PD [10]. T2DM and PD share some common pathophysiological mechanisms mediated by similar detrimental genetic and environmental factors. Recent studies have reported that patients with T2DM have a 30–36% increased risk of developing PD and often display parkinsonian-like symptoms [11]. Reports also suggest that patients with idiopathic PD (IPD) have a genetic susceptibility that puts these individuals at risk for developing both diseases [12]. For example, a single nucleotide polymorphism in Akt gene, encoding Akt kinase that regulates cell growth and survival, has been implicated in the pathogenesis of both T2DM and PD [13]. Also, decreased expressions of (DnaJ-1) DJ-1, encoded by the PD-related park7 gene, were reported in pancreatic islets of patients with T2DM [14]. Recent studies have also reported that exposure to environmental toxins like maneb, paraquat (PQ), and rotenone, including heavy metals like iron, magnesium, and copper, increases the risk of developing T2DM and PD onset [15]. Exposure to pesticides and heavy metals results in loss of β-cell function in T2DM and may put individuals at higher risk of developing PD onset/progression [6].
A previous meta-analysis study based on four prospective studies indicated T2DM as a potential risk factor for the development of PD [5]. Another comprehensive updated meta-analysis on 7 observational cohort studies reported that patients with T2DM were associated with a 38% increase in the risk of developing PD compared to non-diabetic individuals [16]. In yet another meta-analysis that involved case–control studies with a much larger sample size also reported individuals with T2DM may have an increased incidence of PD compared to normal healthy individuals [16]. A longitudinal study reported that hypertension and T2DM as the most frequent comorbidities in patients with PD compared to control subjects [17]. A large prospective cohort study in Finland found an increased risk of 80% of developing PD-like symptoms in patients with T2DM and a similar study in the USA found that patients with T2DM were 40% more likely to develop PD-like symptoms [18]. Observational studies implying different strategies also reported that patients with T2DM had 32% more chances of developing PD in the UK and 23% in Taiwan and 36% in Denmark [4]. Reports also suggest that T2DM may predispose to a PD-like pathology and induce a more aggressive phenotype when coexisting with PD. For example, patients with T2DM had higher Unified Parkinson Disease Rating Scale (UPDRS) motor scores and more severe Hoehn and Yahr staging [19]. Other than the progression, T2DM has also been implicated in posture instability, difficulty in gait strides during the development of PD [20]. Previous studies suggest that these associations between T2DM and PD progression are likely due to more than one mechanism involved. The precise mechanism by which T2DM is related to PD onset and progression is mostly unknown; however, some strikingly common dysregulated pathways like impaired insulin signaling, inflammation, mitochondrial dysfunction, ER stress, autophagy, and dysfunctions in UPS are among some of the shared mechanisms between both chronic diseases [6].
Insulin Resistance in T2DM and PD
Insulin regulates cell growth, gene expression, protein synthesis, mitochondrial function, and autophagy [21]. Insulin acts as a ligand and binds to its receptor resulting in enhanced tyrosine kinase activity that phosphorylates insulin receptor substrate (IRS)-1&-2 that binds to p85-phosphatidylinositol 3-phosphate kinase (PI3K) phosphorylating Akt and glycogen synthase kinase-3beta (GSK3-β), thereby regulating glucose metabolism [21]. Any link between T2DM and PD may relate to dysregulations in the common pathways due to chronic hyperglycemia conditions. People with T2DM suffer from insulin resistance which results in the progressive loss of insulin secretion from pancreatic β-cells, ultimately leading to hyperglycemic conditions in T2DM [22]. PI3K/Akt signaling is inactivated, which inhibits the transport of plasma glucose into cytoplasm resulting in dysregulated blood glucose homeostasis and insulin resistance in T2DM. Chronic hyperglycemia results in the generations of reactive oxygen species (ROS) that have been implicated in the loss of β-cell mass in T2DM [23]. Furthermore, studies have also shown that GLP-1 suppresses glucagon secretion from α-cells and regulates glucose-dependent insulin secretion from pancreatic β-cells in T2DM [24]. This loss in function and mass of β-cell in T2DM may be attributed to the consequence of glucolipotoxicity, accumulation of cholesterol, and inflammation in islets [25].
Insulin also regulates the activation of neural stem cells, cognition, synaptic formation, and neuronal apoptosis [26]. Implications of neuronal insulin resistance are commonly seen in various neurodegenerative diseases [27, 28]. Studies have suggested the role of insulin in regulating DA-ergic activity and a reciprocal insulin regulation by DA-ergic neurons. Recent evidence showed the prevalence of insulin resistance exhibiting impaired glucose tolerance in patients with PD [29]. Interestingly, some drugs used to treat PD, such as levodopa (L-DOPA), induce hyperglycemia conditions indicating insulin resistance may increase the risk of developing PD [29].
Dysregulation of PI3K/Akt signaling pathway may alter the expression of α-synuclein, leading to DA-ergic cell death [30]. Studies have also highlighted the role of leucine-rich repeat kinase 2 (LRRK2) in insulin-dependent intracellular signaling. Reports suggest that phosphorylation of Ras-related protein (Rab10), an endocytic sorting protein, by LRRK2 is crucial for glucose transporters type 4 (GLUT4) translocation to the neuronal plasma membrane is inhibited in PD patients with the LRRK2 (G2019S) mutation [31]. These studies show that insulin resistance can be a potential risk factor for T2DM and the pathologic driver of neurodegeneration in PD.
Inflammation in T2DM and PD Etiopathogenesis
Inflammation is a highly regulated process that protects against tissue damage that has been implicated in the pathogenesis of T2DM and PD [32]. Studies have reported elevated inflammatory response mediated by immune cells, cytokines, and chemokines in the islets of patients with T2DM [33]. Inflammatory response likely causes insulin resistance by activating cytokines which is reciprocally mediated by hyperglycemia conditions thereby promoting severity in T2DM [34]. Insulin resistance causes activation of microglia and inflammation mediated by nuclear factor-κB (NF-κB) and the PI3K/Akt pathway [35, 36]. Inflammatory cytokines like tumor necrosis factor-α (TNF-α) induce the inactivation of insulin receptor substrate-1 (IRS1), which inhibits activation of downstream regulators promoting T2DM conditions [37]. Activated cytokines downregulate anabolic cascades in insulin signaling resulting in insulin sensitivity and dysregulations in glucose metabolism [38]. Several lines of evidence have suggested the role of pro-inflammatory cytokines in regulating insulin resistance and dysregulations in glucose homeostasis in T2DM. Reports suggest that low-grade pancreatic tissue inflammation plays a critical role in the pathophysiology of T2DM [39]. Similarly, studies have shown that a high glucose/cholesterol diet can induce activated microglia mediating pro-inflammatory cytokines—interleukin-6 (IL-6) and TNF-α release in an in vivo model of T2DM [40]. Reports also suggest increased levels of infiltrating macrophages that release IL-1β, which leads to loss of β-cell function and its progressive destruction in T2DM [41]. Studies have also shown that hyperglycemia activates (P2X purinoceptor 7) a P2X7 receptor that induces NOD-, LRR-, and pyrin domain-containing 3 (NLRP3) inflammasome activation to increase IL-1β secretion resulting in pancreatic β-cell dysfunction and death [42]. Treating T2DM rodents with anti-inflammatory drugs like aspirin resulted in the decrease of pro-inflammatory cytokines release, thereby alleviating insulin resistance [43]. Also, these anti-inflammatory drugs have been reported to result in dysregulations in IKK-β signaling pathways that otherwise mediate insulin resistance and T2DM [44]. Similarly, exogenous administration of TNF-α or IL-6 results in insulin resistance and lower levels of cytokines were observed in TNF-α knock-out mice [38]. The newly discovered cytokine IL-37 binds to IL-18 receptor α/β (IL-18Rα) chain and shows anti-inflammatory properties by inhibiting IL-6, IL-18, IL-33, IL-1β, and TNF-α, suggesting a potential therapy for low-grade inflammation in T2DM [39]. Taken together, these observations suggest inflammation as a key feature in the regulation of insulin resistance and the development of T2DM.
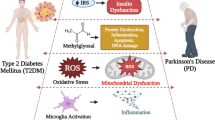
Inflammation also plays a very important role in the pathogenesis of PD. Previous studies report that activated microglia and astrocytes promote neuroinflammation as key pathways of PD progression [45]. Activation of microglia and subsequent microgliosis events may be mediated by the aggregation of α-synuclein released by damaged and injured neurons in PD [46]. Aggregation of misfolded α-synuclein acts as damaged-associated molecular patterns (DAMPs), inducing NLRP3 inflammasome in PD, which then interacts with caspase-1 forming activated NLRP3–caspase-1 complex followed by secretion of IL-1β from microglia [47]. Similarly, activation of reactive astrocytes results in sustained neuroinflammation that mediates degenerations in PD [48]. Elevated levels of pro-inflammatory cytokines, including IL-6, IL-1β, TNF-α, and IFN-γ, have been observed in a 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydrodropyridine (MPTP)–induced mouse model of PD [49]. Elevated concentrations of pro-inflammatory cytokines like IL-1β, IL-6, and TNF-α were observed in the brain, CSF, and blood of PD patients [50]. Reports suggest that activated glial cells secrete various pro-inflammatory cytokines resulting in neuroinflammation and neurodegeneration via NF-kB signaling [51]. The β2-adrenoceptor, a G-GPCR, is expressed in microglia, astrocyte, and postsynaptic neuron surface. Treatment with its agonist clenbuterol and formoterol activates cAMP/PKA/CREB/IkBα pathway that results in inhibition of the NF-kβ inflammatory pathway in the intra-nigral LPS induced rat model of PD [52]. Taken together, these observations suggest that inflammation is a key feature in the regulation of insulin resistance in T2DM and neuronal death in PD. The role of insulin resistance and inflammation in promoting T2DM conditions and neurodegeneration in PD is given in Fig. 1

Diagrammatic representation of the possible role of (A)insulin resistance and (B) inflammation in promoting T2DM conditions and neurodegeneration in PD. Abbreviations: DAMPs, damage-associated molecular patterns; GLP-1, glucagon-like peptide; GSK3β, glycogen synthase kinase 3 beta; IFN-α, interferon alfa; IL-3, interleukin-3; IL-6, interleukin-6; IL-18, interleukin-18; IL-33, interleukin-33; IL-1β, interleukin 1 beta; IRS-1, insulin receptor substrate 1; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NLR family pyrin domain-containing 3; PD, Parkinson’s disease; PI3K, phosphoinositide 3-kinase; TNF-α, tumor necrosis factor alfa; T2DM, type 2 diabetes mellitus
Mitochondrial Dysfunctions in T2DM and PD
Mitochondria is essential for glucose-stimulated insulin secretions from β-cells and its dysfunction has been implicated in T2DM [53]. The prevalence of mitochondrial diabetes constitutes about 0.5–1% of all types of DM mediated by impaired insulin secretions, unlike other T2DM mediated by insulin resistance [54]. Depletions in mtDNA dramatically decrease oxygen consumption and suppress glucose-stimulated insulin secretion in T2DM [54]. Studies on the role of mtDNA in the etiopathogenesis of T2DM are scarce. Yet, a dozen loci in nuclear-encoded mitochondrial genes have been directly related to T2DM by regulating β-cell function [55]. A co-transcription factor peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1-α) and a transcription factor B1 mitochondrial (TFB1M) are essential for the normal functioning of mitochondria. During mitochondrial dysfunction, PGC1-α and TFB1M have been implicated in the pathogenesis of T2DM by decreasing oxygen consumption, ATP production, and concomitantly insulin secretion [56]. Another such gene UCP2, mitochondrial C4 metabolite exchanger, was found to be highly expressed in islets of different animal and human models of T2DM. Hyperglycemia induces glucose oxidation by elevating NADH and pyruvate in mitochondrial complex I, III resulting in ROS production, thereby impairing β-cell function [57].
Mitochondria perform a plethora of functions by regulating various cellular processes like Ca2+ homeostasis, stress response, and apoptotic pathways [58]. Mitochondrial dysfunction has been implicated in the pathogenesis of PD. α-synuclein aggregation in mitochondrial is characterized by the generation of ROS, decrease in the activity of mitochondrial complex I enzyme, decrease in ATP levels, cytochrome-c release, and caspase-3 activation [59]. α-synuclein-mediated mitochondrial dysregulations may be due to α-synuclein-induced p38 MAPK activation, which phosphorylates and activates Drp1 (dynamin-related protein-1), causing mitochondrial fission [60]. Neurons have incredibly high metabolic demands making them highly susceptible to mitochondrial dysfunction [61]. The very first report on the role of mitochondrial dysfunction in PD pathogenesis came from the accidental infusion of the MPTP, which selectively inhibited mitochondrial complex I of electron transport chain [58]. Later studies reported environmental toxins MPP+, rotenone, and paraquat target the mitochondrial respiratory chain resulting in mitochondrial dysfunction in PD (57). Overexpression of mutant A53T or wild-type human SNCA gene in different in vitro and in vivo models of PD resulted in cytochrome c release, increased mitochondrial Ca2+ and nitric oxide (NO) levels, and decreased mitochondrial complex I [62]. Parkin and PINK1 genes together regulate mitochondrial function and biogenesis and their mutations have been reported to result in mitochondrial impairments by reducing complex I activity and increasing mitochondrial ROS production in the different PD models [63]. DJ-1 protein fractionally located at the mitochondria acts as a neuroprotective intracellular redox sensor and its mutation/inactivation/knockout resulted in the decrease of mitochondrial complex I activity, decreased mtDNA levels, respiratory control ratio, and ATP levels in the experimental PD models [64]. Similarly, overexpressions of the LRRK2 gene resulted in decreased mitochondrial membrane potential (MMP) and ATP levels, thereby impairing mitochondrial function in PD patients [65]. These observations indicate that mitochondrial function is paramount for the normal functioning of the cellular processes and its dysregulations are implicated in the etiopathogenesis of T2DM and PD.
There is a strong correlation between neuroinflammation and mitochondrial dysfunction in the pathogenesis of T2DM and PD. In the MPTP-induced PD mice model, neuroinflammation involving microglia activation has been reported in the brain’s striatum and substantia nigra regions. Activated microglia release IL-1β and IL-6 and induce activation of iNOS and NADPH-oxidase [66]. Activated microglia also release excitotoxins like quinolinic acid and glutamate that overstimulate glutamate receptors expressed in neurons, thereby increasing membrane lipid peroxidation, reactive nitrogen species, and mitochondrial damage [67].
In T2DM, inflammation and mitochondrial dysfunction are correlated with each other. Systemic inflammation is also one of the factors responsible for insulin resistance and pathogenesis of T2DM. Damaged mitochondria releases mitochondrial DAMP molecules, such as cytochrome C, mt.DNA, and ATP. When released in circulation, the mitochondrial DAMPs activate systemic inflammatory responses. It has been reported that there is an elevation in circulating cell-free mt.DNA in the plasma of T2DM patients. The circulating cell-free mt.DNA induces AIM2 inflammasome-dependent activation of caspase-I and secretion of IL-18 and IL-1β from macrophages. It concludes in chronic inflammation in T2DM patients [68].
Excessive microglial activation causes loss of mitochondrial membrane potential along with ROS production leading to neurodegeneration [69]. Increased oxidative stress oxidizes lysine, threonine, proline, and arginine residue of mitochondrial proteins, mitochondrial ETC components, and mt.DNA, resulting in mitochondrial dysfunction [70]. It has been reported that the presence of glia increased neurodegeneration in neuron/glia culture from rat mesencephalon in rotenone-induced toxicity associated with O2.− release from microglia. Intranigral LPS-injected rats showed increased microglia activation; increased release of pro-inflammatory cytokines and iNOS, COX-2, and SN DA-ergic neurodegeneration; and reduced striatal DA level. LPS also induced significant nigral and striatal mitochondrial complex I and II in LPS-injected rats. Thus, inflammation causes loss of mitochondrial function, ultimately leading to DA-ergic neurodegeneration [71]. When injected into the rat brain ventricles, DA inhibits mitochondrial complex I, leading to increased cellular ROS and α-synuclein aggregation. Metabolism of DA also generates ROS (H2O2, DA-quinone, and semiquinone) as a by-product. This oxidative stress contributes to inflammatory response, as seen in PD patients [72].
Endoplasmic Reticulum Stress in the Pathogenesis of T2DM and PD
ER plays a crucial role in correct folding and post-translational modification of cell surface receptors, secretory proteins, and integral membrane proteins [8]. Aggregation of unfolded or misfolded proteins in ER lumen induces ER dysfunction resulting in ER stress [73]. There is an initiation of a complex signaling pathway called unfolded protein response (UPR) during the ER stress conditions. There are three specific sensor proteins of ER stress named as follows: Activating transcription factor 6 (ATF6), inositol requiring enzyme 1 α (IRE1α), and eukaryotic pancreatic ER kinase (PKR)–like ER kinase (PERK) [74]. Various studies have proposed ER stress as an underlying mechanism in the pathogenesis of T2DM and PD [75]. Reports suggest that ER stress–mediated β-cell loss in T2DM is regulated by the key transcription factors and gene networks [76]. ER stress mediates islet amyloid polypeptide (IAPP), inducing pancreatic β-cell apoptosis in T2DM. When ER homeostasis is destroyed, then PERK gets activated, and it subsequently triggers phosphorylation of eIF2α, ultimately leading to ATF4 upregulation. However, when UPR stress cannot restore the ER homeostasis, PERK activates downstream effectors and mediators of apoptosis, including caspase-12, CHOP, and Bcl family genes. Activation of the PERK-CHOP pathway also induces activation of NF-κB, resulting in inflammation and aggravating β-cell apoptosis in T2DM [77]. Under normal conditions, ATF6 causes upregulation of Grp78 protein. But diabetes-induced ATF6 is associated with dilation of ER cisterns and an increase in ER volume, reflecting ER’s increased functional capacity [78]. Another study has reported that ER stress induces phosphorylation of IRE1α that further activates its downstream effector XBP-1 and concludes in a series of events leading to UPR response in T2DM [74]. Mutation in EIF2AK3, encoding human eIF2α-kinase, which regulates transitional control in response to ER stress, has been reported in T2DM, suggesting its role in normal β-cell functioning and survival [79]. Similar studies also demonstrated aberrant expression of ER stress biomarkers including p-PERK, p-eIF2α, p-IRE1α, CHOP, BiP, cleaved caspase-3, and caspase-8 in the individuals with T2DM [73]. More evidence for the role of ER stress in T2DM etiology comes from a study where ER stress–mediated overexpressions of BiP, CHOP, and p58 were observed in β-cells of patients with T2DM compared to the healthy non-diabetic patients [80]. A similar study found a fold increase in ER stress in β-cells of T2DM patients compared to the normal, non-diabetic individuals [81]. Studies have shown that prolonged ER stress induces apoptosis by activating the caspase-3 pathway in rats with T2DM. The caspase-mediated apoptotic pathway involves activating caspase-8 that activates caspase3/caspase6/7/PARP (poly ADP-ribose polymerase) signaling resulting in apoptosis in pancreatic β-cell during T2DM [2]. Studies have also shown that genetic variations in Wolfram syndrome gene 1 (WFS1), encoding ER Ca2+ channel, result in selective loss of β-cell in young-onset diabetes and T2DM. This reduction in ER stress–mediated β-cell mass may be due to chronic exposure to gluco- and lipotoxicity [79]. It is very unlikely that loss of β-cell entirely accounts for reduced insulin secretions in T2DM and more detailed studies are required to determine the role of ER stress–mediated reduction in β-cell mass.
Correspondingly in PD, studies have shown that ER stress induces the aggregation of α-synuclein into LB that promotes neurodegeneration [82]. Studies have shown that IRE1α gets activated and induces activation of the caspase-12 mediated apoptotic pathway in the MPP + treated PC12 cells, causing DA-ergic neuronal death. Isorhynchophylline (IRN) treatment inhibits IRE1α-mediated ER stress resulting in the protection of PC-12 cells from caspase-9-dependent apoptosis [83]. Activation of PERK in ER stress conditions phosphorylates eIF2α, activating ATF4. An increased level of ATF4 further elevates transcription of CHOP that downregulates Bcl-2. Studies have shown that β-asarone, a component of Acorus tatarinowii Schott volatile oil, regulates the ER stress–autophagy by inhibiting the PERK/CHOP/Bcl-2/Beclin-1 pathway resulting in neuroprotection in 6-OHDA-induced parkinsonian rats [84]. Activation of the ATF6 pathway in the ER stress causes trafficking and proteolytic processing of ATF6. Activated ATF6 regulates the expression of XBP1 and ER chaperone. A study has shown that aggregation of α-synuclein induces ER stress response that causes activation of PERK and ATF6 pathway in an α-syn-induced rat model of PD. In the absence of re-establishment of ER homeostasis, ATF6 activates proapoptotic CHOP protein in SNpc DA-ergic neurons. However, Grp78 over-expression downregulates ER stress mediators and proapoptotic mediators resulting in DA-ergic neuroprotection [85].
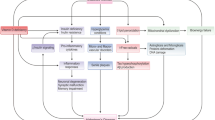
It has also been demonstrated that both HMG–CoA reductase degradation protein-1 (Hrd1) and Parkin-associated endothelin receptor-like receptor (Pael-R) are aberrantly expressed in DA-ergic neurons [75]. Hrd1 is a key enzyme for ER-associated degradation of unfolded or misfolded proteins and Pael-R is a substrate for Parkin, an E3 ubiquitin ligase [86]. Therefore, ER stress caused by the accumulation of Pael-R is relevant to the pathological mechanisms that underlie autosomal recessive PD. However, recent studies have also investigated that Pael-R is ubiquitinated by Hrd1 thereby preventing ER stress–induced neuronal apoptosis in PD [75]. Besides that, an in vivo study has demonstrated that the inhibition of X-box-binding protein 1 (XBP1), a key regulator of the unfolded protein response (UPR), induces chronic ER stress and specific DA-ergic neurodegeneration [87]. Restoring XBP1 levels by gene therapy decreased striatal denervation and neuronal death in the 6-OHDA-induced mice model of PD [88]. In another similar study, activation of XBP1 provided neuroprotection against MPTP-induced DA-ergic loss in in vitro and in vivo models of PD, indicating the crucial role of UPR in the survival of DA-ergic neurons [89]. MPTP-induced ER stress causes dysregulations in ER Ca2+ homeostasis by inhibiting store-operated calcium entry (SOCE) and transient receptor potential channel 1 (TRPC1), resulting in DA-ergic neuronal loss [90]. Recent studies have reported that ER stress–mediated apoptosis was implicated in the PD by activating the apoptotic pathways [91]. These observations suggest that ER stress may be a common pathological pathway and its inhibition can be a common therapeutic target in the treatment strategies against T2DM and PD. Figure 2 depicts the role of mitochondrial dysfunction and endoplasmic reticulum stress in the pathogenesis of T2DM and PD.

Schematic illustration of the role of (A) mitochondrial dysfunction and (B) endoplasmic reticulum stress in the pathogenesis of T2DM and PD. Abbreviations: ATF4, activating transcription factor 4; ATP, adenosine triphosphate; CHOP, C/EBP homologous protein; Bcl, B-cell lymphoma; DA-ergic, dopaminergic; eIF2, eukaryotic initiation factor 2; IRE1α, inositol requiring enzyme-1 alpha; Parkinson’s disease; PERK, protein kinase R-like endoplasmic reticulum kinase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; MAPK, mitogen-activated protein kinase; PD, Parkinson disease; ROS, reactive oxygen species; T2DM, type 2 diabetes mellitus; TFB1M, transcription factor B1 mitochondrial
Impairment of Autophagy in T2DM and PD
Autophagy is a catabolic process that maintains cellular hemostasis by enabling cells to stride past unfavorable stress conditions by regulating the intracellular conditions through cytoplasmic turnover of proteins and organelles [92]. Autophagy, if altered, may be associated with a distinct form of cell death resulting inT2DM and PD onset/progression. Loss of autophagic function by cells results in amyloid aggregation, i.e., amylin protein and α-synuclein in T2DM and PD, respectively [93]. Upregulation of common autophagic markers like LC3-II, p62, beclin-1, p-mTOR, and p-AMPK has been reported in T2DM and PD [94]. Recent investigations have found the role of autophagy in controlling insulin signaling and lipid metabolism. Under normal conditions, autophagy is essential for the survival and maintenance of β-cell function, but during stressful conditions, it prevents β-cell dysfunctions by serving as an adaptive mechanism [95]. Studies have shown the accumulation of autophagosomes and undigested autophagic vacuoles in the pancreatic β-cells in T2DM [96]. Impaired autophagy resulted in dysregulated vascular function and its stimulation by upregulating autophagy markers LC3-II, Beclin-1, and p62 improved vascular function in the mesenteric arteries of T2DM mice [97]. Atg7 and Atg12-Atg5 play central roles in the biogenesis of autophagosomes and their dysregulations increase sensitivity towards insulin and have been therefore implicated in T2DM pathogenesis [98]. Recent work demonstrated upregulating autophagy genes reduced insulin secretion and the incidence of diabetes in STZ-treated β-cells [99]. In another experiment, metformin, a T2DM drug, was shown to decrease the formation of autophagic vacuoles in T2DM by enhancing AMP kinase activity that inhibits the mTOR pathway resulting in the removal of autophagic vacuoles [96]. Exendin-4, an agonist of the glucagon-like peptide GLP-1 receptor (GLP-1R), promotes insulin secretion and regulates autophagic markers, such as mTOR, LC3-II, LAMP1, parkin, Atg7, and p62, further validating the fact that autophagy plays crucial role in regulating T2DM [92].
Similarly, autophagy has been implicated in the etiopathogenesis of PD. Many genes and protein products of PD-related genes, either causative or risk factors for PD, appear to have an essential role in regulating autophagy pathways. α-synuclein, an insoluble aggregated protein implicated in PD, is usually degraded by chaperone-mediated autophagy (CMA) binding to its pentapeptide sequence motif [100]. α-synuclein enters into lysosome by binding to lysosomal-associated membrane protein type 2A (LAMP-2A), where it is degraded by proteases [101]. However, during dysregulated autophagy in PD, LAMP-2A does not take up mutant α-synuclein due to its very high binding affinity, which does not allow proper translocation of α-synuclein into the lysosome and its subsequent degradation [102]. Mutations in LRRK2, one of the major causes of PD, disrupt aggresomal formation and autophagic clearance of accumulated protein aggregates [103]. PINK1 regulates parkin translocation to impaired mitochondria driving their elimination via autophagy. In normal conditions, LRRK2 gets degraded by CMA; however, very high concentrations of LRRK2 during PD can block CMA by inhibiting translocation complex at lysosomal membrane, thereby resulting in dysregulations in autophagy function [104]. The autophagic adapter protein p62, a component of the PINK1-parkin pathway, is involved in the proteasomal degradation of misfolded or unfolded proteins. It interacts with the mitochondrial outer membrane proteins like mitochondrial mitofusin (MFN), VDAC, Fis1, and TOM20 that facilitate its recruitment to LC3, which promotes their incorporation into phagophores [104]. Defective p62 function and mutation in PINK1 lead to the accumulation of ubiquitinated protein aggregates resulting in persistently damaged mitochondria and cell death promoting PD pathology [105]. These observations imply that autophagy plays a crucial role in the pathogenesis of PD and T2DM or even the convergence point for the symptoms of T2DM and PD.
Dysregulated Ubiquitin Proteasomal System in T2DM and PD
Accumulation of damaged protein is harmful to normal cellular functioning; therefore, its degradation is essential [106]. UPS is one of the major pathways that degrade various proteins following polyubiquitination [107]. Studies have shown that UPS plays a significant role in insulin secretion by regulating various key proteins in the β-cell secretory cascade [108]. It also regulates various crucial proteins like IRS-2, MafA, or CREB essential for the β-cell survival [109]. Inhibition in UPS function may cause a decline in the level of insulin secretion in the pancreatic β-cell [110]. Recent studies suggest that the formation of toxic human islet amyloid polypeptide aggregates (h-IAPP) results in the loss of β-cell function leading to T2DM progression [111]. The increased expression of h-IAPP in T2DM downregulates ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), causing alteration in UPS causing deleterious consequences on β-cell function and survival [112]. Studies have also shown that toxic human amylin (hA) protein interacts with 20S core and 19S lid subunit of the β-cell proteasomal complex resulting in decreased proteolytic activity in T2DM [113]. Dysregulations in UPS have been reported to decrease insulin signaling by downregulating insulin receptor and insulin substrate (IRS) proteins in T2DM [114]. The downregulation in IRS proteins is mediated by dysregulations in the suppressor of cytokine signaling 1 (SOCS1) and SOCS3, substrate-recognition modules in elongin BC ubiquitin-ligase complex [115]. Additionally, IRS stability is regulated by a negative feedback mechanism in which insulin-dependent activation of mTOR phosphorylates IRS proteins and their subsequent ubiquitination and degradation of ubiquitin ligase subunit Fbw8 [116].
Similarly, dysregulations in UPS have been implicated in the pathogenesis of PD, leading to aggregation of α-synuclein [117]. It has been observed that alterations and dysfunction in UPS due to over-expression of α-synuclein may lead to DA-ergic neurodegeneration in PD. UPS components with PD-related protein parkin and UCH-L1 are implicated in the degradation of the misfolded α-synuclein protein [118]. Aggregates of α-synuclein may, in turn, selectively bind to 6S subunit of 26S proteasome inhibiting UPS function to induce further neuronal toxicity [119]. Therefore, proteasome and α-synuclein aggregation may reciprocally regulate a feed-forward mechanism and exacerbate the onset and progression of PD. Mutations in the catalytic and autoinhibitory domain Ub ligase Parkin (Park2) and phosphatase and tensin homolog (PTEN) PTEN-induced putative phosphatase 1 (PINK1) interrupt proteasome activity and lead to α-synuclein aggregation [120]. Mutation in DJ-1, a substrate for ubiquitin-like modifier-1 (SUMO-1) conjugation, has been implicated in the onset of PD [121]. Moreover, studies have also shown that deficiency of DA activates PKA (protein kinase A) and α-synuclein aggregate activates MAPK and mitogen and stress-activated kinase-1 (MSK1). The resultant activation of these enzymes causes phosphorylation of tyrosine hydroxylase enzyme that gets degraded by UPS, resulting in degradation and loss of DA-ergic neurons [122]. Protein degradation by UPS in T2DM and PD is crucial in cellular functioning, and evidence suggests its disruption as an important risk factor for T2DM and PD pathogenesis. Illustrative representation of the role of dysregulated autophagy and ubiquitin proteasomal system in promoting T2DM conditions and PD is given in Fig. 3.

Illustrative representation of the role of dysregulated (A) autophagy and (B) ubiquitin proteasomal system in promoting T2DM conditions and PD. Abbreviations: AMP, adenosine monophosphate; CREB, cAMP response element-binding protein; DA-ergic, dopaminergic; h-IAPP, human islet amyloid polypeptide; IRS-2, insulin receptor substrate 2; MafA, MAF BZIP transcription factor A; MAPK1, mitogen-activated protein kinase 1; MSK1, mitogen- and stress-activated kinase 1; PD, Parkinson’s disease; Park2, Parkin RBR E3 ubiquitin-protein ligase 2; T2DM, type 2 diabetes mellitus; UCH-L1, ubiquitin C-terminal hydrolase L1
Memory Impairment in T2DM and PD
The hippocampus region of the brain is susceptible to hyperglycemia associated with diabetes. It also plays a very crucial role in learning and memory. Diabetic animal models have shown that neuronal apoptosis in the hippocampus leads to spatial memory and cognition impairment [123]. Studies have shown that caffeine consumption abrogates memory deficits associated with downregulation of A1 receptors and upregulation of A2A receptors in the hippocampus in the mice model of T2DM [124]. It has been also shown that a high-fat diet (HFD) induces memory loss through microglia activation. Activated microglia increased pro-inflammatory markers like TNF-α, and Iba-1 in the hypothalamus and hippocampus region of the brain, causing inflammation. Treatment with abscisic acid (ABA) rescued all these HFD-induced changes in rats and improved cognitive function [125]. Central neuroinflammation and bleeding can interfere with normal blood–brain barrier (BBB) function and lead to memory and learning disabilities. Studies have shown that long-term use of Mangifera Indica Lin extract decreased microglia-mediated inflammation, limited hippocampal and cortical atrophy, and lowered tau phosphorylation, resulting in improved memory and learning disabilities [126]. It is reported that purinergic receptors play a crucial role in memory impairment in T2DM. DNA damage mediates downregulation of P2X purinoceptor 4 (P2X4R), resulting in increased levels of activated microglia in the hippocampus. These studies show that memory impairment is implicated in the pathogenesis of T2DM [127].
PD patients have comorbidities like-cognitive dysfunction, depression, and anxiety that significantly burden their quality of life [128]. Several studies have shown that PD is associated with impairment in memory [128–130]. Deltamethrin is most commonly used in controlling pests, and its administration causes hyperactivity and impairment in cognition and DA-ergic degeneration [130]. IPD patients have shown memory impairment, but it has been observed in the recall stage with relatively spare recognition. Aarsland et al. conducted a study with 1346 IPD patients without dementia and found that memory deterioration is the most common cognitive dysfunction in these patients, with the prevalence of mild cognitive impairment in 25.8% of patients [131]. The features associated with cognitive dysfunction in PD are impairment in visuo-perceptual processing and working memory. Kawashima et al. have reported that there is reduced activation of middle frontal gyrus (MFG) and inferior parietal lobule (IPL) regions of the brain in PD patients with cognitive impairment, suggesting that these regions may be associated with the pathophysiology of impairment in working memory in PD patients that involves fronto-striatal network dysfunction [129]. PD-induced cognitive dysfunction is associated with alternation in the DA-ergic vs. cholinergic neurotransmission [128].
DA signaling is crucial for normal memory function. In the cortex and medial temporal lobe, mesocorticolimbic DA modulates memory consolidation and retrieval, whereas striatal DA facilitates learning [132]. Studies have shown that etazolate, a pyrazolo pyridine compound, prevents long- and short-term memory deficits in the 6-OHDA-induced rat model of PD by increasing BDNF and antioxidant levels in the hippocampus [128]. The parahippocampal gyrus and salience network are consistently involved during memory retrieval. Other studies have shown that DA-ergic dysfunction in salience network and median temporal lobe leads to memory impairment in PD. It has been reported that rotenone exposure causes neurodegeneration in the cortex and hippocampus, resulting in cognitive dysfunction in the mice model [133]. Studies have shown that microglia-mediated neuroinflammation concluding in neuronal apoptosis is a pathological characteristic in memory impairment associated with PD [133, 134]. It has been reported that microglia-mediated disruption of the blood–brain barrier leads to neurodegeneration, resulting in impairment in memory and learning in rotenone induced mice model of PD [133]. Depletion of norepinephrine (NE) and damage to locus coeruleus (LC) noradrenergic neurons are early events in PD. It exacerbates the DA-ergic neurotoxicity and motor deficits. The studies have shown that damage to LC/NE increased α-synuclein expression, Ser129-phosphorylation, and synaptic loss, causing hippocampal neurodegeneration through microglia-mediated neuroinflammation and ferroptosis, as a result of which learning and memory get impaired in PQ and a maneb-induced mice model of PD [134].
Common Therapeutic Targets Against T2DM and PD
T2DM and PD have shared pathogenesis that leads to the onset and establishment of either disease. It suggests that targeting a common molecular pathway may provide a novel therapeutic target against both diseases. Aggregation of amylin peptide, mitochondrial dysfunction, oxidative stress, inflammation, autophagic dysfunction, and loss of UPS function are some of the etiological factors that play a crucial role in the onset and development of PD. There is a co-existence of insulin receptors and DA-ergic neurons in SNpc in both diseases. Another thing is that reduction in insulin signaling in basal ganglia is associated with depletion of DA in the striatum [135].
Several studies have shown that antidiabetic agents play a neuroprotective role in PD. Metformin has been the most common drug for years for the treatment of T2DM. Studies have shown that metformin has a neuroprotective effect against PD. Alteration in mitochondrial respiration is associated with PD [136]. Metformin can act as an inhibitor of complex I of mitochondria, thereby reducing elevated mitochondrial respiration and maintaining mitochondrial homeostasis [137]. Other studies have shown that it activates AMPK/BDNF and effectively improves motor deficits, and suppresses aging-induced gene activation, reactive astrocytes in a 6-OHDA-induced mice model of PD [138]. It has also been reported that metformin has a role in autophagy. Metformin treatment enhances neuronal autophagy through AMPK activation, increasing expression of LC3-II and decreasing expression of p62 resulting in DA-ergic neuroprotection [139]. It suggests that metformin is an emerging therapeutic drug targeting the shared pathogenesis of both T2DM and PD.
Another emerging therapy is the incretin hormone. It affects insulinotropic secretion and improves insulin resistance. Incretin hormone includes glucose-dependent insulinotropic polypeptide (GIP) and GLP-1. GLP-1R is expressed in different organs like islets, heart, immune cells, kidneys, and intestine. GLP-1 binds to its receptor GLP-1R of islet cells and evokes an antihyperglycemic effect and robust insulin release. It also inhibits apoptosis of β-cell and promotes β-cell proliferation [140]. The synthetic version of GLP1 agonist, exendin-4, is exenatide, which was approved in 2005 for use against T2DM.
Clinical studies have shown that treatment with exenatide improves motor deficits and cognitive dysfunction in PD patients [141]. GLP-1R is also expressed in different regions of the CNS, including the cortex, hypothalamus, striatum, SNpc, hippocampus, brain stem, and in the subventricular zone. The studies have shown that exendin-4 improves motor deficits and protects DA-ergic neurons in the MPTP-induced mice model of PD. Another study involving the LPS and 6-OHDA-induced PD model in SNpc has reported that after 7 days of treatment with exendin-4, amphetamine-induced circling behavior reduced, and DA production increased in basal ganglia [142]. Another GLP-1 analogue, liraglutide, binds to and stimulates GLP-1R, causing an increase in the cAMP. It leads to a cascade of intracellular events mediated by PKB/Akt and MAPK/ERK pathways, such as inhibition of neuronal apoptosis, neuronal cell survival, cell growth, repair, regeneration, and activation of Ca2+ channels. Finally, it concludes with neuroprotection, neuronal development, and memory formation [140, 142].
Insulin signaling is also one of the common molecular targets that gets disrupted in both T2DM and PD. Studies have shown that there is decreased level of insulin signaling markers and increased level of its negative regulator PTEN in the SNpc regions of PD brain compared to control. It suggests that restoring insulin pathway or inhibiting nuclear aggregation of PTEN could be possible therapeutic target against both T2DM and PD [143].
Since there are many shared pathways between T2DM and PD, so, use of anti-parkinsonian therapy for T2DM and antidiabetic therapy for PD or vice versa in which research is going on. For example, DA agonist is most commonly used for the treatment of motor symptoms associated with PD. L-DOPA is the most commonly used DA agonist. Studies have shown that treatment of PD patients with L-DOPA decreased insulin secretion. Bromocriptine is another DA agonist used commonly for treatment of PD. It was the first DA-ergic drug approved for the treatment of T2DM. Studies have shown that its treatment improved glucose tolerance by suppressing growth hormone release from pituitary adenomas in obese T2DM patients as well as in non-diabetic obese human and animals. It resulted in decreasing growth hormone–mediated antagonism action of insulin [144]. Thus, use of DA agonist is also a possible therapeutic strategy against both T2DM and PD.
Pioglitazone, a peroxisome proliferator-activated receptor (PPARs) agonist that improves hyperglycemia and ameliorates insulin resistance, may also be a potential therapeutic agent for PD. It has been reported that a PPAR pathway has a crucial role in the pathogenesis of PD. Studies have shown that pioglitazone rescued DA-ergic neurons through microglia inhibition in a PD model [145, 146]. Other studies have suggested that combinations of pioglitazone and statin modulate several inflammatory pathways (e.g., iNOS expression, cyclooxygenase-2 (COX-2) activity, and inhibition of NF-kB) and offer anti-inflammatory-mediated neuroprotection [146]. Studies have also shown that a combination of pioglitazone and statin lowers the incidence of PD in T2DM patients [145]. This suggests the PPAR pathway as a possible molecular target for therapeutics against both T2DM and PD. Illustrative representation of therapeutic action of antidiabetic agents against PD pathogenesis is given in Fig. 4.

Illustrative representation of therapeutic action of antidiabetic agents against PD pathogenesis. Abbreviations: AMPK, adenosine monophosphate-activated protein kinase; BDNF, brain-derived neurotrophic factor; cAMP, adenosine 3′,5′-cyclic monophosphate; GLP-1, glucagon-like peptide 1; IR, insulin receptor; LC3-II, light chain 3-II; MAPK1, mitogen-activated protein kinase; PKB, protein kinase B; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; PPARs, peroxisome proliferator-activated receptors
Conclusions
In conclusion, implications in insulin signaling dysregulate blood glucose homeostasis and insulin resistance in T2DM and cause DA-ergic cell death in PD. ER stress results in dysregulations in the β-cell functioning and survival in T2DM and impaired Ca2+ hemostasis resulting in DA-ergic neuronal loss in PD. Mitochondrial dysfunction decreases oxygen consumption and suppresses glucose-stimulated insulin secretion in T2DM and decreases mitochondrial complex I activity, MPP, and ATP levels in PD. Dysregulated autophagy results in amyloid aggregation, i.e., amylin protein and α-synuclein in T2DM and PD, respectively. And dysfunctional UPS may decrease insulin secretion in the pancreatic β-cell and neurotoxicity in PD. Thus, T2DM and PD share some common but independent molecular pathways that play a crucial role in their progression and etiopathogenesis and can therefore provide common and alternate therapeutic approaches for treating T2DM and PD.
Data Availability
No data is used in this review.
Abbreviations
- 6-OHDA:
-
6 Hydroxy dopamine
- ABA:
-
Abscisic acid
- AIM2 inflammasome:
-
Absent in melanoma 2 inflammasome
- AKT:
-
Protein kinase B
- AMP:
-
Adenosine monophosphate
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- ATF4:
-
Activating transcription factor 4
- ATF6:
-
Activating transcription factor 6
- Atg:
-
Autophagy related
- BBB:
-
Blood-brain barrier
- Bcl-2:
-
B cell lymphoma 2
- BDNF:
-
Brain-derived neurotrophic factor
- BiP:
-
Immunoglobulin heavy chain binding protein
- cAMP:
-
Cyclic AMP
- CAT:
-
Catalase
- CHOP:
-
C/EBP homologous protein
- CMA:
-
Chaperone-mediated autophagy
- COX-2:
-
Cyclooxygenase-2
- CREB:
-
CAMP response element binding protein
- CSF:
-
Cerebrospinal fluid
- DA:
-
Dopamine
- DA-ergic:
-
Dopaminergic
- DAMP:
-
Damaged-associated molecular pattern
- DJ-1:
-
DnaJ-1
- DM:
-
Diabetes mellitus
- Drp1:
-
Dynamin-related protein-1
- EIF2AK3:
-
Eukaryotic translation initiation factor 2 alpha kinase 3
- eIF2α:
-
Eukaryotic initiation factor 2 alpha
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinases ERK1
- ETC:
-
Electron transport chain
- Fis1:
-
Fission 1
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide–l
- GLP-1R:
-
GLP-1 receptor
- GLUT4:
-
Glucose transporters type 4
- GPCR:
-
G protein-coupled receptor
- Grp78 protein:
-
Glucose-regulated protein 78
- GSK3-β:
-
Glycogen synthase kinase-3beta
- hA:
-
Human amylin
- HFD:
-
High-fat diet
- h-IAPP:
-
Human islet amyloid polypeptide
- Hrd1:
-
HMG–CoA reductase degradation protein-1
- IAPP:
-
Islet amyloid polypeptide
- Iba-1:
-
Ionized calcium binding adaptor molecule 1
- IFN-γ:
-
Interferon gamma
- IkBα:
-
NF-kappa-B inhibitor alpha (I-kappa-B-alpha) (IkB-alpha)
- IKK-β:
-
Inhibitor of nuclear factor-kappaB (IkappaB) kinase beta
- IL:
-
Interleukin
- IL-18R α/β:
-
IL-18 receptor α/β
- iNOS:
-
Inducible nitric oxide synthase
- IPD:
-
Idiopathic PD
- IPL:
-
Inferior parietal lobule
- IRE1α:
-
Inositol requiring enzyme 1 α
- IRN:
-
Isorhynchophylline
- IRS:
-
Insulin receptor substrate
- LAMP1:
-
Lysosomal-associated membrane protein type 1
- LB:
-
Lewy body
- LC3:
-
Microtubule-associated protein 1A/1B-light chain 3
- LC:
-
Locus coeruleus
- L-DOPA:
-
Levodopa
- LRRK2:
-
Leucine-rich repeat kinase 2
- MafA:
-
Musculoaponeurotic fibrosarcoma oncogene family A
- MAPK:
-
Mitogen-activated protein kinase
- MFG:
-
Middle frontal gyrus
- MFN:
-
Mitochondrial mitofusin
- MHC:
-
Major histocompatibility complex
- Miro 1:
-
Mitochondrial rho GTPase 1
- MMP:
-
Mitochondrial membrane potential
- MPP+ :
-
1-Methyl-4-phenylpyridinium
- MPTP:
-
1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydrodropyridine
- MSK1:
-
Mitogen and stress-activated kinase-1
- mTOR:
-
Mechanistic target of rapamycin
- NADH:
-
Nicotinamide adenine dinucleotide (NAD) + hydrogen (H)
- NADPH-oxidase:
-
Nicotinamide adenine nucleotide phosphate oxidase
- NF-ΚB:
-
Nuclear factor-κB
- NLRP3 inflammasome:
-
NOD-, LRR-, and pyrin domain-containing 3 inflammasome
- Nrf2:
-
Nuclear factor erythroid 2–related factor 2
- P2X4R:
-
P2X purinoceptor 4
- P2X7R:
-
P2X purinoceptor 7
- Pael-R:
-
Parkin-associated endothelin receptor-like receptor
- Park2:
-
Ub ligase Parkin
- PARP:
-
Poly ADP-ribose polymerase
- PD:
-
Parkinson’s disease
- PERK:
-
Pancreatic ER kinase (PKR)–like ER kinase
- PGC1-α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1
- PI3K:
-
Phosphatidylinositol 3-phosphate kinase
- PINK1:
-
PTEN-induced putative phosphatase 1
- PKA:
-
Protein kinase A
- PKB:
-
Protein kinase B
- PPARs:
-
Peroxisome proliferator-activated receptors
- PQ:
-
Paraquat
- PTEN:
-
Phosphatase and tensin homolog
- Rab10:
-
Ras-related protein Rab 10
- ROS:
-
Reactive oxygen species
- SNCA:
-
Synuclein alpha
- SNpc:
-
Substantia nigra pars compacta
- SOCE:
-
Store-operated calcium entry (SOCE)
- SOCS:
-
Suppressor of cytokine signaling
- SOD:
-
Superoxide dismutase
- STZ:
-
Streptozotocin
- SUMO-1:
-
Substrate for ubiquitin-like modifier-1
- T2DM:
-
Type 2 diabetes mellitus
- TFB1M:
-
Transcription factor B1 mitochondrial
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumor necrosis factor-α
- TOM20:
-
Translocase of the outer membrane 20
- TRPC1:
-
Transient receptor potential channel 1
- UCH-L1:
-
Ubiquitin carboxy-terminal hydrolase L1
- UCP2:
-
Uncoupling protein 2
- UPDRS:
-
Unified Parkinson Disease Rating Scale
- UPR:
-
Unfolded protein response
- UPS:
-
Ubiquitin proteasome system
- VDAC:
-
Voltage-dependent anion-selective channel
- WFS1:
-
Wolfram syndrome gene 1
- XBP1:
-
X-box-binding protein 1
References
Hong CT, Chen KY, Wang W, Chiu JY, Wu D, Chao TY, Hu CJ, Chau KD, Bamodu OA (2020) Insulin resistance promotes Parkinson’s disease through aberrant expression of α-synuclein, mitochondrial dysfunction, and deregulation of the polo-like kinase 2 signaling. Cells 9(3):740. https://doi.org/10.3390/cells9030740
Xuan WT, Wang H, Zhou P, Ye T, Gao HW, Ye S, Wang JH, Chen ML, Song H, Wang Y, Cai B (2020) Berberine ameliorates rats model of combined s disease and type diabetes mellitus via the suppression of endoplasmic reticulum stress. Biotech 10(8):359. https://doi.org/10.1007/s13205-020-02354-7
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE (2017) Parkinson disease. Nat Rev Dis Primers 3:17013
Yang YW, Hsieh TF, Li CI, Liu CS, Lin WY, Chiang JH, Li TC, Lin CC (2017) Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Medicine (Baltimore) 96(3):e5921. https://doi.org/10.1097/MD.0000000000005921
Cereda E, Barichella M, Pedrolli C, Klersy C, Cassani E, Caccialanza R, Pezzoli G (2011) Diabetes and risk of Parkinson’s disease: a systematic review and meta-analysis. Diabetes Care 34(12):2614–2623. https://doi.org/10.2337/dc11-1584
Santiago JA, Potashkin JA (2013) Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol Med 19(3):176–186. https://doi.org/10.1016/j.molmed.2013.01.002
Katila N, Bhurtel S, Shadfar S, Srivastav S, Neupane S, Ojha U, Jeong GS, Choi DY (2017) Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 125:396–407. https://doi.org/10.1016/j.neuropharm.2017.08.015
Grieco M, Giorgi A, Gentile MC, d’Erme M, Morano S, Maras B, Filardi T (2019) Glucagon-like peptide-1: a focus on neurodegenerative diseases. Front Neurosci 18(13):1112. https://doi.org/10.3389/fnins.2019.01112
Sandyk R (1993) The relationship between diabetes mellitus and Parkinson’s disease. Int J Neurosci 69(1–4):125–130. https://doi.org/10.3109/00207459309003322
Pagano G, Polychronis S, Wilson H, Giordano B, Ferrara N, Niccolini F, Politis M (2018) Diabetes mellitus and Parkinson disease. Neurology 8 90(19):1654–1662. https://doi.org/10.1212/WNL.0000000000005475
XuQ PY, Huang X, Hollenbeck A, Blair A, Schatzkin A, Chen H (2011) Diabetes and risk of Parkinson’s disease. Diabetes Care 34(4):910–915. https://doi.org/10.2337/dc10-1922
Qi L, Cornelis MC, Kraft P, Stanya KJ, Linda Kao WH, Pankow JS, Dupuis J, Florez JC, Fox CS, Paré G, Sun Q, Girman CJ, Laurie CC, Mirel DB, Manolio TA, Chasman DI, Boerwinkle E, Ridker PM, Hunter DJ, Meigs JB, Lee CH, Hu FB, van Dam RM (2010). Meta-Analysis of Glucose and Insulin-related traits Consortium (MAGIC); Diabetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium. Genetic variants at 2q24 are associated with susceptibility to type 2 diabetes. Hum Mol Genet 19 (13):2706–15 https://doi.org/10.1093/hmg/ddq156
Xiromerisiou G, Hadjigeorgiou GM, Papadimitriou A, Katsarogiannis E, Gourbali V, Singleton AB (2008) Association between AKT1 gene and Parkinson’s disease a protective haplotype. Neurosci Lett 436(2):232–4. https://doi.org/10.1016/j.neulet.2008.03.026
Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L, Marchetti P, Lammert E (2012) Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J Mol Cell Biol 4(4):221–230. https://doi.org/10.1093/jmcb/mjs025
Fukushima T, Tan X, Luo Y, Kanda H (2011) Serum vitamins and heavy metals in blood and urine, and the correlations among them in Parkinson’s disease patients in China. Neuroepidemiology 36(4):240–244. https://doi.org/10.1159/000328253
Yue X, Li H, Yan H, Zhang P, Chang L, Li T (2016) Risk of Parkinson disease in diabetes mellitus: an updated meta-analysis of population-based cohort studies. Medicine (Baltimore) 95(18):e3549. https://doi.org/10.1097/MD.0000000000003549
Santos García D, Suárez Castro E, Expósito I, de Deus T, Tuñas C, Aneiros A, López Fernández M, Núñez Arias D, Bermúdez Torres M (2017) Comorbid conditions associated with Parkinson’s disease: a longitudinal and comparative study with Alzheimer disease and control subjects. J Neurol Sci 15(373):210–215. https://doi.org/10.1016/j.jns.2016.12.046
Cheong JLY, de Pablo-Fernandez E, Foltynie T, Noyce AJ (2020) The association between type 2 diabetes mellitus and Parkinson’s disease. J Parkinsons Dis 10(3):775–789. https://doi.org/10.3233/JPD-191900
Cereda E, Barichella M, Cassani E, Caccialanza R, Pezzoli G (2012) Clinical features of Parkinson disease when onset of diabetes came first a case control study. Neurology 78(19):1507–11. https://doi.org/10.1212/WNL.0b013e3182553cc9
Bohnen NI, Kotagal V, Müller ML, Koeppe RA, Scott PJ, Albin RL, Frey KA, Petrou M (2014) Diabetes mellitus is independently associated with more severe cognitive impairment in Parkinson disease. Parkinsonism Relat Disord 20(12):1394–8. https://doi.org/10.1016/j.parkreldis.2014.10.008
Hölscher C (2020) Brain insulin resistance: role in neurodegenerative disease and potential for targeting. Expert Opin Investig Drugs 29(4):333–348. https://doi.org/10.1080/13543784.2020.1738383
Sirangelo I, Borriello M, Vilasi S, Iannuzzi C (2020) Hydroxytyrosol inhibits protein oligomerization and amyloid aggregation in human insulin. Int J Mol Sci 21(13):4636. https://doi.org/10.3390/ijms21134636
Cho JH, Kim JW, Shin JA, Shin J, Yoon KH (2011) β-cell mass in people with type 2 diabetes. J Diabetes Investig 2(1):6–17. https://doi.org/10.1111/j.2040-1124.2010.00072.x
Love KM, Liu J, Regensteiner JG, Reusch JEB, Liu Z (2020) GLP-1 and insulin regulation of skeletal and cardiac muscle microvascular perfusion in type 2 diabetes. J Diabetes 12(7):488–498. https://doi.org/10.1111/1753-0407.13045
Mukherjee A, Morales-Scheihing D, Butler PC, Soto C (2015) Type 2 diabetes as a protein misfolding disease. Trends Mol Med 21(7):439–449. https://doi.org/10.1016/j.molmed.2015.04.005
Bosco D, Plastino M, Cristiano D, Colica C, Ermio C, De Bartolo M, Mungari P, Fonte G, Consoli D, Consoli A, Fava A (2012) Dementia is associated with insulin resistance in patients with Parkinson’s disease. J Neurol Sci 315(1–2):39–43. https://doi.org/10.1016/j.jns.2011.12.008
Ahmad MH, Fatima M, Mondal AC (2019) Role of hypothalamic-pituitary-adrenal axis, hypothalamic-pituitary-gonadal axis and insulin signaling in the pathophysiology of Alzheimer’s disease. Neuropsychobiology 77(4):197–205. https://doi.org/10.1159/000495521
Hong CT, Chen KY, Wang W, Chiu JY, Wu D, Chao TY, Hu CJ, Chau KD, Bamodu OA (2020) Insulin resistance promotes Parkinson’s disease through aberrant expression of α-synuclein, mitochondrial dysfunction, and deregulation of the polo-like kinase 2 signaling. Cells 9(3):740. https://doi.org/10.3390/cells9030740
Craft S, Watson GS (2004) Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 3(3):169–78. https://doi.org/10.1016/S1474-4422(04)00681-7
Sekar S, Taghibiglou C (2018) Elevated nuclear phosphatase and tensin homolog (PTEN) and altered insulin signaling in substantia nigral region of patients with Parkinson’s disease. Neurosci Lett 666:139–143. https://doi.org/10.1016/j.neulet.2017.12.049
Athauda D, Foltynie T (2016) Insulin resistance and Parkinson’s disease: a new target for disease modification? Prog Neurobiol 145–146:98–120. https://doi.org/10.1016/j.pneurobio.2016.10.001
Funk N, Munz M, Ott T, Brockmann K, Wenninger-Weinzierl A, Kühn R, Vogt-Weisenhorn D, Giesert F, Wurst W, Gasser T, Biskup S (2019) The Parkinson’s disease-linked leucine-rich repeat kinase 2 (LRRK2) is required for insulin-stimulated translocation of GLUT4. Sci Rep 9(1):4515. https://doi.org/10.1038/s41598-019-40808-y
Wang T, Yuan F, Chen Z, Zhu S, Chang Z, Yang W, Deng B, Que R, Cao P, Chao Y, Chan L, Pan Y, Wang Y, Xu L, Lyu Q, Chan P, Yenari MA, Tan EK, Wang Q. Vascular, inflammatory and metabolic risk factors in relation to dementia in Parkinson’s disease patients with type 2 diabetes mellitus. Aging (Albany NY) 12(15):15682–15704. https://doi.org/10.18632/aging.103776.
Yuan T, Yang T, Chen H, Fu D, Hu Y, Wang J, Yuan Q, Yu H, Xu W, Xie X (2019) New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol 20:247–260. https://doi.org/10.1016/j.redox.2018.09.025
Ahmad MH, Rizvi MA, Fatima M, Mondal AC (2021) Pathophysiological implications of neuroinflammation mediated HPA axis dysregulation in the prognosis of cancer and depression. Mol Cell Endocrinol 520:111093. https://doi.org/10.1016/j.mce.2020.111093
Lontchi-Yimagou E, Sobngwi E, Matsha TE, Kengne AP (2013) Diabetes mellitus and inflammation. Curr Diab Rep 13(3):435–444. https://doi.org/10.1007/s11892-013-0375-y
Subba R, Sandhir R, Singh SP, Mallick BN, Mondal AC (2021) Pathophysiology linking depression and type 2 diabetes: Psychotherapy, physical exercise, and fecal microbiome transplantation as damage control. Eur J Neurosci 53(8):2870–2900. https://doi.org/10.1111/ejn.15136
Kim DS, Choi HI, Wang Y, Luo Y, Hoffer BJ, Greig NH (2017) A new treatment strategy for Parkinson’s disease through the gut-brain axis: the glucagon-like peptide-1 receptor pathway. Cell Transplant 26(9):1560–1571. https://doi.org/10.1177/0963689717721234
Badawi A, Klip A, Haddad P, Cole DE, Bailo BG, El-Sohemy A, Karmali M (2010) Type 2 diabetes mellitus and inflammation: prospects for biomarkers of risk and nutritional intervention. Diabetes Metab Syndr Obes 3:173–186. https://doi.org/10.2147/dmsott.s9089
Conti P, Ronconi G, Kritas SK, Caraffa A, Theoharides TC (2018) Activated mast cells mediate low-grade inflammation in type 2 diabetes: interleukin-37 could be beneficial. Can J Diabetes 42(5):568–573. https://doi.org/10.1016/j.jcjd.2018.01.008
Cheng KT, Ong HL, Liu X, Ambudkar IS (2013) Contribution and regulation of TRPC channels in store-operated Ca2+ entry. Curr Top Membr 71:149–179. https://doi.org/10.1016/B978-0-12-407870-3.00007-X.PMID:23890115;
Wang J, Li Y, Lai K, Zhong Q, Demin KA, Kalueff AV, Song C (2020) High-glucose/high-cholesterol diet in zebrafish evokes diabetic and affective pathogenesis: the role of peripheral and central inflammation, microglia and apoptosis. Prog Neuropsychopharmacol Biol Psychiatry 96:109752. https://doi.org/10.1016/j.pnpbp.2019.109752
Wang D, Wang H, Gao H, Zhang H, Zhang H, Wang Q, Sun Z (2011) P2X7 receptor mediates NLRP3 inflammasome activation in depression and diabetes. Cell Biosci 10:28. https://doi.org/10.1186/s13578-020-00388-1
Sun X, Han F, Yi J, Han L, Wang B (2011) Effect of aspirin on the expression of hepatocyte NF-κB and serum TNF-α in streptozotocin-induced type 2 diabetic rats. J Korean Med Sci. 26(6):765–70. https://doi.org/10.3346/jkms.2011.26.6.765
Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, Shoelson SE, Shulman GI (2001) Prevention of fat-induced insulin resistance by salicylate. J Clin Invest 108(3):437–446. https://doi.org/10.1172/JCI11559
Pajares M, Rojo IA, Manda G, Boscá L, Cuadrado A (2020) Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells 9(7):1687. https://doi.org/10.3390/cells9071687
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7(4):354–365. https://doi.org/10.1016/j.nurt.2010.05.014
Chatterjee K, Roy A, Banerjee R, Choudhury S, Mondal B, Halder S, Basu P, Shubham S, Dey S, Kumar H (2020) Inflammasome and α-synuclein in Parkinson’s disease: a cross-sectional study. J Neuroimmunol 338:577089. https://doi.org/10.1016/j.jneuroim.2019.577089
Philips T, Robberecht W (2011) Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol 10(3):253–263. https://doi.org/10.1016/S1474-4422(11)70015-1
Rodríguez-Cruz A, Romo-Mancillas A, Mendiola-Precoma J, Escobar-Cabrera JE, García-Alcocer G, Berumen LC (2019) Effect of valerenic acid on neuroinflammation in a MPTP-induced mouse model of Parkinson’s disease. IBRO Rep 8:28–35. https://doi.org/10.1016/j.ibror.2019.12.002
Chen H, O’Reilly EJ, Schwarzschild MA, Ascherio A (2008) Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am J Epidemiol 167(1):90–95. https://doi.org/10.1093/aje/kwm260
Lee S, Lee Y, Ha S, Chung HY, Kim H, Hur JS, Lee J (2020) Anti-inflammatory effects of usnic acid in an MPTP-induced mouse model of Parkinson’s disease. Brain Res 1730:146642. https://doi.org/10.1016/j.brainres.2019.146642
O’Neill E, Yssel JD, McNamara C, Harkin A (2020) Pharmacological targeting of β2-adrenoceptors is neuroprotective in the LPS inflammatory rat model of Parkinson’s disease. Br J Pharmacol 177(2):282–297. https://doi.org/10.1111/bph.14862
Las G, Oliveira MF, Shirihai OS (2020) Emerging roles of β-cell mitochondria in type-2-diabetes. Mol Aspects Med 71:100843. https://doi.org/10.1016/j.mam.2019.100843
Kajihara N, Kukidome D, Sada K, Motoshima H, Furukawa N, Matsumura T, Nishikawa T, Araki E (2017) Low glucose induces mitochondrial reactive oxygen species via fatty acid oxidation in bovine aortic endothelial cells. J Diabetes Investig 8(6):750–761. https://doi.org/10.1111/jdi.12678
Sharoyko VV, Abels M, Sun J, Nicholas LM, Mollet IG, Stamenkovic JA, Göhring I, Malmgren S, Storm P, Fadista J, Spégel P, Metodiev MD, Larsson NG, Eliasson L, Wierup N, Mulder H (2014) Loss of TFB1M results in mitochondrial dysfunction that leads to impaired insulin secretion and diabetes. Hum Mol Genet 23(21):5733–5749. https://doi.org/10.1093/hmg/ddu288
Luo X, Li R, Yan LJ (2015) Roles of pyruvate, NADH, and mitochondrial complex I in redox balance and imbalance in β cell function and dysfunction. J Diabetes Res 2015:512618. https://doi.org/10.1155/2015/512618
Bose A, Beal MF (2016) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139(Suppl 1):216–231. https://doi.org/10.1111/jnc.13731
Park JS, Davis RL, Sue CM (2018) Mitochondrial dysfunction in Parkinson’s disease: new mechanistic insights and therapeutic perspectives. Curr Neurol Neurosci Rep 18(5):21. https://doi.org/10.1007/s11910-018-0829-3
Filichia E, Hoffer B, Qi X, Luo Y (2016) Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci Rep 6:32656. https://doi.org/10.1038/srep32656
Valdinocci D, Simões RF, Kovarova J, Cunha-Oliveira T, Neuzil J, Pountney DL (2019) Intracellular and intercellular mitochondrial dynamics in Parkinson’s disease. Front Neurosci 13:930. https://doi.org/10.3389/fnins.2019.00930
Subramaniam SR, Chesselet MF (2013) Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol 106–107:17–32. https://doi.org/10.1016/j.pneurobio.2013.04.004
Schapira AH (2007) Mitochondrial dysfunction in Parkinson’s disease. Cell Death Differ 14(7):1261–1266. https://doi.org/10.1038/sj.cdd.4402160
Lev N, Roncevic D, Ickowicz D, Melamed E, Offen D (2006) Role of DJ-1 in Parkinson’s disease. J Mol Neurosci 29(3):215–225. https://doi.org/10.1385/jmn:29:3:215
Martin I, Kim JW, Dawson VL, Dawson TM (2014) LRRK2 pathobiology in Parkinson’s disease. J Neurochem 131(5):554–565. https://doi.org/10.1111/jnc.12949
Beal MF (2003) Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci 991:120–131. https://doi.org/10.1111/j.1749-6632.2003.tb07470.x
Kones R (2010) Parkinson’s disease: mitochondrial molecular pathology, inflammation, statins, and therapeutic neuroprotective nutrition. Nutr Clin Pract 25(4):371–389. https://doi.org/10.1177/0884533610373932
Bae JH, Jo SI, Kim SJ, Lee JM, Jeong JH, Kang JS, Cho NJ, Kim SS, Lee EY, Moon JS (2019) Circulating cell-free mtDNA contributes to AIM2 inflammasome-mediated chronic inflammation in patients with type 2 diabetes. Cells 8(4):328. https://doi.org/10.3390/cells8040328
Rasheed M, Liang J, Wang C, Deng Y, Chen Z (2021) Epigenetic regulation of neuroinflammation in Parkinson’s disease. Int J Mol Sci 22(9):4956. https://doi.org/10.3390/ijms22094956
Naoi M, Maruyama W, Shamoto-Nagai M (2020) Rasagiline and selegiline modulate mitochondrial homeostasis, intervene apoptosis system and mitigate α-synuclein cytotoxicity in disease-modifying therapy for Parkinson’s disease. J Neural Transm (Vienna) 127(2):131–147. https://doi.org/10.1007/s00702-020-02150-w
Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC, Cass WA, Sullivan PG, Bing G (2007) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem 100(5):1375–1386. https://doi.org/10.1111/j.1471-4159.2006.04327.x
Whitton PS (2007) Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol 150(8):963–976. https://doi.org/10.1038/sj.bjp.0707167
Wu Y, Reece EA, Zhong J, Dong D, Shen WB, Harman CR, Yang P (2016) Type 2 diabetes mellitus induces congenital heart defects in murine embryos by increasing oxidative stress, endoplasmic reticulum stress, and apoptosis. Am J Obstet Gynecol 215(3):366.e1-366.e10. https://doi.org/10.1016/j.ajog.2016.03.036
Choi SK, Lim M, Yeon SI, Lee YH (2016) Inhibition of endoplasmic reticulum stress improves coronary artery function in type 2 diabetic mice. Exp Physiol 101(6):768–777. https://doi.org/10.1113/EP085508
Nomura J, Hosoi T, Kaneko M, Ozawa K, Nishi A, Nomura Y (2016) Neuroprotection by endoplasmic reticulum stress-induced HRD1 and chaperones: possible therapeutic targets for Alzheimer’s and Parkinson’s disease. Med Sci (Basel) 4(3):14. https://doi.org/10.3390/medsci4030014
Fonseca SG, Gromada J, Urano F (2011) Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol Metab 22(7):266–274. https://doi.org/10.1016/j.tem.2011.02.008
Hu Y, Liu J, Yuan Y, Chen J, Cheng S, Wang H, Xu Y (2018) Sodium butyrate mitigates type 2 diabetes by inhibiting PERK-CHOP pathway of endoplasmic reticulum stress. Environ Toxicol Pharmacol 64:112–121. https://doi.org/10.1016/j.etap.2018.09.002
Natrus LV, Osadchuk YS, Lisakovska OO, Labudzinskyi DO, Klys YG, Chaikovsky YB (2022) Effect of propionic acid on diabetes-induced impairment of unfolded protein response signaling and astrocyte/microglia crosstalk in rat ventromedial nucleus of the hypothalamus. Neural Plast 22(2022):6404964. https://doi.org/10.1155/2022/6404964
Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29(1):42–61. https://doi.org/10.1210/er.2007-0015
Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ (2007) Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50(4):752–763. https://doi.org/10.1007/s00125-006-0590-z
Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, Filipponi F, Weir GC, Eizirik DL, Cnop M (2007) The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 50(12):2486–2494. https://doi.org/10.1007/s00125-007-0816-8
Farombi EO, Awogbindin IO, Olorunkalu PD, Ogbuewu E, Oyetunde BF, Agedah AE, Adeniyi PA (2020) Kolaviron protects against nigrostriatal degeneration and gut oxidative damage in a stereotaxic rotenone model of Parkinson’s disease. Psychopharmacology 237(11):3225–3236. https://doi.org/10.1007/s00213-020-05605-w
Li XM, Zhang XJ, Dong MX (2017) Isorhynchophylline attenuates MPP+-induced apoptosis through endoplasmic reticulum stress- and mitochondria-dependent pathways in PC12 cells: involvement of antioxidant activity. Neuromolecular Med 19(4):480–492. https://doi.org/10.1007/s12017-017-8462
Ning B, Zhang Q, Wang N, Deng M, Fang Y (2019) β-Asarone regulates ER stress and autophagy via inhibition of the PERK/CHOP/Bcl-2/Beclin-1 pathway in 6-OHDA-induced parkinsonian rats. Neurochem Res 44(5):1159–1166. https://doi.org/10.1007/s11064-019-02757-w
Gorbatyuk MS, Shabashvili A, Chen W, Meyers C, Sullivan LF, Salganik M, Lin JH, Lewin AS, Muzyczka N, Gorbatyuk OS (2012) Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol Ther 20(7):1327–1337. https://doi.org/10.1038/mt.2012.28
Omura T, Kaneko M, Okuma Y, Orba Y, Nagashima K, Takahashi R, Fujitani N, Matsumura S, Hata A, Kubota K, Murahashi K, Uehara T, Nomura Y (2006) A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of Parkin. J Neurochem 99(6):1456–1469. https://doi.org/10.1111/j.1471-4159.2006.04155.x
Tong Y, Mukhamejanova Z, Zheng Y, Wen T, Xu F, Pang J (2021) Marine-derived xyloketal compound ameliorates MPP+-induced neuronal injury through regulating of the IRE1/XBP1 signaling pathway. ACS Chem Neurosci 12(16):3101–3111. https://doi.org/10.1021/acschemneuro.1c00362
Valdés P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, Martinez A, Galleguillos D, Armentano D, Schneider BL, Hetz C (2014) Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci U S A 111(18):6804–6809. https://doi.org/10.1073/pnas.1321845111
Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, Mizuguchi H, Momota H, Kishibuchi R, Hashimoto T, Wada D, Kitagawa H, Watanabe TK (2009) Protective effect against Parkinson’s disease-related insults through the activation of XBP1. Brain Res 1257:16–24. https://doi.org/10.1016/j.brainres.2008.11.104
Sun Y, Selvaraj S, Pandey S, Humphrey KM, Foster JD, Wu M, Watt JA, Singh BB, Ohm JE (2018) MPP+ decreases store-operated calcium entry and TRPC1 expression in mesenchymal stem cell derived dopaminergic neurons. Sci Rep 8(1):11715. https://doi.org/10.1038/s41598-018-29528-x
Wang T, Li X, Yang D, Zhang H, Zhao P, Fu J, Yao B, Zhou Z (2015) ER stress and ER stress-mediated apoptosis are involved in manganese-induced neurotoxicity in the rat striatum in vivo. Neurotoxicology 48:109–119. https://doi.org/10.1016/j.neuro.2015.02.007
Saha S, Panigrahi DP, Patil S, Bhutia SK (2018) Autophagy in health and disease: a comprehensive review. Biomed Pharmacother 104:485–495. https://doi.org/10.1016/j.biopha.2018.05.007
Rocha M, Apostolova N, Diaz-Rua R, Muntane J, Victor VM (2020) Mitochondria and T2D: role of autophagy, ER stress, and inflammasome. Trends Endocrinol Metab 31(10):725–741. https://doi.org/10.1016/j.tem.2020.03.004
Marasco MR, Linnemann AK (2018) β-Cell autophagy in diabetes pathogenesis. Endocrinology 159(5):2127–2141. https://doi.org/10.1210/en.2017-03273
Cheng Y, Ren X, Hait WN, Yang JM (2013) Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev 65(4):1162–1197. https://doi.org/10.1124/pr.112.007120
Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, Marselli L, Masiello P, Marchetti P (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52(6):1083–1086. https://doi.org/10.1007/s00125-009-1347-2
Choi SK, Kwon Y, Byeon S, Lee YH (2020) Stimulation of autophagy improves vascular function in the mesenteric arteries of type 2 diabetic mice. Exp Physiol 105(1):192–200. https://doi.org/10.1113/EP087737
Quan W, Jung HS, Lee MS (2013) Role of autophagy in the progression from obesity to diabetes and in the control of energy balance. Arch Pharm Res 36(2):223–229. https://doi.org/10.1007/s12272-013-0024-7
Wang Y, He D, Ni C, Zhou H, Wu S, Xue Z, Zhou Z (2016) Vitamin D induces autophagy of pancreatic β-cells and enhances insulin secretion. Mol Med Rep 14(3):2644–2650. https://doi.org/10.3892/mmr.2016.5531
Olanow CW, Brundin P (2013) Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord 28(1):31–40. https://doi.org/10.1002/mds.25373
Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK (2019) How is alpha-synuclein cleared from the cell? J Neurochem 150(5):577–590. https://doi.org/10.1111/jnc.14704
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305(5688):1292–1295. https://doi.org/10.1126/science
Bang Y, Kim KS, Seol W, Choi HJ (2016) LRRK2 interferes with aggresome formation for autophagic clearance. Mol Cell Neurosci 75:71–80. https://doi.org/10.1016/j.mcn.2016.06.007
Yue Z, Yang XW (2013) Dangerous duet: LRRK2 and α-synuclein jam at CMA. Nat Neurosci 16(4):375–377. https://doi.org/10.1038/nn.3361
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147(4):893–906. https://doi.org/10.1016/j.cell.2011.10.018
Bozi LHM, Campos JC (2017) Targeting the ubiquitin proteasome system in diabetic cardiomyopathy. J Mol Cell Cardiol 109:61–63. https://doi.org/10.1016/j.yjmcc.2017.06.009
McKinnon C, De Snoo ML, Gondard E, Neudorfer C, Chau H, Ngana SG, O’Hara DM, Brotchie JM, Koprich JB, Lozano AM, Kalia LV, Kalia SK (2020) Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol Commun 8(1):17. https://doi.org/10.1186/s40478-020-0894-0
Kawaguchi M, Minami K, Nagashima K, Seino S (2006) Essential role of ubiquitin-proteasome system in normal regulation of insulin secretion. J Biol Chem 281(19):13015–13020. https://doi.org/10.1074/jbc.M601228200
Costes S, Vandewalle B, Tourrel-Cuzin C, Broca C, Linck N, Bertrand G, Kerr-Conte J, Portha B, Pattou F, Bockaert J, Dalle S (2009) Degradation of cAMP-responsive element-binding protein by the ubiquitin-proteasome pathway contributes to glucotoxicity in beta-cells and human pancreatic islets. Diabetes 58(5):1105–1115. https://doi.org/10.2337/db08-0926
Gao C, Huang W, Kanasaki K, Xu Y (2014) The role of ubiquitination and sumoylation in diabetic nephropathy. Biomed Res Int 2014:160692. https://doi.org/10.1155/2014/160692
Haataja L, Gurlo T, Huang CJ, Butler PC (2008) Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev 29(3):303–316. https://doi.org/10.1210/er.2007-0037
Costes S (2018) Targeting protein misfolding to protect pancreatic beta-cells in type 2 diabetes. Curr Opin Pharmacol 43:104–110. https://doi.org/10.1016/j.coph.2018.08.016
Singh S, Trikha S, Sarkar A, Jeremic AM (2016) Proteasome regulates turnover of toxic human amylin in pancreatic cells. Biochem J 473(17):2655–2670. https://doi.org/10.1042/BCJ20160026
White MF (2002) IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab 283(3):E413–E422. https://doi.org/10.1152/ajpendo.00514.2001
Rui L, Yuan M, Frantz D, Shoelson S, White MF (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 277(44):42394–42398. https://doi.org/10.1074/jbc.C200444200
Kim SJ, DeStefano MA, Oh WJ, Wu CC, Vega-Cotto NM, Finlan M, Liu D, Su B, Jacinto E (2012) mTOR complex 2 regulates proper turnover of insulin receptor substrate-1 via the ubiquitin ligase subunit Fbw8. Mol Cell 48(6):875–887. https://doi.org/10.1016/j.molcel.2012.09.029
Pelzer C, Cabalzar K, Wolf A, Gonzalez M, Lenz G, Thome M (2013) The protease activity of the paracaspase MALT1 is controlled by monoubiquitination. Nat Immunol 14(4):337–345. https://doi.org/10.1038/ni.2540
Joshi N, Raveendran A, Nagotu S (2020) Chaperones and proteostasis: role in Parkinson’s disease. Diseases 8(2):24. https://doi.org/10.3390/diseases8020024
Momtaz S, Memariani Z, El-Senduny FF, Sanadgol N, Golab F, Katebi M, Abdolghaffari AH, Farzaei MH, Abdollahi M (2020) Targeting ubiquitin-proteasome pathway by natural products: novel therapeutic strategy for treatment of neurodegenerative diseases. Front Physiol 28(11):361. https://doi.org/10.3389/fphys.2020.00361
Spratt DE, Martinez-Torres RJ, Noh YJ, Mercier P, Manczyk N, Barber KR, Aguirre JD, Burchell L, Purkiss A, Walden H, Shaw GS (2013) A molecular explanation for the recessive nature of parkin-linked Parkinson’s disease. Nat Commun 4:1983. https://doi.org/10.1038/ncomms2983
Giguère N, Pacelli C, Saumure C, Bourque MJ, Matheoud D, Levesque D, Slack RS, Park DS, Trudeau LÉ (2018) Comparative analysis of Parkinson’s disease-associated genes in mice reveals altered survival and bioenergetics of Parkin-deficient dopamine neurons. J Biol Chem 293(25):9580–9593. https://doi.org/10.1074/jbc.RA117.000499
Kawahata I, Fukunaga K (2020) Degradation of tyrosine hydroxylase by the ubiquitin-proteasome system in the pathogenesis of Parkinson’s disease and dopa-responsive dystonia. Int J Mol Sci 21(11):3779. https://doi.org/10.3390/ijms21113779.PMID:32471089
Yun JH, Lee DH, Jeong HS, Kim HS, Ye SK, Cho CH (2021) STAT3 activation in microglia exacerbates hippocampal neuronal apoptosis in diabetic brains. J Cell Physiol 236(10):7058–7070. https://doi.org/10.1002/jcp.30373
Cunha RA, Agostinho PM (2010) Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. J Alzheimers Dis 20(Suppl 1):S95-116. https://doi.org/10.3233/JAD-2010-1408
Sánchez-Sarasúa S, Moustafa S, García-Avilés Á, López-Climent MF, Gómez-Cadenas A, Olucha-Bordonau FE, Sánchez-Pérez AM (2016) The effect of abscisic acid chronic treatment on neuroinflammatory markers and memory in a rat model of high-fat diet induced neuroinflammation. Nutr Metab (Lond) 26(13):73. https://doi.org/10.1186/s12986-016-0137-3
Infante-Garcia C, Jose Ramos-Rodriguez J, Marin-Zambrana Y, Teresa Fernandez-Ponce M, Casas L, Mantell C, Garcia-Alloza M (2017) Mango leaf extract improves central pathology and cognitive impairment in a type 2 diabetes mouse model. Brain Pathol 27(4):499–507. https://doi.org/10.1111/bpa.12433
Zhang PA, Sun Q, Li YC, Weng RX, Wu R, Zhang HH, Xu GY (2020) Overexpression of purinergic P2X4 receptors in hippocampus rescues memory impairment in rats with type 2 diabetes. Neurosci Bull 36(7):719–732. https://doi.org/10.1007/s12264-020-00478-7
Alzoubi KH, Mokhemer E, Abuirmeileh AN (2018) Beneficial effect of etazolate on depression-like behavior and learning, and memory impairment in a model of Parkinson’s disease. Behav Brain Res 17(350):109–115. https://doi.org/10.1016/j.bbr.2018.05.004
Kawashima S, Shimizu Y, Ueki Y, Matsukawa N (2021) Impairment of the visuospatial working memory in the patients with Parkinson’s disease: an fMRI study. BMC Neurol 21(1):335. https://doi.org/10.1186/s12883-021-02366-7
Souza MF, Medeiros KAAL, Lins LCRF, Bispo JMM, Gois AM, Freire MAM, Marchioro M, Santos JR (2020) Intracerebroventricular injection of deltamethrin increases locomotion activity and causes spatial working memory and dopaminergic pathway impairment in rats. Brain Res Bull 154:1–8. https://doi.org/10.1016/j.brainresbull.2019.10.002
Hanoğlu L, Ercan FB, Mantar N, Helvacı Yılmaz N, Sitrava S, Özer F, Yuluğ B (2019) Accelerated forgetting and verbal memory consolidation process in idiopathic nondement Parkinson’s disease. J Clin Neurosci 70:208–213. https://doi.org/10.1016/j.jocn.2019.08.012
Christopher L, Duff-Canning S, Koshimori Y, Segura B, Boileau I, Chen R, Lang AE, Houle S, Rusjan P, Strafella AP (2015) Salience network and parahippocampal dopamine dysfunction in memory-impaired Parkinson disease. Ann Neurol 77(2):269–280. https://doi.org/10.1002/ana.24323
Guo Z, Ruan Z, Zhang D, Liu X, Hou L, Wang Q (2022) Rotenone impairs learning and memory in mice through microglia-mediated blood brain barrier disruption and neuronal apoptosis. Chemosphere 291(Pt 2):132982. https://doi.org/10.1016/j.chemosphere.2021.132982
Hou L, Sun F, Sun W, Zhang L, Wang Q (2019) Lesion of the locus coeruleus damages learning and memory performance in paraquat and maneb-induced mouse Parkinson’s disease model. Neuroscience 1(419):129–140. https://doi.org/10.1016/j.neuroscience.2019.09.006
Wang SY, Wu SL, Chen TC, Chuang CS (2020) Antidiabetic agents for treatment of Parkinson’s disease: a meta-analysis. Int J Environ Res Public Health 17(13):4805. https://doi.org/10.3390/ijerph17134805
Ahmad MH, Fatima M, Ali M, Rizvi MA, Mondal AC (2021) Naringenin alleviates paraquat-induced dopaminergic neuronal loss in SH-SY5Y cells and a rat model of Parkinson's disease. Neuropharmacology 201:108831. https://doi.org/10.1016/j.neuropharm.2021.108831
Mor DE, Sohrabi S, Kaletsky R, Keyes W, Tartici A, Kalia V, Miller GW, Murphy CT (2020) Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc Natl Acad Sci U S A 117(42):26438–26447. https://doi.org/10.1073/pnas.2009838117
Ryu YK, Go J, Park HY, Choi YK, Seo YJ, Choi JH, Rhee M, Lee TG, Lee CH, Kim KS (2020) Metformin regulates astrocyte reactivity in Parkinson’s disease and normal aging. Neuropharmacology 15(175):108173. https://doi.org/10.1016/j.neuropharm.2020.108173
Paudel YN, Angelopoulou E, Piperi C, Shaikh MF, Othman I (2020) Emerging neuroprotective effect of metformin in Parkinson’s disease: a molecular crosstalk. Pharmacol Res 152:104593. https://doi.org/10.1016/j.phrs.2019.104593
Li Y, Li L, Hölscher C (2016) Incretin-based therapy for type 2 diabetes mellitus is promising for treating neurodegenerative diseases. Rev Neurosci 27(7):689–711. https://doi.org/10.1515/revneuro-2016-0018
Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T (2013) Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest 123(6):2730–2736. https://doi.org/10.1172/JCI68295
Hölscher C (2014) Drugs developed for treatment of diabetes show protective effects in Alzheimer’s and Parkinson’s diseases. Sheng Li Xue Bao 66(5):497–510
Sekar S, Taghibiglou C (2018) Elevated nuclear phosphatase and tensin homolog (PTEN) and altered insulin signaling in substantia nigral region of patients with Parkinson’s disease. Neurosci Lett 14(666):139–143. https://doi.org/10.1016/j.neulet.2017.12.049
Lopez Vicchi F, Luque GM, Brie B, Nogueira JP, Garcia Tornadu I, Becu-Villalobos D (2016) Dopaminergic drugs in type 2 diabetes and glucose homeostasis. Pharmacol Res 109:74–80. https://doi.org/10.1016/j.phrs.2015.12.029
Chang YH, Yen SJ, Chang YH, Wu WJ, Lin KD (2021) Pioglitazone and statins lower incidence of Parkinson disease in patients with diabetes mellitus. Eur J Neurol 28(2):430–437. https://doi.org/10.1111/ene.14542
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem 88(2):494–501. https://doi.org/10.1046/j.1471-4159.2003.02210.x
Struck LK, Rodnitzky RL, Dobson JK (1990) Stroke and its modification in Parkinson’s disease. Stroke 21(10):1395–1399. https://doi.org/10.1161/01.str.21.10.1395
Levine RL, Jones JC, Bee N (1992) Stroke and Parkinson’s disease. Stroke 23(6):839–42. https://doi.org/10.1161/01.str.23.6.839
Pressley JC, Louis ED, Tang MX, Cote L, Cohen PD, Glied S, Mayeux R (2003) The impact of comorbid disease and injuries on resource use and expenditures in parkinsonism. Neurology 14 60(1):87–93. https://doi.org/10.1212/wnl.60.1.87.24
Guttman M, Slaughter PM, Theriault ME, DeBoer DP, Naylor CD (2004) Parkinsonism in Ontario: comorbidity associated with hospitalization in a large cohort. Mov Disord 19(1):49–53. https://doi.org/10.1002/mds.10648
Powers KM, Smith-Weller T, Franklin GM, Longstreth WT Jr, Swanson PD, Checkoway H (2006) Diabetes, smoking, and other medical conditions in relation to Parkinson’s disease risk. Parkinsonism Relat Disor 12(3):185–189. https://doi.org/10.1016/j.parkreldis.2005.09.004
Leibson CL, Maraganore DM, Bower JH, Ransom JE, O’brien PC, Rocca WA, (2006) Comorbid conditions associated with Parkinson’s disease: a population-based study. Mov Disord 21(4):446–455. https://doi.org/10.1002/mds.20685
Scigliano G, Musicco M, Soliveri P, Piccolo I, Ronchetti G, Girotti F (2006) Reduced risk factors for vascular disorders in Parkinson disease patients: a case-control study. Stroke 37(5):1184–1188. https://doi.org/10.1161/01.STR.0000217384.03237.9c
Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J (2007) Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 30(4):842–847. https://doi.org/10.2337/dc06-2011
Arvanitakis Z, Wilson RS, Bienias JL, Bennett DA (2007) Diabetes and parkinsonian signs in older persons. Alzheimer Dis Assoc Disord 21(2):144–149. https://doi.org/10.1097/WAD.0b013e31805ba768
Moran, L.B. and M.B.J.N. Graeber, Towards a pathway definition of Parkinson’s disease: a complex Moran LB, Graeber MB (2008). Towards a pathway definition of Parkinson’s disease: a complex disorder with links to cancer, diabetes and inflammation. Neurogenetics. 2008 Feb;9(1):1–13. https://doi.org/10.1007/s10048-007-0116-y.
Driver JA, Smith A, Buring JE, Gaziano JM, Kurth T, Logroscino G (2008) Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 31(10):2003–2005. https://doi.org/10.2337/dc08-0688
D’Amelio M, Ragonese P, Callari G, Di Benedetto N, Palmeri B, Terruso V, Salemi G, Famoso G, Aridon P, Savettieri G (2009) Diabetes preceding Parkinson’s disease onset. A case-control study Parkinsonism Relat Disord 15(9):660–664. https://doi.org/10.1016/j.parkreldis.2009.02.013
Menon R, Farina C (2011) Shared molecular and functional frameworks among five complex human disorders: a comparative study on interactomes linked to susceptibility genes. PLoS ONE 6(4):e18660. https://doi.org/10.1371/journal.pone.0018660
Palacios N, Gao X, McCullough ML, Jacobs EJ, Patel AV, Mayo T, Schwarzschild MA, Ascherio A (2011) Obesity, diabetes, and risk of Parkinson’s disease. Mov Disord 26(12):2253–2259. https://doi.org/10.1002/mds.23855
Schernhammer E, Hansen J, Rugbjerg K, Wermuth L, Ritz B (2011) Diabetes and the risk of developing Parkinson’s disease in Denmark. Diabetes Care. 34(5):1102–8. https://doi.org/10.2337/dc10-1333
Sun Y, Chang YH, Chen HF, Su YH, Su HF, Li CY (2012) Risk of Parkinson disease onset in patients with diabetes: a 9-year population-based cohort study with age and sex stratifications. Diabetes Care 35(5):1047–1049. https://doi.org/10.2337/dc11-1511
Wahlqvist ML, Lee MS, Hsu CC, Chuang SY, Lee JT, Tsai HN (2012) Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat Disord 18(6):753–758. https://doi.org/10.1016/j.parkreldis.2012.03.010
Santiago JA, Potashkin JA (2013) Integrative network analysis unveils convergent molecular pathways in Parkinson’s disease and diabetes. PLoS ONE 8(12):e83940. https://doi.org/10.1371/journal.pone.0083940
Santiago JA, Potashkin JA (2014). System-based approaches to decode the molecular links in Parkinson’s disease and diabetes. Neurobiol Dis 72 Pt A:84–91. https://doi.org/10.1016/j.nbd.2014.03.019.
Kotagal V, Albin RL, Müller ML, Koeppe RA, Frey KA, Bohnen NI (2013) Diabetes is associated with postural instability and gait difficulty in Parkinson disease. Parkinsonism Relat Disord 19(5):522–526. https://doi.org/10.1016/j.parkreldis.2013.01.016
De Pablo-Fernandez E, Goldacre R, Pakpoor J, Noyce AJ, Warner TT (2018) Association between diabetes and subsequent Parkinson disease: a record-linkage cohort study. Neurology 91(2):e139–e142. https://doi.org/10.1212/WNL.0000000000005771
Zhu Y, Pu J, Chen Y, Zhang B (2020) Decreased risk of Parkinson’s disease in diabetic patients with thiazolidinediones therapy: an exploratory meta-analysis. PLoS ONE 14(10):e0224236. https://doi.org/10.1371/journal.pone.0224236
Jeong SM, Han K, Kim D, Rhee SY, Jang W, Shin DW (2020) Body mass index, diabetes, and the risk of Parkinson’s disease. Mov Disord 35(2):236–244. https://doi.org/10.1002/mds.27922
Acknowledgements
The authors would like to acknowledge the School of Life Sciences, JNU.
Funding
ACM highly acknowledges the financial supports from DBT (BT/PR32907/MED/122/227/2019), Ministry of Science and Technology (Govt. of India). Institutional umbrella grants like DBT-BUILDER-Level-III (BT/INF/22/SP45382/2022), and DST-FIST-II to School of Life Sciences, JNU, New Delhi. Mr. Sandeep acknowledges the financial support from CSIR Grant (09/263(1219)/2019-EMR-I. MHA acknowledges the financial support from ICMR-SRF (45/07/2019/MP/BMS).
Author information
Authors and Affiliations
Contributions
ACM conceptualized, framed, and finally edited the manuscript; Sandeep, MHA, and LR wrote the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Ethical approval is not required.
Consent for Publication
The authors have given consent for publication.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
XXXX, S., Ahmad, M.H., Rani, L. et al. Convergent Molecular Pathways in Type 2 Diabetes Mellitus and Parkinson’s Disease: Insights into Mechanisms and Pathological Consequences. Mol Neurobiol 59, 4466–4487 (2022). https://doi.org/10.1007/s12035-022-02867-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02867-7