
Abstract
At present, chronic post-surgical pain (CPSP) is difficult to prevent and cure clinically because of our lack of understanding of its mechanisms. Surgical injury induces the upregulation of voltage-gated sodium channel Nav1.7 in dorsal root ganglion (DRG) neurons, suggesting that Nav1.7 is involved in the development of CPSP. However, the mechanism leading to persistent dysregulation of Nav1.7 is largely unknown. Given that nerve growth factor (NGF) induces a long-term increase in the neuronal hyperexcitability after injury, we hypothesized that NGF might cause the long-term dysregulation of Nav1.7. In this study, we aimed to investigate whether Nav1.7 regulation by NGF is involved in CPSP and thus contributes to the specific mechanisms involved in the development of CPSP. Using conditional nociceptor-specific Nav1.7 knockout mice, we confirmed the involvement of Nav1.7 in NGF-induced pain and identified its role in the maintenance of pain behavior during long-term observations (up to 14 days). Using western blot analyses and immunostaining, we showed that NGF could trigger the upregulation of Nav1.7 expression and thus support the development of CPSP in rats. Using pharmacological approaches, we showed that the increase of Nav1.7 might be partly regulated by an NGF/TrkA-SGK1-Nedd4-2-mediated pathway. Furthermore, reversing the upregulation of Nav1.7 in DRG could alleviate spinal sensitization. Our results suggest that the maintained upregulation of Nav1.7 triggered by NGF contributes to the development of CPSP. Attenuating the dysregulation of Nav1.7 in peripheral nociceptors may be a strategy to prevent the transition from acute post-surgical pain to CPSP.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Currently, chronic post-surgical pain (CPSP) is still hard to prevent and cure in clinic and has a strong effect on the quality of life of patients [1, 2]. According to epidemiology, there are many risk factors associated with the incidence and severity of CPSP, including preoperative, intraoperative, and postoperative factors [2]. Since the mechanisms for pain chronification after surgery are complicated, the pathogenesis underlying the transition of acute post-surgical pain to CPSP is not entirely clear to date. However, it is generally believed that peripheral sensitization caused by surgical injury is the primary factor affecting the transition process [2]. Consistently, some studies revealed that the severity of acute postoperative pain was a strong risk factor for the development of CPSP [3, 4].
Peripheral sensitization is primarily related to the changes in proinflammatory molecules and ion channels. In particular, the voltage-gated sodium channels (Navs) play a key role in the sensitization of nociceptors, as they regulate the rising phase of the action potential and resting membrane potential of nociceptors [5]. Among the sodium channels, Nav1.7 is the most abundant one in primary nociceptors, and enhanced expression of Nav1.7 has been demonstrated to result in chronic neuronal hyperexcitability [6]. Several studies suggested the involvement of Nav1.7 in many different chronic pain syndromes [7,8,9]. Our research group has previously shown that genetic polymorphisms in SCN9A, which encodes Nav1.7, could influence postoperative pain sensitivity in patients [10]. In addition, our recent study has demonstrated that the upregulation of Nav1.7 contributes to the development of acute post-surgical pain [11]. Furthermore, a current study demonstrated that increased Nav1.7 played a central role in CPSP [12]. However, the mechanism underlying the dysregulation of Nav1.7 after surgery remains unclear.
After tissue or nerve injury, the “inflammatory soup”, which includes several factors secreted by nociceptive neurons and recruited by non-neural cells, affects the chemical environment surrounding nociceptive neurons [13]. These mediators can lead to rapid or long-term post-translational modification (PTM) of the expression and function of ion channels in nociceptive neurons, consequently leading to nociceptor hyperexcitability. The long-term changes may be partially explained by the phenomenon that pronociceptive molecules, such as nerve growth factor (NGF), persist around the site of injury [14, 15]. NGF has been previously demonstrated to induce marked long-term changes in the excitability of neurons [16] and has been considered one of the critical factors in the initiation of peripheral sensitization and subsequent persistent pain [17, 18]. In addition, accumulating studies, including our previous studies, have demonstrated the upregulation of NGF in surgical pain models [12, 19,20,21,22].
Moreover, NGF has been shown to upregulate the expressions and currents of Navs and prolong the period of increased sensitivity after binding to tropomyosin receptor kinase A [16, 23,24,25,26,27]. A very recent study from Atmaramani et al. showed that the treatment of NGF and IL-6 could increase the excitability of adult DRG neurons, which was attenuated by Nav1.7 and Nav1.8 antagonist [28]. But, in contrast to several prior reports, their results showed that the increase in excitability induced by NGF and IL-6 unlikely related to the increase of the expression of Nav1.7 and Nav1.8 [28]. Therefore, the exact effect of NGF on Nav1.7 and the potential mechanism by which NGF regulates Navs urgently need to be investigated. PTM is a well-described process that modifies the expression and function of Navs [29]. NGF can trigger the PTMs of Navs, resulting in long-term increased hyperexcitability in nociceptors [30]. Ubiquitylation is an important PTM that negatively affects the function and expression of Navs [31]. Neuronal precursor cell-expressed developmentally downregulated 4-2 (Nedd4-2), a member of the E3 ubiquitin ligase, increases the rate of internalization and degradation of Navs. Previous in vitro experiments have demonstrated that Nedd4-2 negatively regulates Navs [32, 33]. In addition, the expression of Nav1.7 in the sciatic nerve and the expression of Nav1.8 in the DRG in Nedd4-2 knockout mice were increased, suggesting that Nedd4-2 is essential for the downregulation of Navs protein expression [34]. Importantly, NGF is known to induce the phosphorylation of Nedd4-2, resulting in decreased ubiquitylating efficiency [35].
The central nervous system is another important neurobiological basis for the transition of chronic postoperative pain, and the changes within the central nervous system contribute to the maintenance of chronic pain [36]. The hyperexcitability of primary nociceptors after surgery may result in increased release of excitatory transmitters and enhance the synaptic connections within the spinal dorsal horn, ultimately leading to a state of central sensitization and chronic pain. Alleviating the peripheral sensitization may prevent the initiation and maintenance of central sensitization and CPSP. While peripheral sensitization has been shown to be associated with Nav1.7, whether Nav1.7 contributes to the development of central sensitization remains unstudied. The present study aimed to test the hypothesis that sustained increased NGF after surgery may induce dysregulation of Nav1.7, and preventing the dysregulated Nav1.7 in primary nociceptors may alleviate central sensitization and prevent the transition to CPSP.
Methods
Animals
Animals were housed and kept in controlled conditions (temperature: 22–24 °C; humidity: 50–60%; light: 12-h light/dark cycle) in the animal center of Tongji Hospital. All animals were group-housed in cages (2–5 per cage) with standard rodent chow and water ad libitum. Nav1.7 conditional knock-out (cKO) mice, which were presented by Professor Stephen G. Waxman (Yale University School of Medicine, USA), were generated as described previously [27] and were bred at our facility in Wuhan. PCR genotyping was performed as described previously [37]. Adult male Nav1.7 cKO mice, and wild-type littermate mice were used for the NGF intraplantar injection experiments. The skin/muscle incision and retraction (SMIR) model was established using adult male Sprague Dawley rats (weight: 250–300 g). All experiments were performed in a blinded manner.
Behavioral Testing
Three days before the behavioral test, all animals were individually placed in plastic chambers, which were elevated by a wire mesh or glass floor, for 30 min daily to habituate to the testing environment. Before each test, animals were allowed to habituate for 30 min. For rats, mechanical sensitivity was assessed by an electronic von Frey filament (Model 38450; Ugo Basile, Gemonio, Italy), as described previously [22]. The average was calculated from three tests. For mice, mechanical sensitivity was assessed using the up-down method with grade-strength von Frey monofilaments (0.008, 0.02, 0.04, 0.07, 0.16, 0.4, 0.6, 1.0, 1.4, and 2.0 g), as previously described [38]. The paw withdrawal threshold (PWT) was measured by applying the filaments to the mid-plantar surface of the ipsilateral hind paw vertically. Rapid withdrawal, shaking, or licking of the paw was regarded as a positive response. All stimuli applications were performed in a calm state, not during grooming or sleeping. The baseline mechanical sensitivity was calculated from the average of the three baseline measurements that were conducted on separate days before the procedure. Behavioral tests were performed between 8:00 and 12:00.
NGF Administration
NGF (Sigma) was dissolved in sterile saline to a concentration of 0.15 μg/μl. After anesthetizing with 2% isoflurane through a nose cone, wild-type and Nav1.7 cKO mice were administered 20 μl NGF in hind paws using a microsyringe, while controls were administered 20 μl of saline. Mechanical sensitivity tests were performed at 4 h and on days 1, 3, 5, 7, 10, and 14 after NGF administration depending on acute or chronic studies [39].
Establishment of Skin/Muscle Incision and Retraction (SMIR) Model
SMIR was performed as described previously [40]. General anesthesia was predominantly used to alleviate pain during the experiments. Briefly, after anesthesia with pentobarbital sodium intraperitoneally at a dose of 50 mg/kg, the medial region of the right thigh was shaved, and the animals were laid on the back. After sterilizing with sterile alcohol, a 1.5–2-cm incision about 4 mm medial to the saphenous vein was made on the skin to reveal the muscle of the thigh, and a 7–10-mm incision was then made on the superficial muscle layer. The superficial muscle was subsequently parted by spreading blunt scissors to insert a microdissection retractor (NO. R22009–02, RWD Life Science Inc.). The incision of the thigh was retracted by 2 cm to reveal the fascia underlying adductor muscles, and the process of retraction was maintained for 1 h. The surgical area was covered with sterile gauze to prevent dehydration of the surgical site. After retraction, the skin and muscle were closed respectively. Sham-operated rats underwent the same procedure, except for retraction. Mechanical sensitivity tests were performed on days 1, 3, 5, 7, 10, 14, and 21 after surgery.
Intrathecal Catheter Implantation
On a preoperative day, a polyethylene-10 (PE-10) intrathecal catheter was implanted to the subarachnoid space as described previously [41]. Briefly, after anesthesia with pentobarbital sodium intraperitoneally at a dose of 50 mg/kg, an incision about 2 cm was made in the center of the backside, and laminectomy was then performed at the L5 vertebra of animals. After the intravertebral space between L4 and L5 was exposed, the spinal dura mater was punctured by a blunt needle, and the catheter was then implanted into the subarachnoid space to reach the lumbar enlargement level. The catheter was fixed by medical silk, and the incision was closed layer by layer. Rats were housed individually after surgery, and the location of the catheter was determined by intrathecal injection of lidocaine (10 μl, 2%). The rats with transient hind paw paralysis after lidocaine injection were used for further experiments, while the rats that failed to show any paralysis or showed any signs of nerve damage were excluded from the experiments. There were at least 5 days for rats to recover before further experiments.
Drug Administration
GW441756 (Selleck Chemicals, Houston, TX, USA), a validated and specific tropomyosin receptor kinase A (TrkA) inhibitor (IC50 = 2 nM), displays a higher selectivity on TrkA than other inhibitors do. GW441756 was dissolved in dimethyl sulfoxide (DMSO) and diluted with normal saline. The highly selective serum and glucocorticoid-inducible kinase (SGK)1 inhibitor EMD638683 (IC50 = 3 μM) (MedChem Express, Monmouth Junction, NJ, USA) was dissolved in DMSO and diluted with normal saline. The specificity and selectivity of EMD638683 have been demonstrated previously [42]. For acute treatment, a single dose of GW441756 (10 μl, 1 μM/10 μM/100 μM) or EMD638683 (10 μl, 50 μM) was administered on day 10 after the establishment of the surgical pain model through the intrathecal catheter with a microsyringe. Behavioral tests were conducted before administration and 1, 2, 4, and 6 h after administration. For chronic treatment, GW441756 (10 μl, 1 μM/10 μM/100 μM)) or EMD638683 (10 μl, 50 μM) was administered once daily from day 10 to day 14. The dose of drugs was chosen according to our preliminary results and the findings in previous studies [43,44,45]. Mechanical sensitivity tests were performed before administration and from day 10 to day 14 after administration.
Western Blotting
Animals were sacrificed by cervical dislocation after being anesthetized with pentobarbital sodium (60 mg/kg, i.p.), and tissue samples were collected during daytime hours for further analyses. Following decapitation, the spinal column was opened to expose the lumbar spinal cord. The L3–L5 segments of the DRG and lumbar enlargement of the spinal cord were collected rapidly. The collected samples were homogenized in radioimmunoprecipitation assay lysis buffer. The homogenates were centrifuged at 12,000×g for 15 min at 4 °C, and the supernatants were then collected. Protein concentrations were determined by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). The supernatants were boiled at 100 °C in loading sample buffer for 5 min. Equal amounts of proteins (30–60 μg) were electrophoresed on 8–12% SDS/PAGE gel and then transferred onto polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). After blocking with 5% (w/v) defatted milk in tris-buffered saline with 0.1% Tween 20 (TBST; 2 mmol/L Tris–HCl, 50 mmol/L NaCl, pH 7.4) for 2 h at room temperature, the membranes were incubated overnight at 4 °C with specific primary antibodies for NGF (1:1000; ab52918; Abcam), TrkA (1:200, BA0404; Boster), Nav1.7 (1:500; ASC-008; Alomone), SGK1 (1:500; BM4452; Boster), Nedd4-2 (1:1000; 4013S; CST), phospho-Nedd4-2(ser342) (1:1000; 12146S; CST), β-Tubulin (1:2000; AC021; ABclonal), brain-derived neurotrophic factor (BDNF; 1:500; DF6387; affinity), vesicular glutamate transporter 2 (VGLUT2; 1:2000; A15177; ABclonal), chemokine ligand 21 (CCL21; 1:1000; DF6681; Affinity), or β-actin (1:2000; BM0627; Boster). After washing with TBST three times, membranes were incubated with HRP-conjugated goat anti-rabbit (1:5000; AS1107; Aspen) or goat anti-mouse secondary antibody (1:5000; AS1106; Aspen) for 2 h at room temperature. After further washing with TBST three times, the blots were visualized using the SuperLumia ECL Plus HRP Substrate Kit (K22030; Abbkine). The protein bands were quantified based on the gray values using an image analysis system (Bio-Rad, ChemiDoc XRS+, USA), and the gray values were normalized to those of β-actin or β-tubulin.
Immunohistochemistry
Rats were deeply anesthetized with pentobarbital sodium (60 mg/kg, i.p.) during daytime hours and subsequently perfused transcardially with 0.1-M PBS, followed by 4% ice-cold paraformaldehyde in PBS. The L3–L5 segments of DRGs and spinal cords were collected and post-fixed in 4% paraformaldehyde overnight and were cryoprotected overnight in 0.1-M PBS containing 30% sucrose. The embedded DRGs and spinal cords were respectively cut at 14-μm and 20-μm thickness using a cryostat (CM1900, Leica, Germany). Immunohistochemistry was performed as described previously [46]. Briefly, the sections were penetrated with 0.3% Triton X-100 for 15 min and blocked with 10% sheep serum (AR0009; Boster) for 1 h at room temperature. The sections were then incubated with primary antibodies overnight at 4 °C. Primary antibodies included rabbit anti-TrkA antibody (1:100; BA0404; Boster), rabbit anti-SGK1 antibody (1:100; BM4452; Boster), rabbit anti-phosphor-Nedd4-2 (1:200; 12146S; CST), rabbit anti-Nav1.7 (1:200; ASC-008; Alomone), mouse anti-CGRP antibody (1:250; ab81887; Abcam), rabbit anti-BDNF antibody (1:200; DF6387; Affinity), or rabbit anti-VGLUT2 antibody (1:200; A15177; ABclonal). After washing three times in PBS, the sections were incubated with Alexa Fluor 594-conjugated goat anti-rabbit secondary antibody (1:200; A23220; Abbkine) or Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (1:200; A23420; Abbkine) for 2 h at room temperature. Images were captured using a fluorescence microscope (DM2500, Leica, Germany).
Statistical Analyses
GraphPad Prism 5.01 software (GraphPad Software, Inc., La Jolla, CA, USA, RRID:SCR-002798) was used for statistical analyses. Data are expressed as mean ± standard deviation (SD). Analyses of behavioral tests were performed using a two-way analysis of variance (ANOVA) with repeated measures, followed by the Bonferroni post hoc test. The expression values of proteins were analyzed using Student’s unpaired t test or one-way ANOVA followed by Bonferroni post hoc test. A p value of < 0.05 was considered statistically significant.
Results
Nav1.7 Was Required for NGF-Induced Long-Term Pain Behaviors, and NGF Upregulated the Expression of Nav1.7
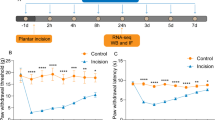
Nav1.7 cKO mice were generated by crossing the floxed (SCN9A) Nav1.7 mice with the mice in which Cre expression was driven by the Nav1.8 promotor. These mice have been demonstrated to show tissue-specific ablation of Nav1.7 in nociceptive neurons expressing Nav1.8 [27, 37]. We used western blotting to examine the expression of Nav1.7 in the DRG of Nav1.7 cKO mice to confirm the efficacy of deletion. As shown in Fig. 1a, the expression level of Nav1.7 was lower in Nav1.7 cKO mice than in wild-type mice. To test whether Nav1.7 is involved in NGF-induced pain behaviors, we tested the mechanical sensitivity in Nav1.7 cKO mice and wild-type mice after NGF injection. As shown in Fig. 1b, no significant differences were found at baseline among the three groups. Nav1.7 cKO mice displayed hypersensitivity to mechanical stimuli at the early stage after NGF injection; however, at the late stage, Nav1.7 cKO mice displayed a recovery in mechanical hypersensitivity and did not develop long-lasting hypersensitivity after NGF injection. In addition, the expression of Nav1.7 was increased in the DRG in wild-type mice after intraplantar NGF injection (Fig. 1c). The experimental timeline was shown in Fig. 1d. These findings suggested that Nav1.7 expressed in Nav1.8-positive neurons in the DRG was required for the development of NGF-induced long-term pain behaviors, and NGF was able to upregulate the expression of Nav1.7 as upstream signaling.

Nav1.7 is required for NGF-induced long-term pain behavior. a Deletion of Nav1.7 in the DRG was verified by western blotting. The expression of Nav1.7 in the DRG significantly decreased in Nav1.7 cKO mice compared with WT mice. *p < 0.05 vs WT mice; n = 3 mice per group. b Intraplantar NGF-induced long-term mechanical hypersensitivity in WT mice. In contrast, the mechanical hypersensitivity in Nav1.7 cKO mice was significantly recovered in the later stage. ***p < 0.001 vs the WT + NS group; #p < 0.05, ##p < 0.01 vs the WT + NGF group; n = 4–6 mice per group. c Western blotting analyses showed that intraplantar NGF administration upregulated the expression of Nav1.7 in the DRG in WT mice. ***p < 0.001 vs the WT + NS group; n = 4–6 mice per group. Each value was presented as mean ± SD. d Timeline for the experiments. Behavioral tests were performed 3 days before experiments. Wild-type and Nav1.7 cKO mice were administered NGF intraplantarly, while control mice were administered normal saline. Mechanical sensitivity tests were performed at 4 h and on days 1, 3, 5, 7, 10, and 14 after NGF or normal saline administration. WT: wild-type; NS: normal saline
Pain-Related Behavior in Rats Following SMIR Surgery
In this study, we used the SMIR rat model, a well-described CPSP rat model, which is a reliable tool for studying CPSP. The PWTs were assessed at baseline and on days 3, 5, 7, 10, 14, and 21 after surgery to observe the development of mechanical hyperalgesia. As shown in Fig. 2a, baseline mechanical sensitivity was similar in the SMIR and sham groups. The mechanical withdrawal threshold significantly decreased from 5 days after surgery in the SMIR group, while no significant changes were observed in the sham group. The findings suggest that SMIR surgery evoked a persistent mechanical hypersensitivity.

NGF negatively regulates ubiquitination of Nav1.7 in rats with SMIR-induced post-surgical pain. a The time course of the mechanical paw withdrawal threshold in sham and SMIR rats. ***p < 0.001 vs the sham group; n = 7 rats per group. b The time course of NGF expressions in the skin in SMIR rats (normalized by β-actin). c The time course of NGF expressions in the DRG in SMIR rats (normalized by tubulin). d The expressions of Nav1.7 in the DRG were upregulated from day 7 to day 21 after surgery in SMIR rats (normalized by tubulin). e Nedd4-2 in the DRG significantly decreased in the later stage in the SMIR model (normalized by Tubulin). f P-Nedd4-2 increased in the DRG in SMIR rats (normalized by tubulin). g The expression of TrkA in the DRG increased in SMIR rats during the observation period (normalized by tubulin). h The expression of SGK1 in the DRG was upregulated from day 7 to day 21 after surgery in SMIR rats. *p < 0.05, **p < 0.01, ***p < 0.001 vs the sham group, n = 4–6 rats per group. Each value is presented as mean ± SD. i Timeline for the experiments. Rats underwent SMIR surgery or a sham-operated procedure. Behavioral tests were performed on days 1, 3, 5, 7, 10, 14, and 21 after the surgery
Dynamic Expression of NGF and Nav1.7 in the DRG After SMIR Surgery
To further explore the correlation between NGF and Nav1.7 in CPSP, we examined their expression patterns in SMIR rats. Using western blotting, we examined the expression of NGF in the skin around the incision and the DRG. The results showed that the expression of NGF in the skin increased in the early stage and then declined (Fig. 2b), and the protein level of NGF in the DRG persistently increased until day 21 (Fig. 2c). In addition, we found that the expression of Nav1.7 in DRG increased from day 7 until day 21 in the DRG in SMIR rats (Fig. 2d). The changes in NGF expression preceded the increase in Nav1.7 expression. Therefore, we speculate that the increase in Nav1.7 after SMIR surgery may be related to the upregulation of NGF.
P-Nedd4-2 Was Overexpressed in the DRG in SMIR Rats
It has been demonstrated that ubiquitin ligases regulate the internalization and degradation of Navs in the sensory nervous system [29]. Therefore, we examined the expression of Nedd4-2 in SMIR rats, which plays a key role in downregulating the expression of Navs. We found that the expression of Nedd4-2 in the DRG decreased after SMIR surgery (Fig. 2e). In contrast, the protein levels of p-Nedd4-2 significantly increased in the DRG in SMIR rats compared with sham-operated rats (Fig. 2f). The phosphorylated modification influences the efficiency of Nedd4-2. It suggested that the increase of Nav1.7 might be related to the phosphorylated modification of Nedd4-2. A study by Arevalo et al. showed that NGF could trigger the phosphorylation of Nedd4–2 [35]. However, the underlying mechanism remained unclear. To explore the underlying mechanism/signal pathway, we examined the expression of SGK1 kinase, which can phosphorylate Nedd4–2 in a PY motif-dependent manner. We found that SGK1 obviously increased in the DRG in SMIR rats (Fig. 2h). Based on the findings in previous studies and our results, we speculate that increased NGF in SMIR rats may trigger the phosphorylation of Nedd4-2 by activating SGK1. The experimental timeline was shown in Fig. 2i.
Analgesic Effects of GW441756 and EMD638683 in SMIR Rats
To determine whether upregulated Nav1.7 is modulated by the NGF-SGK1-Nedd4-2-mediated pathway, we intrathecally administrated inhibitors of TrkA and SGK1 in SMIR rats. GW441756 and EMD638683 are specific TrkA and SGK1 inhibitors, respectively. The rats were administrated intrathecally a single dose of GW441756 (10 μl, 1 μM/10 μM/100 μM) or EMD638683 (10 μl, 50 μM) on day 10 after surgery to determine whether GW441756 and EMD638683 could alleviate pain in the advanced phase of SMIR. Behavioral tests were conducted before and at 1, 2, 4, and 6 h after drug administration. As shown in Fig. 3a, intrathecal administration of low-dose TrkA inhibitor (10 μl, 1 μM GW441756) had no significant effects on PWTs. On the other hand, intrathecal administration of 10 μl of 10 μM or 100 μM GW441756 markedly reduced mechanical hyperalgesia dose-dependently. The upregulation of PWTs peaked at 1 h and lasted for 6 h. As shown in Fig. 3d, intrathecal injection of 10 μl of 50 μM EMD638683 had a significant effect on PWTs. The upregulation of PWTs began at 1 h, peaked at 2 h, and lasted for 6 h.

Analgesic effects of intrathecal (i.t.) administration of TrkA inhibitor or SGK1 inhibitor in SMIR rats. a A single dose of TrkA inhibitor (10 μl, 1 μM/10 μM/100 μM) was intrathecally administered on day 10 after SMIR surgery. Behavioral tests were performed at 1, 2, 4, and 6 h after drug administration. b–c For chronic treatment, the TrkA inhibitor (10 μl, 1 μM/10 μM/100 μM) was administrated from day 10 to day 14. Behavioral tests were conducted at 1 h before daily treatment and 1 h after daily treatment. d A single dose of SGK1 inhibitor (10 μl, 50 μM) was intrathecally administered on day 10 after SMIR surgery. Behavioral tests were performed at 1, 2, 4, and 6 h after drug administration. e–f SGK1 inhibitor was administered for 5 days (from days 10 to 14). Behavioral tests were conducted at 1 h before daily drug injection and 2 h after daily treatment. **p < 0.01, ***p < 0.001 vs the sham group; #p < 0.05, # #p < 0.01, ###p < 0.001 vs the SMIR + vehicle group; $$p < 0.01, $$$p < 0.001 vs the SMIR +10 μM TrkA inhibitor group; n = 6 rats per group. Each value was presented as mean ± SD. g Eight days before experiments, intrathecal catheters were implanted into the subarachnoid space of rats. The location of the catheter was determined by intrathecal injection of lidocaine. TrkA inhibitor was administered on day 10 through the intrathecal catheter. Behavioral tests were conducted before administration and at 1, 2, 4, and 6 h after administration. TrkA inhibitor was administered once daily from day 10 to day 14. Mechanical sensitivity tests were performed before administration and from day 10 to day 14 after administration. h Intrathecal catheters were implanted into the subarachnoid space of rats 8 days before experiments. SGK1 inhibitor was administered on day 10 through the intrathecal catheter. Behavioral tests were conducted before administration and at 1, 2, 4, and 6 h after administration. SGK1 inhibitor was administered once daily from day 10 to day 14. Behavioral tests were performed before administration and from day 10 to day 14 after administration
To address whether repetitive treatment with GW441756 or EMD638683 has cumulative analgesic effects, we administered GW441756 or EMD638683 once daily from day 10 to day 14. Behavioral tests were performed before drug administration and at 1 h after GW441756 administration (10 μl, 1 μM/10 μM/100 μM) or 2 h after EMD638683 administration (10 μl, 50 μM) from day 10 to day 14. As shown in Fig. 3b, c, e, and f, repeated injection of GW441756 (10 μl, 100 μM) and EMD638683 (10 μl, 50 μM) markedly reversed the mechanical allodynia in SMIR rats without tolerance. These results show that consecutive treatments with GW441756 (10 μl, 100 μM) and EMD638683 (10 μl, 50 μM) have cumulative analgesic effects in rats with chronic surgical pain. The experimental timeline was shown in Fig. 3g and h.
GW441756 and EMD638683 Suppressed the Increase of Nav1.7 in SMIR Rats
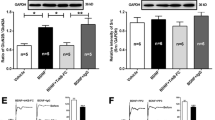
To investigate whether the increase of Nav1.7 after SMIR surgery was induced by NGF/TrkA pathway, we used western blotting and immunohistochemistry to test the expression of Nav1.7 after TrkA inhibitor administration. We found that increased expressions of Nav1.7 could be reversed by repeated treatment with the TrkA inhibitor GW441756 (10 μl, 100 μM) (Fig. 4d and e). In addition, the expressions of SGK1 and p-Nedd4-2 were also decreased. Moreover, increased expression of p-Nedd4-2 was also inhibited by repeated treatment with the SGK1 inhibitor EMD638683 (10 μl, 50 μM) (Fig. 5b and d). Altogether, these findings suggest that the NGF/TrkA-SGK1 pathway in the DRG may be involved in the development of SMIR pain.

TrkA inhibitor GW441756 attenuates increased expressions of Nav1.7 in SMIR rats. a–d Representative bands showed the expression levels of TrkA, SGK1, p-Nedd4-2, and Nav1.7 in different groups. Histograms showed semi-quantitative analyses after normalization to corresponding Tubulin. Western blot analyses showed that repeated GW441756 (10 μl, 100 μM) restore the protein expressions of TrkA, SGK1, p-Nedd4-2, and Nav1.7; *p < 0.05, **p < 0.01, ***p < 0.001 vs the sham group or SMIR + vehicle group; n = 4–6 rats per group. Each value was presented as mean ± SD. e Immunofluorescence showed that immunoactivities of TrkA, SGK1, p-Nedd4-2, and Nav1.7 were suppressed by GW441756 (10 μl, 100 μM)

SGK1 inhibitor EMD638683 inhibits increased expressions of p-Nedd4-2 and Nav1.7 in SMIR rats. a–c Representative bands showed the expressions of TrkA, SGK1, p-Nedd4-2, Nedd4-2, and Nav1.7 in different groups. Histograms showed the quantitative analysis after normalization to corresponding Tubulin. The protein expressions of TrkA, SGK1, p-Nedd4-2, and Nav1.7 were significantly lower in EMD638683 (10 μl, 50 μM) repeatedly treated SMIR rats than in vehicle-treated SMIR rats. d Immunofluorescence showed that immunoactivities of TrkA, SGK1, p-Nedd4-2, and Nav1.7 were suppressed by repeated EMD638683 (10 μl, 50 μM). Scale bar (a–i) = 100 μm. *p < 0.05, **p < 0.01, ***p < 0.001 vs the sham group or SMIR + vehicle group; n = 4–6 rats per group
Nav1.7 Was Required in NGF-Induced Central Sensitization
Increased outputs from primary afferent neurons increase the excitability of secondary neurons and enhance the release of the central mediators. The activation of glutamatergic synaptic transmission, glial signaling, and neuronal plasticity in the spinal cord play critical roles in the initiation and maintenance of central sensitization and chronic pain [47,48,49,50]. Notably, VGLUT2, BDNF, and CCL21 play key roles in the activation of glutamatergic synaptic activity, neuronal plasticity, and glial signaling in the spinal cord [49, 51, 52]. Therefore, we examined the level of central sensitization-related mediators in the spinal cord in Nav1.7 cKO mice after intraplantar NGF injection to investigate the role of Nav1.7 in spinal sensitization. As shown in Fig. 6, the expressions of VGLUT2, BDNF, and CCL21 increased in the wild-type + NGF group compared to the wild-type + normal saline (NS) group. However, the increased expressions were significantly attenuated in the Nav1.7 cKO group compared with the wild-type + NGF group. It is well-known that the increased peripheral input induced by hyperexcitability nociceptors leads to central sensitization in the spinal cord [53]. Importantly, the increase in sodium channels and increased conductance can cause neuronal hyperexcitability [54]. Therefore, these results suggest that Nav1.7 is involved in NGF-induced central sensitization.

Nav1.7 is involved in NGF-induced central sensitization. a–c Representative bands showed the expressions of CCL21, VGLUT2, and BDNF in different groups. Histograms showed the quantitative analysis after normalization to corresponding β-actin. The protein expressions of CCL21, VGLUT2, and BDNF significantly increased in the WT + NGF group but decreased in the Nav1.7 cKO group. *p < 0.05, **p < 0.01, ***p < 0.001 vs the WT + NS group or WT + NGF group; n = 3–4 mice per group. Each value was presented as mean ± SD. WT: wild-type; NS: normal saline; SC: spinal cord
Upregulation of Nav1.7 Induced by NGF Contributed to Central Sensitization in the Spinal Cord in SMIR Rats
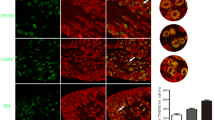
To further determine the role of Nav1.7 in surgery-induced central sensitization, we examined the expressions of central sensitization mediators in the spinal cord in SMIR rats. As shown in Fig. 7, the expressions of VGLUT2, BDNF, and CCL21 increased in the spinal cord in SMIR rats. Then, the inhibitors of TrkA or SGK1 were intrathecally administered. Western blot analyses showed that the upregulated expressions of VGLUT2, BDNF, and CCL21 were reduced by repeated treatment with GW441756 (10 μl, 100 μM) (Fig. 8a–c) and EMD638683 (10 μl, 50 μM) (Fig. 9a–c). Similarly, immunofluorescence showed that the immunoactivity of CGRP, VGLUT2, and BDNF was suppressed by repeated GW441756 (10 μl, 100 μM) (Fig. 8d) or EMD638683 (10 μl, 50 μM) (Fig. 9d). Therefore, the inhibition of the NGF/TrkA-SGK1 pathway could suppress central sensitization in the spinal cord in SMIR rats. Taken together, these results suggest that increased Nav1.7 induced by NGF contributes to surgery-related central sensitization, and downregulation of Nav1.7 could alleviate central sensitization.

Upregulation of CCL21, VGLUT2, and BDNF contributes to central sensitization in the spinal cord in SMIR rats. a–c Western blot analyses showed significantly increased CCL21, VGLUT2, and BDNF levels in the spinal cord in the SMIR model (normalized by β-actin). *p < 0.05, **p < 0.01, ***p < 0.001 vs the sham group; n = 4–6 rats per group. Each value was presented as mean ± SD. SC: spinal cord

TrkA inhibitor GW441756 inhibits central sensitization in the spinal cord in SMIR rats. a–c Histograms showed the semi-quantitative analyses after normalization to corresponding β-actin. Western blot analyses showed that the upregulation of CCL21, VGLUT2, and BDNF was restored by chronic GW441756 (10 μl, 100 μM). *p < 0.05, **p < 0.01 vs the sham group or SMIR + vehicle group; n = 4–6 rats per group. Each value represented mean ± SD. SC: spinal cord. (D) Immunofluorescence showed suppressed immunoactivity of CGRP, VGLUT2, and BDNF by GW441756 (10 μl, 100 μM). Scale bar (a–i) = 200 μm

SGK1 inhibitor EMD638683 inhibits central sensitization in the spinal cord in SMIR rats. a–c Histograms showed the semi-quantitative analyses after normalization to corresponding β-actin. Western blot analyses showed that the upregulation of CCL21, VGLUT2, and BDNF was restored by repeated EMD638683 (50 μM). *p < 0.05, **p < 0.01, ***p < 0.001 vs the sham group or SMIR + vehicle group; n = 4–6 rats per group. Each value represented mean ± SD. SC: spinal cord. d Immunofluorescence showed suppressed immunoactivity of CGRP, VGLUT2, and BDNF by EMD638683 (50 μM). Scale bar (a–i) = 200 μm
Discussion
The principal findings in this study were as follows: (1) Nav1.7 was involved in NGF-induced long-term pain, and the expression of Nav1.7 was increased after NGF application; (2) the increased expression of Nav1.7 in SMIR rats was suppressed by repetitive TrkA and SGK1 inhibitor treatment; (3) conditional deletion or pharmacological inhibition of Nav1.7 alleviated the development of spinal sensitization. Taken together, we speculate that the upregulation of Nav1.7 in SMIR rats may be attributed to the persistence of NGF in the DRG, which triggers SGK1-dependent phosphorylation of Nedd4-2. Therefore, attenuating the upregulation of Nav1.7 may prevent the development of central sensitization and the transition of CPSP.
Both NGF and Nav1.7 play crucial roles in the peripheral sensitization, as well as long-term generation and maintenance of chronic pain [6, 55]. To date, the association between NGF and Nav1.7 remains unclear. Nassar et al. found a reduction in acute thermal hypersensitivity within 8 h after intraplantar injection of 500 ng NGF in Nav1.7 cKO mice but did not investigate long-term changes [27]. We found that Nav1.7 cKO mice displayed reduced hypersensitivity at the late stage after NGF administration and did not develop long-lasting mechanical hypersensitivity, suggesting that Nav1.7 is required for NGF-induced chronic pain. There are three possible reasons for the mechanical hypersensitivity in Nav1.7 cKO mice at the early stage after NGF injection. First, the initiation of hyperalgesia may be induced by NGF in Nav1.8-negative DRG neurons, which remain intact within Nav1.7 cKO mice. Second, at the early stage, NGF-induced pain is mainly evoked by the activation of transient receptor potential vanilloid family member (TRPV) 4 or TRPV1 after binding to TrkA, suggesting that Nav1.7 is associated with these mechanosensitive channels in the DRG [56, 57]. Third, Nav1.7 may play a downstream role in mechanotransduction through the mechanically activated currents in sensory neurons [58]. In addition, after NGF injection, we found that the expression of Nav1.7 in the DRG was significantly increased. These findings suggest that NGF may be an upstream regulator of Nav1.7 and upregulate Nav1.7 expressions.
After confirming the association between NGF and Nav1.7, we used the SMIR model, which is a CPSP rat model created by Flatters et al. in 2008, to investigate the underlying mechanism. Our results showed the expression of NGF increased in the skin at the early stage of SMIR, while it sustainably increased in the DRG. This finding suggests that NGF around nociceptors might be more critical for the development of CPSP. In addition, our results showed that the upregulation of NGF preceded expression changes of Nav1.7. Furthermore, intrathecal administration of the inhibitors of TrkA attenuated the upregulation of Nav1.7 in SMIR rats. Based on our results and the findings in previous reports, we speculate that the increased NGF in the DRG might upregulate Nav1.7 in SMIR rats.
As one of the important neurotrophic factors, NGF is known to be involved in a wide range of biological processes related to viability, plasticity, neuronal growth, and neuronal protection [59, 60]. For examples, Su et al. demonstrated that NGF played a protective role in Aβ25–35-induced injury through the Nrf2/HO-1 pathway [61]. In addition, a recent study from Dai et al. showed that the inhibitory effect of curcumin on oxidative stress was attributed to the activation of NGF/Akt and Nrf2/HO-1 pathways [62]. Although many studies have demonstrated the neuroprotective effect of NGF in several biological processes [63, 64], accumulating researches have indicated many negative neurological effects of NGF, such as inflammation, hyperexcitability, oxidative stress, and neurodegeneration [65,66,67]. The effect of oxidative stress induced by oxidants on Navs has been verified [68, 69]. A recent study from Friederike et al. showed that oxidation enhanced the function of Nav1.7 and Nav1.8 [70]. The relevance of neuroprotection and neurotoxicity of molecules may be associated with the principles of hormesis [71, 72]. A variety of endogenous cellular activities and biochemical signaling were regulated by hormetic does responses [73, 74]. Preconditioning signal leading to cellular protection through hormesis is an important redox-dependent aging-associated neurodegenerative/ neuroprotective issue. Therefore, it may be that the one of possible mechanisms underlying the effect of NGF on the modification of Nav1.7 and chronic pain may be associated with hormetic responses. Hormetic responses occur in a wide range of biological models for a large and diverse array of endpoints. However, further investigation is needed to evaluate the quantitative features of the dose-response relationships and underlying mechanisms that can account for the biphasic nature of the hormetic response after exposure to NGF.
Nevertheless, the mechanism by which NGF regulates Nav1.7 remains to be further investigated. Brackenbury et al. showed that NGF positively regulated total Navs protein expression but did not affect the Nav1.7 mRNA level in the Mat-LyLu rat prostate cancer cell line, suggesting that the regulation of Nav1.7 by NGF might be post-translational [26]. Ubiquitylation is a well-described post-translational modification of Nav1.7. After ubiquitylation by Nedd4-2, the rate of internalization of Nav1.7 increases. Subsequently, the internalized Nav1.7 protein is degraded or recycled, resulting in reduced expressions. In contrast, the reduction of Nedd4-2 increases the expression of membrane proteins with a PY motif [75]. Consistent with our results, Laedermann et al. showed that the expression of Nedd4-2 in DRG cells was decreased in neuropathic pain model [34]. In addition, accumulating evidence has established that the downregulation of Nedd4-2 is implicated in chronic pain [76]. However, the mechanism underlying the downregulation of Nedd4-2 in chronic pain condition remains unclear. A previous study showed that SGK1 increased Nav1.5 mediated sodium current and expression in Xenopus oocytes by inactivating NEDD4-2 [77]. Importantly, it has been demonstrated that the SGK1 inhibits Nedd4-2 function by increasing phosphorylation of Nedd4-2 [78]. SGK1 is a serine-threonine protein kinase and is regulated by diverse hormones, growth factors, and cytokines [79, 80]. It is reported that NGF could trigger the phosphorylation of Nedd4-2 after binding to the TrkA receptor [35]. After binding to the TrkA receptor, three major signaling cascades including phospholipase C-γ (PLC-γ), mitogen-activated protein kinase (MAPK), and phosphatidylinositol 3-kinase (PI3K) are activated depending on different cell physiological conditions [81]. SGK1 is a critical downstream regulator of PI3K activation and is involved in the regulation of cellular activity by phosphorylating target proteins [82]. It is well-known that the phosphorylation of protein could trigger the degradation of protein [83,84,85]. Together, we speculated that the long-term phosphorylation of Nedd4-2 mediated by SGK1 may result in subsequently degradation of Nedd4-2. Thus, it may be a possible mechanism underlying the decreased expression of Nedd4-2 in the condition of CPSP.
Recently, the role of SGK1 in pain perception has been demonstrated. Peng et al. found that SGK1 contributed to post-inflammatory hyperalgesia, which is involved in specific glutamatergic receptors trafficking [86]. In addition, the inhibitors of SGK1 alleviate mechanical allodynia induced by neuropathic pain [87, 88]. Therefore, we speculate that sustainably increased NGF after surgery might upregulate the expression of Nav1.7 through a Trk receptor-mediated mechanism, which activates SGK1 and promotes the phosphorylation of Nedd4-2. We found that after the administration of the TrkA inhibitor, the expression of SGK1 and p-Nedd4-2 was suppressed. Moreover, the inhibitor of SGK1 also could attenuate the upregulation of p-Nedd4-2. Furthermore, repetitive administration of the inhibitors of TrkA and SGK1 attenuated the upregulation of Nav1.7 and reversed mechanical hyperalgesia in SMIR rats. Although many other complicated mechanisms may also be involved in this process, our results partly suggested that the NGF-induced upregulation of Nav1.7 via the activation of SGK1 and phosphorylation of Nedd4-2 might contribute to hyperalgesia in SMIR rats.
Increased outputs from primary afferent neurons increase the excitability of secondary neurons and enhance the release of the central mediators. To determine the role of Nav1.7 in spinal sensitization, we examined some relevant mediators in the spinal cord in Nav1.7 cKO mice after NGF injection. The activation of glutamatergic synaptic transmission, glial signaling, and neuronal plasticity in the spinal cord play critical roles in the initiation and maintenance of central sensitization and chronic pain [47,48,49,50]. Notably, VGLUT2 controls glutamatergic synaptic activity through loading glutamate into synaptic vesicles [49]. The upregulation of VGLUT2 leads to increased glutamatergic activity in the spinal cord. BDNF has been found to be involved in chronic pain and is identified as a driving force behind neuronal plasticity in central sensitization [51]. The activation of glial signaling, especially microglia activation, has been demonstrated to be important in spinal sensitization after incision [47, 89, 90]. Importantly, CCL21 is considered a potent modulator of microglial activation. Zhao et al. demonstrated the critical role of CCL21 in microglial activation and neuronal hyperexcitability in the spinal cord injury model [52]. We found that the expression levels of these mediators were significantly reduced in the spinal cord in Nav1.7 cKO mice after NGF injection, suggesting Nav1.7 in nociceptors plays a vital role in the development of spinal sensitization. We found that these central sensitization mediators were upregulated during the development of CPSP. After Nav1.7 was suppressed by TrkA or SGK1 inhibitor in SMIR rats, these mediators were decreased in the spinal cord. Therefore, the findings suggest that attenuation of the upregulation of Nav1.7 in the DRG alleviates spinal sensitization. Since Nav1.7 plays a critical role in both surgery-related peripheral sensitization and spinal sensitization, Nav1.7 may be implicated in the transition of long-term post-surgical pain. Navs specially Nav1.7 are required for the normal function of sensory neurons, but also for pain processing and an increase of neural excitability. Thus, the modification of Nav1.7 results in the development of CPSP. This condition linked protein homeostasis with health of the organism [74]. Given polyphenol compounds, redox status, and the vitagene network play critical role neuroprotection and neurodegeneration [91,92,93], the interaction of these factors may be also involved in the development of chronic pain. It is also known that polyphenols are strong antioxidants and metal chelators, with characteristics that are of beneficial therapeutic values for their development as candidates targeting neurodegenerative diseases [94]. Thus, interplay and coordination and redox interactions of polyphenols are an emerging field of research interest in anticancer, antidegenerative, and analgesic therapeutics.
However, this study has some limitations. First, we did not investigate the specific mechanism by which NGF activates SGK1, which will be addressed in our future investigations. Second, whether the upregulation of Nav1.7 on the cell membrane is due to internalization inhibition needs to further verify by ubiquitin assay in DRG primary culture. And because our primary aim was to establish the possible association of Nav1.7 with NGF in CPSP, we did not investigate the function of Nav1.7 channel and specific mechanism of internalization or degradation, which will be examined in future studies. Third, we did not investigate the role of the supraspinal mechanism in CPSP in this study. Because of the key role of descending modulation in the development of chronic pain, future studies are needed to investigate the supraspinal mechanism. Fourth, sex/gender-dependent differences are important in pain syndromes; however, we only used male mice and rats in this study. Therefore, further studies are needed to confirm the present findings in female rodents.
In conclusion, this study indicates that sustained NGF in the DRG may upregulate Nav1.7 partly through activating SGK1 to phosphorylate Nedd4-2. In addition, the upregulation of Nav1.7 in the DRG may facilitate nociceptive transmission to the spinal cord, leading to the development of spinal sensitization involved in the transition of CPSP (Fig. 10). Therefore, the attenuation of dysregulation of Nav1.7 in peripheral nociceptors may prevent the transition of CPSP. Our findings would improve the understanding of the underlying mechanism of CPSP.

Diagram of the mechanisms by which peripheral sensitization induced by upregulation of Nav1.7 in nociceptors after surgery leads to spinal sensitization and persistent post-surgical pain. Damage to tissues or nerves during surgery results in increased release of local NGF. NGF binds to the extracellular TrkA receptor and then triggers downstream intracellular signaling pathways. The NGF-TrkA complex is endocytosed and retrogradely transported to the cell bodies of nociceptors, leading to an increase in the concentration of peptides and post-translational modification of Nav1.7. Nedd4-2 binds directly to the PY-motifs of Nav1.7 and causes the ubiquitination of Nav1.7. SGK1, activated by NGF, can provoke Nedd4-2 phosphorylation. Phosphorylated Nedd4-2 cannot bind to Nav1.7. Therefore, ubiquitination and degradation of Nav1.7 induced by Nedd4-2 at the cell surface are decreased, leading to an increase of Nav1.7 and persistent hyperexcitability of nociceptors. Excitatory outputs from primary afferent terminals increase the excitability of secondary neurons in the spinal cord. The activity of nociceptive neurons in the spinal cord is also regulated by resident cells such as astrocytes, microglia, and interneurons. The enhanced excitability of peripheral nerves increases the release of CGRP, BDNF, and glutamate, which further sensitizes the neurons and helps to maintain the state of central sensitization. U: ubiquitin; BDNF: brain-derived neurotrophic factor; TrkB: tyrosine kinase receptor B; NMDA: N-methyl-D-aspartate receptor; AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CGRP: calcitonin gene-related peptide
Abbreviations
- CPSP:
-
chronic post-surgical pain
- Navs:
-
voltage-gated sodium channels
- NGF:
-
nerve growth factor
- DRG:
-
dorsal root ganglion
- PTM:
-
post-translational modification
- Nedd4-2:
-
neuronal precursor cell-expressed developmentally downregulated 4-2
- SGK1:
-
serum and glucocorticoid-inducible kinase
- cKO:
-
conditional knock-out
- SMIR:
-
skin/muscle incision and retraction
- PWT:
-
paw withdrawal threshold
- DMSO:
-
dimethyl sulfoxide
- PLC-γ:
-
phospholipase C-γ
- MAPK:
-
mitogen-activated protein kinase
- PI3K:
-
phosphatidylinositol 3-kinase
- VGLUT2:
-
vesicular glutamate transporter 2
- BDNF:
-
brain-derived neurotrophic factor
- CCL21:
-
chemokine ligand 21
- TrkB:
-
tyrosine kinase receptor B
- CGRP:
-
calcitonin gene-related peptide
- TRPV:
-
transient receptor potential vanilloid family member
- NMDA:
-
N-methyl-D-aspartate receptor
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
References
Wang L, Chang Y, Kennedy SA, Hong PJ, Chow N, Couban RJ, McCabe RE, Bieling PJ et al (2018) Perioperative psychotherapy for persistent post-surgical pain and physical impairment: a meta-analysis of randomised trials. Br J Anaesth 120(6):1304–1314. https://doi.org/10.1016/j.bja.2017.10.026
Chapman CR, Vierck CJ (2017) The transition of acute postoperative pain to chronic pain: an integrative overview of research on mechanisms. J Pain 18(4):359 e351–359 e338. https://doi.org/10.1016/j.jpain.2016.11.004
Poleshuck EL, Katz J, Andrus CH, Hogan LA, Jung BF, Kulick DI, Dworkin RH (2006) Risk factors for chronic pain following breast cancer surgery: a prospective study. J Pain 7(9):626–634. https://doi.org/10.1016/j.jpain.2006.02.007
Pluijms WA, Steegers MA, Verhagen AF, Scheffer GJ, Wilder-Smith OH (2006) Chronic post-thoracotomy pain: a retrospective study. Acta Anaesthesiol Scand 50(7):804–808. https://doi.org/10.1111/j.1399-6576.2006.01065.x
Rush AM, Cummins TR, Waxman SG (2007) Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol 579(Pt 1):1–14. https://doi.org/10.1113/jphysiol.2006.121483
Dib-Hajj SD, Yang Y, Black JA, Waxman SG (2013) The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14(1):49–62. https://doi.org/10.1038/nrn3404
Waxman SG, Merkies ISJ, Gerrits MM, Dib-Hajj SD, Lauria G, Cox JJ, Wood JN, Woods CG et al (2014) Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol 13(11):1152–1160. https://doi.org/10.1016/S1474-4422(14)70150-4
Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG (2004) Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108(3):237–247. https://doi.org/10.1016/j.pain.2003.12.035
Casals-Diaz L, Casas C, Navarro X (2015) Changes of voltage-gated sodium channels in sensory nerve regeneration and neuropathic pain models. Restor Neurol Neurosci 33(3):321–334. https://doi.org/10.3233/RNN-140444
Duan G, Xiang G, Zhang X, Yuan R, Zhan H, Qi D (2013) A single-nucleotide polymorphism in SCN9A may decrease postoperative pain sensitivity in the general population. Anesthesiology 118(2):436–442. https://doi.org/10.1097/ALN.0b013e31827dde74
Sun J, Li N, Duan G, Liu Y, Guo S, Wang C, Zhu C, Zhang X (2018) Increased Nav1.7 expression in the dorsal root ganglion contributes to pain hypersensitivity after plantar incision in rats. Mol Pain 14:1744806918782323. https://doi.org/10.1177/1744806918782323
Li Z, Li Y, Cao J, Han X, Cai W, Zang W, Xu J, Zhang W (2017) Membrane protein Nav1.7 contributes to the persistent post-surgical pain regulated by p-p65 in dorsal root ganglion (DRG) of SMIR rats model. BMC Anesthesiol 17(1):150. https://doi.org/10.1186/s12871-017-0438-8
Basbaum AI, Bautista DM, Scherrer G, Julius D (2009) Cellular and molecular mechanisms of pain. Cell 139(2):267–284. https://doi.org/10.1016/j.cell.2009.09.028
Gaudet AD, Popovich PG, Ramer MS (2011) Wallerian degeneration: Gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation 8:110. https://doi.org/10.1186/1742-2094-8-110
Leung L, Cahill CM (2010) TNF-alpha and neuropathic pain--a review. J Neuroinflammation 7:27. https://doi.org/10.1186/1742-2094-7-27
Baldelli P, Forni PE, Carbone E (2000) BDNF, NT-3 and NGF induce distinct new Ca2+ channel synthesis in developing hippocampal neurons. Eur J Neurosci 12(11):4017–4032
Wu C, Erickson MA, Xu J, Wild KD, Brennan TJ (2009) Expression profile of nerve growth factor after muscle incision in the rat. Anesthesiology 110(1):140–149. https://doi.org/10.1097/ALN.0b013e318190bc84
Sahbaie P, Shi X, Guo TZ, Qiao Y, Yeomans DC, Kingery WS, Clark JD (2009) Role of substance P signaling in enhanced nociceptive sensitization and local cytokine production after incision. Pain 145(3):341–349. https://doi.org/10.1016/j.pain.2009.06.037
Wu C, Boustany L, Liang H, Brennan TJ (2007) Nerve growth factor expression after plantar incision in the rat. Anesthesiology 107(1):128–135. https://doi.org/10.1097/01.anes.0000267512.08619.bd
Spofford CM, Brennan TJ (2012) Gene expression in skin, muscle, and dorsal root ganglion after plantar incision in the rat. Anesthesiology 117(1):161–172. https://doi.org/10.1097/ALN.0b013e31825a2a2b
Banik RK, Subieta AR, Wu C, Brennan TJ (2005) Increased nerve growth factor after rat plantar incision contributes to guarding behavior and heat hyperalgesia. Pain 117(1–2):68–76. https://doi.org/10.1016/j.pain.2005.05.017
Liu B, Liu Y, Li N, Zhang J, Zhang X (2018) Oxycodone regulates incision-induced activation of neurotrophic factors and receptors in an acute post-surgery pain rat model. J Pain Res 11:2663–2674. https://doi.org/10.2147/JPR.S180396
Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I et al (1997) Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A 94(4):1527–1532
Fjell J, Cummins TR, Dib-Hajj SD, Fried K, Black JA, Waxman SG (1999) Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Brain Res Mol Brain Res 67(2):267–282
Fjell J, Cummins TR, Davis BM, Albers KM, Fried K, Waxman SG, Black JA (1999) Sodium channel expression in NGF-overexpressing transgenic mice. J Neurosci Res 57(1):39–47. https://doi.org/10.1002/(SICI)1097-4547(19990701)57:1<39::AID-JNR5>3.0.CO;2-M
Brackenbury WJ, Djamgoz MB (2007) Nerve growth factor enhances voltage-gated Na+ channel activity and transwell migration in Mat-LyLu rat prostate cancer cell line. J Cell Physiol 210(3):602–608. https://doi.org/10.1002/jcp.20846
Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, Wood JN (2004) Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A 101(34):12706–12711. https://doi.org/10.1073/pnas.0404915101
Atmaramani RR, Black BJ, de la Pena JB, Campbell ZT, Pancrazio JJ (2020) Conserved expression of Nav1.7 and Nav1.8 contribute to the spontaneous and thermally evoked excitability in IL-6 and NGF-sensitized adult dorsal root ganglion neurons in vitro. Bioengineering (Basel) 7(2). https://doi.org/10.3390/bioengineering7020044
Laedermann CJ, Abriel H, Decosterd I (2015) Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front Pharmacol 6:263. https://doi.org/10.3389/fphar.2015.00263
Dib-Hajj SD, Cummins TR, Black JA, Waxman SG (2010) Sodium channels in normal and pathological pain. Annu Rev Neurosci 33:325–347. https://doi.org/10.1146/annurev-neuro-060909-153234
Staub O, Rotin D (2006) Role of ubiquitylation in cellular membrane transport. Physiol Rev 86(2):669–707. https://doi.org/10.1152/physrev.00020.2005
van Bemmelen MX, Rougier JS, Gavillet B, Apotheloz F, Daidie D, Tateyama M, Rivolta I, Thomas MA et al (2004) Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res 95(3):284–291. https://doi.org/10.1161/01.RES.0000136816.05109.89
Fotia AB, Ekberg J, Adams DJ, Cook DI, Poronnik P, Kumar S (2004) Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J Biol Chem 279(28):28930–28935. https://doi.org/10.1074/jbc.M402820200
Laedermann CJ, Cachemaille M, Kirschmann G, Pertin M, Gosselin RD, Chang I, Albesa M, Towne C et al (2013) Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J Clin Invest 123(7):3002–3013. https://doi.org/10.1172/JCI68996
Arevalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV (2006) Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 50(4):549–559. https://doi.org/10.1016/j.neuron.2006.03.044
Deumens R, Steyaert A, Forget P, Schubert M, Lavand'homme P, Hermans E, De Kock M (2013) Prevention of chronic postoperative pain: cellular, molecular, and clinical insights for mechanism-based treatment approaches. Prog Neurobiol 104:1–37. https://doi.org/10.1016/j.pneurobio.2013.01.002
Shields SD, Cheng X, Uceyler N, Sommer C, Dib-Hajj SD, Waxman SG (2012) Sodium channel Na(v)1.7 is essential for lowering heat pain threshold after burn injury. J Neurosci 32(32):10819–10832. https://doi.org/10.1523/JNEUROSCI.0304-12.2012
Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53(1):55–63
Mills CD, Nguyen T, Tanga FY, Zhong C, Gauvin DM, Mikusa J, Gomez EJ, Salyers AK et al (2013) Characterization of nerve growth factor-induced mechanical and thermal hypersensitivity in rats. Eur J Pain 17(4):469–479. https://doi.org/10.1002/j.1532-2149.2012.00202.x
Flatters SJ (2008) Characterization of a model of persistent postoperative pain evoked by skin/muscle incision and retraction (SMIR). Pain 135(1–2):119–130. https://doi.org/10.1016/j.pain.2007.05.013
Storkson RV, Kjorsvik A, Tjolsen A, Hole K (1996) Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods 65(2):167–172
Ackermann TF, Boini KM, Beier N, Scholz W, Fuchss T, Lang F (2011) EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell Physiol Biochem 28(1):137–146. https://doi.org/10.1159/000331722
Shukla S, Shariat-Madar Z, Walker LA, Tekwani BL (2016) Mechanism for neurotropic action of vorinostat, a pan histone deacetylase inhibitor. Mol Cell Neurosci 77:11–20. https://doi.org/10.1016/j.mcn.2016.09.003
Kudo TA, Kanetaka H, Mochizuki K, Tominami K, Nunome S, Abe G, Kosukegawa H, Abe T et al (2015) Induction of neurite outgrowth in PC12 cells treated with temperature-controlled repeated thermal stimulation. PLoS One 10(4):e0124024. https://doi.org/10.1371/journal.pone.0124024
Liu G, Honisch S, Liu G, Schmidt S, Pantelakos S, Alkahtani S, Toulany M, Lang F et al (2015) Inhibition of SGK1 enhances mAR-induced apoptosis in MCF-7 breast cancer cells. Cancer Biol Ther 16(1):52–59. https://doi.org/10.4161/15384047.2014.986982
Sun R, Liu Y, Hou B, Lei Y, Bo J, Zhang W, Sun Y, Zhang Y et al (2019) Perioperative activation of spinal alpha7 nAChR promotes recovery from preoperative stress-induced prolongation of postsurgical pain. Brain Behav Immun 79:294–308. https://doi.org/10.1016/j.bbi.2019.02.017
Ying YL, Wei XH, Xu XB, She SZ, Zhou LJ, Lv J, Li D, Zheng B et al (2014) Over-expression of P2X7 receptors in spinal glial cells contributes to the development of chronic postsurgical pain induced by skin/muscle incision and retraction (SMIR) in rats. Exp Neurol 261:836–843. https://doi.org/10.1016/j.expneurol.2014.09.007
Peters CM, Eisenach JC (2010) Contribution of the chemokine (C-C motif) ligand 2 (CCL2) to mechanical hypersensitivity after surgical incision in rats. Anesthesiology 112(5):1250–1258. https://doi.org/10.1097/ALN.0b013e3181d3d978
Izumi Y, Sasaki M, Hashimoto S, Sawa T, Amaya F (2015) mTOR signaling controls VGLUT2 expression to maintain pain hypersensitivity after tissue injury. Neuroscience 308:169–179. https://doi.org/10.1016/j.neuroscience.2015.09.013
Li CQ, Xu JM, Liu D, Zhang JY, Dai RP (2008) Brain derived neurotrophic factor (BDNF) contributes to the pain hypersensitivity following surgical incision in the rats. Mol Pain 4:27. https://doi.org/10.1186/1744-8069-4-27
Nijs J, Meeus M, Versijpt J, Moens M, Bos I, Knaepen K, Meeusen R (2015) Brain-derived neurotrophic factor as a driving force behind neuroplasticity in neuropathic and central sensitization pain: a new therapeutic target? Expert Opin Ther Targets 19(4):565–576. https://doi.org/10.1517/14728222.2014.994506
Zhao P, Waxman SG, Hains BC (2007) Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci 27(33):8893–8902. https://doi.org/10.1523/JNEUROSCI.2209-07.2007
Woolf CJ (2011) Central sensitization: implications for the diagnosis and treatment of pain. Pain 152(3 Suppl):S2–S15. https://doi.org/10.1016/j.pain.2010.09.030
Matzner O, Devor M (1992) Na+ conductance and the threshold for repetitive neuronal firing. Brain Res 597(1):92–98
Hoffmann T, Sharon O, Wittmann J, Carr RW, Vyshnevska A, Col R, Nassar MA, Reeh PW et al (2018) NaV1.7 and pain: Contribution of peripheral nerves. Pain 159(3):496–506. https://doi.org/10.1097/j.pain.0000000000001119
Girard BM, Merrill L, Malley S, Vizzard MA (2013) Increased TRPV4 expression in urinary bladder and lumbosacral dorsal root ganglia in mice with chronic overexpression of NGF in urothelium. J Mol Neurosci 51(2):602–614. https://doi.org/10.1007/s12031-013-0033-5
Shu X, Mendell LM (2001) Acute sensitization by NGF of the response of small-diameter sensory neurons to capsaicin. J Neurophysiol 86(6):2931–2938. https://doi.org/10.1152/jn.2001.86.6.2931
Drew LJ, Rohrer DK, Price MP, Blaver KE, Cockayne DA, Cesare P, Wood JN (2004) Acid-sensing ion channels ASIC2 and ASIC3 do not contribute to mechanically activated currents in mammalian sensory neurones. J Physiol 556(Pt 3):691–710. https://doi.org/10.1113/jphysiol.2003.058693
Canu N, Amadoro G, Triaca V, Latina V, Sposato V, Corsetti V, Severini C, Ciotti MT et al (2017) The intersection of NGF/TrkA signaling and amyloid precursor protein processing in Alzheimer’s disease neuropathology. Int J Mol Sci 18(6). https://doi.org/10.3390/ijms18061319
Isaev NK, Stelmashook EV, Genrikhs EE (2017) Role of nerve growth factor in plasticity of forebrain cholinergic neurons. Biochemistry (Mosc) 82(3):291–300. https://doi.org/10.1134/S0006297917030075
Su R, Su W, Jiao Q (2019) NGF protects neuroblastoma cells against beta-amyloid-induced apoptosis via the Nrf2/HO-1 pathway. FEBS Open Bio 9(12):2063–2071. https://doi.org/10.1002/2211-5463.12742
Dai C, Xiao X, Zhang Y, Xiang B, Hoyer D, Shen J, Velkov T, Tang S (2020) Curcumin attenuates colistin-induced peripheral neurotoxicity in mice. ACS Infect Dis 6(4):715–724. https://doi.org/10.1021/acsinfecdis.9b00341
Zhao G, Ding X, Guo Y, Chen W (2014) Intrathecal lidocaine neurotoxicity: combination with bupivacaine and ropivacaine and effect of nerve growth factor. Life Sci 112(1–2):10–21. https://doi.org/10.1016/j.lfs.2014.07.003
Ferreira RS, Dos Santos NAG, Martins NM, Fernandes LS, Dos Santos AC (2018) Caffeic acid phenethyl ester (CAPE) protects PC12 cells from cisplatin-induced neurotoxicity by activating the NGF-signaling pathway. Neurotox Res 34(1):32–46. https://doi.org/10.1007/s12640-017-9849-z
Cabrera JR, Viejo-Borbolla A, Alcami A, Wandosell F (2016) Secreted herpes simplex virus-2 glycoprotein G alters thermal pain sensitivity by modifying NGF effects on TRPV1. J Neuroinflammation 13(1):210. https://doi.org/10.1186/s12974-016-0677-5
Eskander MA, Ruparel S, Green DP, Chen PB, Por ED, Jeske NA, Gao X, Flores ER et al (2015) Persistent nociception triggered by nerve growth factor (NGF) is mediated by TRPV1 and oxidative mechanisms. J Neurosci 35(22):8593–8603. https://doi.org/10.1523/JNEUROSCI.3993-14.2015
Capsoni S, Brandi R, Arisi I, D'Onofrio M, Cattaneo A (2011) A dual mechanism linking NGF/proNGF imbalance and early inflammation to Alzheimer’s disease neurodegeneration in the AD11 anti-NGF mouse model. CNS Neurol Disord Drug Targets 10(5):635–647. https://doi.org/10.2174/187152711796235032
Kassmann M, Hansel A, Leipold E, Birkenbeil J, Lu SQ, Hoshi T, Heinemann SH (2008) Oxidation of multiple methionine residues impairs rapid sodium channel inactivation. Pflugers Arch 456(6):1085–1095. https://doi.org/10.1007/s00424-008-0477-6
Jeong EM, Liu M, Sturdy M, Gao G, Varghese ST, Sovari AA, Dudley SC Jr (2012) Metabolic stress, reactive oxygen species, and arrhythmia. J Mol Cell Cardiol 52(2):454–463. https://doi.org/10.1016/j.yjmcc.2011.09.018
Schluter F, Leffler A (2016) Oxidation differentially modulates the recombinant voltage-gated Na(+) channel alpha-subunits Nav1.7 and Nav1.8. Brain Res 1648(Pt A):127–135. https://doi.org/10.1016/j.brainres.2016.07.031
Calabrese EJ, Calabrese V, Tsatsakis A, Giordano JJ (2020) Hormesis and Ginkgo biloba (GB): numerous biological effects of GB are mediated via hormesis. Ageing Res Rev:101019. https://doi.org/10.1016/j.arr.2020.101019
Calabrese V, Santoro A, Monti D, Crupi R, Di Paola R, Latteri S, Cuzzocrea S, Zappia M et al (2018) Aging and Parkinson's disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med 115:80–91. https://doi.org/10.1016/j.freeradbiomed.2017.10.379
Calabrese V, Santoro A, Trovato Salinaro A, Modafferi S, Scuto M, Albouchi F, Monti D, Giordano J et al (2018) Hormetic approaches to the treatment of Parkinson's disease: perspectives and possibilities. J Neurosci Res 96(10):1641–1662. https://doi.org/10.1002/jnr.24244
Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP (2010) Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid Redox Signal 13(11):1763–1811. https://doi.org/10.1089/ars.2009.3074
Abriel H, Kamynina E, Horisberger JD, Staub O (2000) Regulation of the cardiac voltage-gated Na+ channel (H1) by the ubiquitin-protein ligase Nedd4. FEBS Lett 466(2–3):377–380
Cachemaille M, Laedermann CJ, Pertin M, Abriel H, Gosselin RD, Decosterd I (2012) Neuronal expression of the ubiquitin ligase Nedd4-2 in rat dorsal root ganglia: modulation in the spared nerve injury model of neuropathic pain. Neuroscience 227:370–380. https://doi.org/10.1016/j.neuroscience.2012.09.044
Boehmer C, Wilhelm V, Palmada M, Wallisch S, Henke G, Brinkmeier H, Cohen P, Pieske B et al (2003) Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc Res 57(4):1079–1084. https://doi.org/10.1016/s0008-6363(02)00837-4
Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC (2004) cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J Biol Chem 279(44):45753–45758. https://doi.org/10.1074/jbc.M407858200
Lang F, Cohen P (2001) Regulation and physiological roles of serum- and glucocorticoid-induced protein kinase isoforms. Sci STKE 2001(108):re17. https://doi.org/10.1126/stke.2001.108.re17
Lang F, Stournaras C, Zacharopoulou N, Voelkl J, Alesutan I (2018) Serum- and glucocorticoid-inducible kinase 1 and the response to cell stress. Cell Stress 3(1):1–8. https://doi.org/10.15698/cst2019.01.170
Marlin MC, Li G (2015) Biogenesis and function of the NGF/TrkA signaling endosome. Int Rev Cell Mol Biol 314:239–257. https://doi.org/10.1016/bs.ircmb.2014.10.002
Frodin M, Antal TL, Dummler BA, Jensen CJ, Deak M, Gammeltoft S, Biondi RM (2002) A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J 21(20):5396–5407
Nakagawa T, Yokoe S, Asahi M (2016) Phospholamban degradation is induced by phosphorylation-mediated ubiquitination and inhibited by interaction with cardiac type Sarco(endo)plasmic reticulum Ca(2+)-ATPase. Biochem Biophys Res Commun 472(3):523–530. https://doi.org/10.1016/j.bbrc.2016.03.009
Ni W, Yao S, Zhou Y, Liu Y, Huang P, Zhou A, Liu J, Che L et al (2019) Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m(6)A reader YTHDF3. Mol Cancer 18(1):143. https://doi.org/10.1186/s12943-019-1079-y
Li X, Zhong L, Wang Z, Chen H, Liao D, Zhang R, Zhang H, Kang T (2018) Phosphorylation of IRS4 by CK1gamma2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics 8(13):3643–3653. https://doi.org/10.7150/thno.26021
Peng HY, Chen GD, Hsieh MC, Lai CY, Huang YP, Lin TB (2012) Spinal SGK1/GRASP-1/Rab4 is involved in complete Freund's adjuvant-induced inflammatory pain via regulating dorsal horn GluR1-containing AMPA receptor trafficking in rats. Pain 153(12):2380–2392. https://doi.org/10.1016/j.pain.2012.08.004
Peng HY, Chen GD, Lai CY, Hsieh MC, Lin TB (2013) Spinal serum-inducible and glucocorticoid-inducible kinase 1 mediates neuropathic pain via kalirin and downstream PSD-95-dependent NR2B phosphorylation in rats. J Neurosci 33(12):5227–5240. https://doi.org/10.1523/JNEUROSCI.4452-12.2013
Koyanagi S, Kusunose N, Taniguchi M, Akamine T, Kanado Y, Ozono Y, Masuda T, Kohro Y et al (2016) Glucocorticoid regulation of ATP release from spinal astrocytes underlies diurnal exacerbation of neuropathic mechanical allodynia. Nat Commun 7:13102. https://doi.org/10.1038/ncomms13102
Wen YR, Suter MR, Ji RR, Yeh GC, Wu YS, Wang KC, Kohno T, Sun WZ et al (2009) Activation of p38 mitogen-activated protein kinase in spinal microglia contributes to incision-induced mechanical allodynia. Anesthesiology 110(1):155–165. https://doi.org/10.1097/ALN.0b013e318190bc16
Ito N, Obata H, Saito S (2009) Spinal microglial expression and mechanical hypersensitivity in a postoperative pain model: comparison with a neuropathic pain model. Anesthesiology 111(3):640–648. https://doi.org/10.1097/ALN.0b013e3181b05f42
Di Rosa G, Brunetti G, Scuto M, Trovato Salinaro A, Calabrese EJ, Crea R, Schmitz-Linneweber C, Calabrese V et al (2020) Healthspan enhancement by olive polyphenols in C. elegans wild type and Parkinson's models. Int J Mol Sci 21(11). https://doi.org/10.3390/ijms21113893
Pilipenko V, Narbute K, Amara I, Trovato A, Scuto M, Pupure J, Jansone B, Poikans J et al (2019) GABA-containing compound gammapyrone protects against brain impairments in Alzheimer's disease model male rats and prevents mitochondrial dysfunction in cell culture. J Neurosci Res 97(6):708–726. https://doi.org/10.1002/jnr.24396
Miquel S, Champ C, Day J, Aarts E, Bahr BA, Bakker M, Banati D, Calabrese V et al (2018) Poor cognitive ageing: Vulnerabilities, mechanisms and the impact of nutritional interventions. Ageing Res Rev 42:40–55. https://doi.org/10.1016/j.arr.2017.12.004
Bowtell J, Kelly V (2019) Fruit-derived polyphenol supplementation for athlete recovery and performance. Sports Med 49(Suppl 1):3–23. https://doi.org/10.1007/s40279-018-0998-x
Acknowledgments
We thank Professor Stephen G. Waxman (Yale University School of Medicine, USA) for kindly providing Nav1.7 cKO mice. We would like to thank Editage (www.editage.cn) for the English language editing.
Availability of Data and Material
All data of the study can be acquired from corresponding author through email.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant number: 81271235).
Author information
Authors and Affiliations
Contributions
Study concept and supervision: X.W.Z; study design: X.W.Z and B.W.L; acquisition and analysis: B.W.L, J.Z, Y.S.H, N.B.L, Y.L, M.Z, W.Y.W and H.Z; manuscript preparation: X.W.Z, B.W.L, and A.L.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
All experiments were approved by the ethical committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (IRB ID: TJ-A20180801) and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and followed the guidelines of the International Association for the Study of Pain.
Consent to Participate
Not applicable.
Consent for Publication
All authors consent to the publication of current data.
Code Availability
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, BW., Zhang, J., Hong, YS. et al. NGF-Induced Nav1.7 Upregulation Contributes to Chronic Post-surgical Pain by Activating SGK1-Dependent Nedd4-2 Phosphorylation. Mol Neurobiol 58, 964–982 (2021). https://doi.org/10.1007/s12035-020-02156-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02156-1