
Abstract
Cisplatin is a highly effective chemotherapeutic drug that is toxic to the peripheral nervous system. Findings suggest that axons are early targets of the neurotoxicity of cisplatin. Although many compounds have been reported as neuroprotective, there is no effective treatment against the neurotoxicity of cisplatin. Caffeic acid phenethyl ester (CAPE) is a propolis component with neuroprotective potential mainly attributed to antioxidant and anti-inflammatory mechanisms. We have recently demonstrated the neurotrophic potential of CAPE in a cellular model of neurotoxicity related to Parkinson’s disease. Now, we have assessed the neurotrophic and neuroprotective effects of CAPE against cisplatin-induced neurotoxicity in PC12 cells. CAPE (10 μM) attenuated the inhibition of neuritogenesis and the downregulation of markers of neuroplasticity (GAP-43, synapsin I, synaptophysin, and 200-kD neurofilament) induced by cisplatin (5 μM). This concentration of cisplatin does not affect cell viability, and it was used in order to assess the early neurotoxic events triggered by cisplatin. When a lethal dose of cisplatin was used (IC50 = 32 μM), CAPE (10 μM) increased cell viability. The neurotrophic effect of CAPE is not dependent on NGF nor is it additive to the effect of NGF, but it might involve the activation of the NGF-high-affinity receptors (trkA). The involvement of other neurotrophin receptors such as trkB and trkC is unlikely. This is the first study to demonstrate the protective potential of CAPE against the neurotoxicity of cisplatin and to suggest the involvement of trkA receptors in the neuroprotective mechanism of CAPE. Based on these findings, the beneficial effect of CAPE on cisplatin-induced peripheral neuropathy should be further investigated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cisplatin (cis-diamminedichloroplatinum II) is an effective broad-spectrum chemotherapeutic agent. One of the major side effects that limit the success of cisplatin chemotherapy is the peripheral sensory neuropathy that occurs in 50% of patients and for which there is no effective therapy (Burton et al. 2007; Albers et al. 2014; Hausheer et al. 2006). Studies have suggested that thiol compounds (amifostine and glutathione), vitamin E, calcium, and magnesium are chemoprotective (Albers et al. 2014; Cascinu et al. 2002; Cascinu et al. 1995; Pace et al. 2003; Planting et al. 1999; Avan et al. 2015). However, there is no solid evidence of the efficacy and safety of these agents that support their therapeutic use against cisplatin-induced peripheral neuropathy (Avan et al. 2015; Albers et al. 2014).
Caffeic acid phenethyl ester (CAPE) is a natural bioactive compound that is extracted from the propolis of honeybee hives (Murtaza et al. 2014). The pharmacological activities of CAPE have been associated with the inhibition of the transcription factor NF-kB, which is involved in inflammation and oxidative stress (Natarajan et al. 1996; van den Berg et al. 2001). In vitro and in vivo studies have suggested that CAPE might be beneficial in neurodegenerative diseases (Bak et al. 2016; Barros Silva et al. 2013; Huang et al. 2013; Santos et al. 2014). Different molecular targets of CAPE have been identified (for review, refer to Tolba et al. 2013), and they are all related to anti-inflammatory and antioxidant mechanisms.
We have previously demonstrated the beneficial effect of CAPE in a rat model of Parkinson’s disease induced by the dopaminergic neurotoxin 6-hydroxidopamine (Barros Silva et al. 2013). We have also demonstrated the involvement of neuritogenesis in the neuroprotective effect of CAPE in a PC12 model treated with 1-methyl-4-phenylpyridinium iodide (MPP+ iodide), a neurotoxicity model related to Parkinson’s disease (Santos et al. 2014). Additionally, we have recently demonstrated some pathways and early targets that might be involved in the neurotoxicity of cisplatin (Ferreira et al. 2016b). Now, we have used the same model (PC12 cells treated with non-cytotoxic concentrations of cisplatin) to investigate (i) the neuroprotective effects of CAPE on the modulators of neuritogenesis that are affected by cisplatin, (ii) the role of NGF itself, and (iii) the involvement of the NGF-triggered pathway in the mechanism of neuroprotection of CAPE. Currently, there are no studies addressing the involvement of neuroplasticity-related pathways in the neuroprotective mechanism of CAPE against the neurotoxicity of cisplatin.
Materials and Methods
Chemicals
High-purity reagents (analytical grade minimum) were obtained from Sigma-Aldrich® (St. Louis, MO, USA), unless differently stated. Cell culture media were purchased from Invitrogen (Carlsbad, CA). Reagents for Western blot analyses were obtained from Bio-Rad Laboratories, Inc. (Hercules, CA, USA). The solutions were prepared with ultra-pure water (type I) purified by a Milli-Q Gradient system (Millipore, Bedford, USA). The stock solution of cisplatin (3.3 mM) was prepared in 0.9% saline solution, and the stock solution of CAPE (100 mM) was prepared in DMSO and stored in a freezer (− 20 °C). The working solutions were prepared immediately before the assays. The working solution of CAPE was prepared by diluting the stock solution in the differentiation medium (F-12K Nutrient Mixture Kaighn’s Modification), while the working solutions of cisplatin were prepared in saline.
Cell Cultures
PC12 and SH-SY5Y cells were obtained from the American Type Culture Collection (ATCC) (Rockville, MD).
PC12 cells were cultured in 75 cm2 culture flasks, at 37 °C, under a humidified atmosphere containing 5% CO2 and 95% air. The growth medium was Dulbecco’s modified Eagle medium (DMEM; GIBCO®) supplemented with 10% heat-inactivated horse serum (GIBCO®), 5% heat-inactivated fetal bovine serum (FBS; GIBCO®), and 1% antibiotic mixture (5 mg/mL penicillin, 5 mg/mL streptomycin, and 10 mg/mL neomycin, PNS GIBCO®). Medium was renewed every 3 days. To harvest PC12 cells, medium was removed and cells were detached with trypsin/EDTA solution (GIBCO®). Trypsin was inactivated by the addition of supplemented medium, and after centrifugation (1000 rpm, 5 min), cells were suspended in the growth medium and plated at the density required by each assay.
SH-SY5Y cells were cultured in 75-cm2 culture flasks, with F12 nutrient mixture (F12 HAM; Sigma Cell Culture, St. Louis, MO) supplemented with 15% FBS (GIBCO) and 1% antibiotic mixture (PNS), at 37 °C, under a humidified atmosphere containing 5% CO2 and 95% air. The growth medium was renewed every day. Confluent cultures were detached with trypsin/EDTA solution (GIBCO®), which was inactivated with growth medium. After centrifugation (1000 rpm, 5 min), cells were subcultured (1:2; every 2–3 days). Third-passage cells with 80% confluence were used in the experiments.
Working Concentrations of Cisplatin
The working concentrations of cisplatin were previously determined (Ferreira et al. 2016b). Thus, for the cell viability assay, the toxicity was induced with 32 μM of cisplatin (IC50), while that in the neurite outgrowth assay, we used a non-cytotoxic concentration of cisplatin (5 μM).
Cell Viability—MTT Assay
PC12 cells were seeded into 96-well plates (2.0 × 104 cells/well) and cultured for 24 h, then incubated with CIS (32 μM, IC50) and CAPE (1, 5, 10, 25, 50, and 100 μM) for 24 h. The protective effect of CAPE against cisplatin-induced cytotoxicity was measured using the MTT colorimetric assay (Mosmann 1983). After 24 h of treatment, plates were incubated with 20 μL of MTT solution (5 mg/mL) for 3 h at 37 °C. Then, plates were centrifuged (1000 rpm, 5 min), the supernatant was discarded, and 200 μL of DMSO was added to dissolve the formazan crystals. The absorbance was measured at 570 nm on a microplate reader (Multiskan FC, Thermo Scientific). Cell viability was expressed as a percentage of the control.
Determination of the Working Concentration of CAPE
PC12 cells were seeded into 24-well plates (2.0 × 105 cells/well) coated with poly-l-lysine (Sigma-Aldrich®, St. Louis, MO, USA) and incubated for 24 h for better adhesion. After this, the medium was replaced by F-12K Nutrient Mixture Kaighn’s Modification (GIBCO®) supplemented with 1% horse serum, 1% antibiotic mixture, and NGF (100 ng/mL). Cells were exposed to 5 μM CIS + serial dilutions of CAPE (1, 5, 10, 25, and 50 μM) for 72 h. Untreated cells were used as controls. The concentration of CAPE that showed the highest ability to minimize the inhibition of cellular differentiation induced by the sublethal concentration of cisplatin (5 μM) was selected as the working concentration.
Quantitative Neurite Outgrowth Assay in PC12 Cells
PC12 cells (2.0 × 105 cells/well) were seeded in 24-well poly-l-lysine-coated plates (Sigma-Aldrich®, St. Louis, MO, USA) and incubated for 24 h for adhesion. Then, the growth medium was replaced by the differentiation medium F-12K Nutrient Mixture Kaighn’s Modification (GIBCO®) supplemented with 1% horse serum and 1% antibiotic mixture. Cells were then incubated (37 °C, 72 h) with one of the following additions: NGF 100 ng/mL, CAPE 10 μM, CAPE 10 μM + NGF 100 ng/mL, CIS 5 μM + NGF 100 ng/mL, or CAPE 10 μM + CIS 5 μM + NGF 100 ng/mL. Untreated cells were used as controls. The morphometric analysis was performed on the images obtained under inverted-phase-contrast microscopy (Carl Zeiss Axio Observer A1 inverted microscope, ×400 magnification), after 72 h of incubation. The percentage of differentiated cells was determined by using the ImageJ open-source software (Rasband, 1997–2014). Cells with at least one neurite with a length equal to (or higher than) the cell body diameter were considered as differentiated (Das et al. 2004).
Quantitative Neurite Outgrowth Assay in SH-SY5Y Neuroblastoma Cells
SH-SY5Y neuroblastoma cells were incubated in 24-well plates (3.0 × 104 cells/well) in the F12 HAM medium, supplemented with 15% FBS and 1% antibiotic mixture (penicillin/streptomycin/neomycin, PSN GIBCO ®). After 24 h, the medium was replaced by a low-serum medium (F12 HAM supplemented with 1% FBS, 1% PSN). Then, cells were incubated (37 °C, 72 h) with one of the following additions: CAPE 10 μM, retinoic acid (RA) 10 μM, or CAPE 10 μM + RA 10 μM. Untreated cells were used as controls. The morphometric analysis was performed on the images obtained under inverted-phase-contrast microscopy (Carl Zeiss Axio Observer A1 inverted microscope, ×400 magnification), after 72 h of incubation. The percentage of differentiated cells was determined by using the ImageJ open-source software (Rasband, 1997–2014). Cells with at least one neurite with a length equal to (or higher than) the cell body diameter were considered as differentiated (Das et al. 2004).
Western Blot Analysis for GAP-43, Synapsin I, and Synaptophysin
After the additions described for the neurite outgrowth assay, cells were incubated for 72 h. To prepare cell lysates, PC12 cells (2.0 × 105 cells/well) were detached with trypsin, transferred to conical microtubes, and centrifuged (1000 rpm, 5 min, 4 °C). The supernatant was discarded and the cell pellet was suspended in 40 μL Tris–Triton lysis buffer (10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, Triton X-100 1%, 10% glycerol, 0.1% SDS, 0.5% deoxycholate, 1:200 protease inhibitor cocktail, and 1% phosphatase inhibitor cocktail). The lysis procedure was performed in tubes placed on ice to reduce the activity of proteases. After 10 min, the cell lysate was centrifuged at 12,000 rpm for 10 min at 4 °C and the supernatant was stored in a freezer (− 80 °C) until the assays. Cell lysate (10 μL) was assayed for protein content by the Bradford method. The Protein Assay Dye Reagent (Bio-Rad) was used according to the manufacturer’s instructions. Lysates and color reagent were diluted with water (1:5) and a calibration curve of BSA (40, 100, 200, and 400 μg/mL) was assayed. The absorbance (595 nm) was determined in a microplate reader (Multiskan FC, Thermo Scientific). The concentration of protein was calculated based on the calibration curve and multiplied by the dilution factor (5). For the SDS-PAGE (SDS-polyacrylamide gel electrophoresis), samples were added to an equal volume of Laemmli sample buffer (65.8 mM Tris, pH 6.8, 26.3% glycerol, 2.1% SDS, 0.01% bromophenol blue, 5% β-mercaptoethanol) and heated to 98 °C for 5 min. Aliquots of 35 μL containing 10 μg total protein were applied to 10% polyacrylamide gel (10 wells) and separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes (1 h, 0.37 A, Tris/glycine buffer). Prior to the immune reaction, the membranes were blocked (30 min, room temperature, 300 rpm) with 5% non-fat milk or 5% BSA in Tween 20/TBS buffer (TTBS). The blocked membranes were incubated with the primary antibodies: anti-GAP-43 (1:1250), anti-synaptophysin (1:400), or anti-synapsin I (1:1000), overnight, at 4 °C, 300 rpm. Then, the membranes were washed with TTBS and incubated (1 h, room temperature, 300 rpm) with the secondary antibody conjugated with horseradish peroxidase (anti-mouse or anti-rabbit IgG-HRP. 1:20,000). After this, the membranes were washed with TTBS and TBS and treated with 3 mL of chemiluminescence enhancer detection reagent (1:1). Images were captured by using ChemiDoc system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Quantitation was performed on the images based on the optical densitometry (OD) of the bands by using the open-source software ImageJ (Rasband, 1997–2014). Finally, the membranes were stripped (2% SDS, 62.5 mM Tris, pH 6.8, and 100 mM mercaptoethanol) and reprobed for loading control (anti-β-actin 1:3000). OD values of GAP-43, synapsin I, and synaptophysin bands were divided by the OD values of β-actin for normalization of the results (L’Episcopo et al. 2011).
TrkA Receptor Inhibition Assay
PC12 cells were seeded in 24-well plates coated with poly-l-lysine (Sigma-Aldrich®, St. Louis, MO, USA) at a density of 2.0 × 105 cells/well and incubated for 24 h, at 37 °C for adhesion. Then, the medium was replaced by F-12K Nutrient Mixture Kaighn’s Modification (GIBCO®) supplemented with 1% horse serum and 1% antibiotic mixture (penicillin/streptomycin/neomycin, GIBCO®). Cells were then incubated (37 °C, 168 h), with one of the following additions: NGF 100 ng/mL, CAPE 10 μM, K252a 100 nM + NGF 100 ng/mL, and K252a 100 nM + CAPE 10 μM. Untreated cells were used as controls. K252a (Sigma-Aldrich®, St. Louis, MO, USA) is a specific inhibitor of trk (tyrosine kinase) receptors, which selectively blocks the effect of nerve growth factor (NGF) and agonists of trkA in PC12 cells (Tapley et al. 1992). The neurite outgrowth was assessed by inverted-phase-contrast microscopy (Carl Zeiss Axio Observer A1 inverted microscope, ×400 magnification). Phase-contrast photomicrographs of four fields per well were taken after incubation for 24, 48, 72, 96, 120, 144, and 168 h. The percentage of cells with neurites was determined in digitalized images by using the ImageJ open-source software (Rasband, 1997–2014). Only those cells with at least one neurite with a length equal to or greater than the diameter of the cell body were considered differentiated (Das et al. 2004).
Determination of NGF Levels
PC12 cells (2.0 × 105 cell/well) were seeded in 24-well plates and incubated at 37 °C for 24 h prior to additions. Untreated cells were used as controls. Then, cells were incubated at 37 °C for 72 h. After treatment, an aliquot of 100 μL of culture supernatant was analyzed for the content of NGF by using enzyme-linked immunosorbent assay kit RAB0381 (Sigma-Aldrich®, St. Louis, MO, USA) as recommended by the manufacturer. The absorbance (450 nm) was determined in a microplate reader (Multiskan FC, Thermo Scientific).
Determination of Neurofilaments (NF-200) by Immunofluorescence
Immunofluorescence staining of neurofilament-200 was performed according to a previously described procedure (Schimmelpfeng et al. 2004) with minor modifications. PC12 cells were seeded (2.0 × 105 cells/well) in 12-well plates containing one sterilized coverslip coated with poly-l-lysine per well (Sigma-Aldrich®, St. Louis, MO, USA). After a 24-h incubation, cells were treated with one of the following additions: NGF 100 ng/mL, NGF 100 ng/mL + CIS 5 μM, CAPE 10 μM, CAPE 10 μM + NGF 100 ng/mL, or CAPE 10 μM + CIS 5 μM + NGF 100 ng/mL, and incubated (37 °C, 5% CO2, 72 h). On the day of the experiment, the medium of each well was removed and the cells were fixed in 4% paraformaldehyde (20 min at room temperature). Cells were washed twice with PBS (phosphate-buffered saline) and permeabilized for 10 min at room temperature with PBS containing 0.2% Triton X-100. Subsequently, proteins were blocked for 30 min (room temperature) with a PBS solution containing 1% BSA and 0.1% Tween 20. Then, cells were incubated (4 °C, overnight) with the primary antibody, anti-neurofilament-200 produced in rabbit (Sigma-Aldrich®, St. Louis, MO, USA, 1:80 dilution in PBS containing 3% BSA). The following day, cells were carefully washed twice with PBS followed by the incubation (room temperature, 1 h in the dark) with the fluorophore-conjugated secondary antibody (anti-rabbit IgG-fluorescein isothiocyanate, FITC), produced in sheep (Sigma-Aldrich®, St. Louis, MO, USA, 1:80 dilution in PBS containing 3% BSA). The washing procedure was repeated and the nuclei were stained with 20 μM Hoechst 33342 (Sigma-Aldrich®, St. Louis, MO, USA) for 2 min. Finally, coverslips were mounted on slides and observed under fluorescence microscopy using FITC and Hoechst filters. Images were captured with Standard Cell Sense software. Corrected total cell fluorescence (CTCF) was calculated by using the ImageJ open-source software (Rasband, 1997–2014) and the following formula CTCF = Integrated density − (area of selected cell × mean fluorescence of background readings).
Statistical Analysis
Data were expressed as mean ± standard error of mean (SEM). Statistical analysis was carried out by using one-way ANOVA (analysis of variance) for multiple comparisons, followed by the Bonferroni post-test (GraphPad Prism Software, version 5.0 for Windows, San Diego, CA, USA). Values of p < 0.05 were considered significant. Experiments were repeated three times using cells from different cultures. The experiment of each day was performed in triplicate.
Results
CAPE Protected Against the Cell Death and the Inhibition of Cell Differentiation Induced by Cisplatin
The viability of cells incubated with the IC50 (32 μM) of cisplatin decreased (39.11 ± 1.9%) approximately 50% as compared to controls (86.39 ± 2.5%), which is in line with our previous findings (Ferreira et al. 2016b). Concentrations of CAPE from 10 to 100 μM (10 μM, 50.97 ± 2.8%; 25 μM, 51.24 ± 1.3%; 50 μM, 64.75 ± 2.1%; 100 μM, 88.78 ± 4.1%) protected against the cytotoxicity induced by 32 μM cisplatin (39.11 ± 1.9%). Concentrations of 1 and 5 μM CAPE had no beneficial effects (Fig. 1).

Effect of different concentrations of CAPE on the viability of PC12 cells exposed to cisplatin (IC50 = 32 μM). Additions: NGF 100 ng/mL; CIS 32 μM; CAPE (1 to 50 μM). Bars represent means ± SEM of three independent experiments; each experiment was performed in triplicate. *Significantly different from control (p < 0.05); #significantly different from CIS 32 μM (p < 0.05). CIS cisplatin, NS not significantly different for p < 0.05
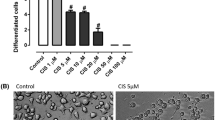
Five different concentrations of CAPE (1, 5, 10, 25, and 50 μM) were evaluated for their potential to minimize the inhibition of cellular differentiation induced by 5 μM cisplatin, which according to our previous findings is the lowest concentration of cisplatin that impairs neurite outgrowth without causing cell death (Ferreira et al. 2016b). All the tested concentrations of CAPE protected against the inhibition of cellular differentiation induced by CIS (5 μM); however, the lowest concentration of CAPE with the highest effectiveness was 10 μM (Fig. 2a). The percentage of differentiation was significantly higher (10.85 ± 0.31%) in cells treated with CIS (5 μM) and CAPE (10 μM) as compared to cells treated with CIS (5 μM) alone (6.12 ± 0.29%). Therefore, as CAPE (10 μM) protected against the cell death induced by the IC50 of CIS (Fig. 1) and against the inhibition of cellular differentiation induced by CIS (5 μM), this concentration of CAPE (10 μM) was selected to investigate the mechanisms of neuroprotection. The bar graph showing the effects of all concentrations of CAPE is presented in Fig. 2a and the phase-contrast photomicrographs of the selected concentration are presented in Fig. 2b.

Effect of different concentrations of CAPE on cisplatin-induced inhibition of cell differentiation. Additions: NGF 100 ng/mL, CIS 5 μM, CAPE (1 to 50 μM). a Bar graph. Each bar represents the mean ± SEM obtained from three independent experiments; each experiment was performed in triplicate. b Phase-contrast photomicrographs showing the protective effect of CAPE (10 μM) against the inhibition of the differentiation induced by cisplatin (5 μM) in NGF-stimulated PC12 cells. Cells with at least one neurite with a length equal to or higher than the cell body were considered differentiated and expressed as a percentage of the total cells in the field. *Significantly different from the controls (p < 0.05); #significantly different from 5 μM CIS (p < 0.05); NS, not significantly different for p < 0.05
CAPE (10 μM) Minimized the Inhibitory Effect of CIS (5 μM) on the Expression of GAP-43, Synapsin I, and Synaptophysin
The expression of GAP-43 (0.48 ± 0.07), synapsin I (1.01 ± 0.04), and synaptophysin (1.36 ± 0.17) was significantly increased in the group CIS (5 μM) + CAPE (10 μM) as compared to the group exposed to CIS (5 μM) alone (0.17 ± 0.07, 0.45 ± 0.16, and 0.57 ± 0.05, respectively). The protein bands are presented in Fig. 3a, and bar graphs of each protein are presented in Fig. 3b–d.

Effect of CAPE on the expression of axonal proteins in PC12 cells treated with CIS. Additions: NGF 100 ng/mL, CIS 5 μM, CAPE (10 μM). a Western blot bands. b, c, d Bar graphs of β-actin-normalized optical densities (OD) of GAP-43, synapsin I, and synaptophysin. Additions: NGF 100 ng/mL, cisplatin 5 μM, and CAPE 10 μM. Bars indicate means ± SEM of three independent experiments performed in triplicates. *Significantly different from controls (p < 0.05); #significantly different from CIS 5 μM (p < 0.05)
The Cellular Differentiation Induced by CAPE in PC12 Cells Was Inhibited by K-252a; CAPE Did Not Induce Differentiation in SH-SY5Y Cells
The percentage of differentiated cells was significantly higher in the group treated with NGF (100 ng/mL) as well as in the group exposed to CAPE (10 μM) as compared to controls, during the whole period of incubation (24 to 168 h). The inhibitory effect of K252a was observed from 72 h of incubation in group K252a (100 nM) + NGF (100 ng/mL) and from 96 h of incubation in group K252a (100 nM) + CAPE (10 μM) in relation to groups NGF or CAPE, respectively. There was no significant difference in the number of PC12 cells with neurites when comparing group K252a (100 nM) and controls of all periods of incubation (Fig. 4a–h).


Effect of CAPE on the differentiation of PC12 cells treated with K252a (trkA inhibitor). Additions: NGF 100 ng/mL, CAPE (10 μM), K252a 100 nM. PC12 cells were incubated with NGF or CAPE in the presence/absence of K252a. a–g Bar graphs after different periods of incubation. a 24 h. b 48 h. c 72 h. d 96 h. e 120 h. f 144 h. g 168 h. h Phase-contrast photomicrographs showing the morphological changes in PC12 cells after 168 h of incubation. Cells with at least one neurite with a length equal to or higher than the cell body were counted and expressed as a percentage of total cells in the field. Results are expressed as mean ± SEM of three different experiments performed in triplicates. *Significantly different from the negative control (p < 0.05); #significantly different from NGF 100 ng/mL (p < 0.05); &significantly different from CAPE 10 μM (p < 0.05); NS, not significantly different for p < 0.05
The percentage of differentiation in SH-SY5Y cells (which do not express trkA receptors) in the group treated with 10 μM retinoic acid (RA, 44.99 ± 1.75%) was significantly higher than that in controls (5.21 ± 0.85%). CAPE (10 μM) did not increase the differentiation (5.64 ± 0.48%) in SH-SY5Y cells as compared to controls (5.21 ± 0.85%). In the group CAPE (10 μM) + RA (10 μM), the percentage of differentiated cells was reduced (37.62 ± 1.52%) as compared to the group treated only with RA (44.99 ± 1.75%). The bar graph is presented in Fig. 5a, and the phase-contrast photomicrographs are presented in Fig. 5b.

Effect of CAPE on the differentiation of SH-SY5Y cells. Additions: RA (10 μM), CAPE (10 μM). a Bar graph. b Photomicrographs of the four groups after 72 h of incubation. Results are expressed as mean ± SEM of three different experiments performed in triplicates. *Significantly different from the control (p < 0.05); #significantly different from RA (p < 0.05). RA, retinoic acid
Both findings (Figs. 4 and 5) indicate the involvement of the NGF-selective receptors (trkA) in the mechanism by which CAPE induces neuritogenesis.
CAPE Did Not Increase the Levels of NGF in PC12 Cells; However, It Increased the Expression of Axonal Proteins and Induced Differentiation in PC12 Cells Even in the Absence of NGF
There was no significant difference in the levels of NFG between the groups CAPE (97.53 ± 3.26%) and control (97.35 ± 1.23%) (Fig. 6). Additionally, no significant difference was observed on the expression of GAP-43 (1.71 ± 0.10), synapsin I (1.27 ± 0.02), and synaptophysin (1.22 ± 0.10) between the groups CAPE and CAPE + NGF (1.59 ± 0.04, 1.27 ± 0.05, and 1.37 ± 0.05, respectively), showing that the effects of both are not additive (Fig. 7a–d).

Effect of CAPE (10 μM) on the levels of NGF in PC12 cells. No significant difference was observed between the effects of CAPE and control. Data are expressed as mean ± SEM of three experiments performed in triplicates. NS, not significantly different from control for p < 0.05

Effect of the association of CAPE and NGF on the expression of axonal proteins in PC12 cells. a Western blot bands and bar graphs of the normalized optical densities (OD) of b GAP-43, c synapsin I, and d synaptophysin. Bars indicate means ± SEM of three independent experiments performed in triplicates. *Significantly different from NGF 100 ng/mL
CAPE induced differentiation in PC12 cells either in the absence (13.21 ± 0.293) or in the presence of NGF (14.02 ± 0.525) as compared to controls (3.09 ± 0.091). No significant difference was observed between the effects of CAPE in the presence or in the absence of NGF (Fig. 8a, b). Altogether, the findings presented in Figs. 6, 7, and 8 suggest that the effect of CAPE on the differentiation of PC12 cells is not dependent on NGF nor is it additive to NGF effect.

Effect of CAPE on the differentiation of PC12 cells in the presence and in the absence of NGF. a Bar graph showing the means ± SEM of three different experiments performed in triplicates. Additions: NGF 100 ng/mL and CAPE 10 μM. b Phase-contrast photomicrographs of the four groups of treatment. Cells with at least one neurite with a length equal to or higher than the cell body were considered differentiated and expressed as a percentage of the total cells in the field. *Significantly different from the control group (p < 0.05); #significantly different from the NGF 100 ng/mL group (p < 0.05)
CAPE (10 μM) Minimized the Inhibitory Effect of CIS (5 μM) on Neurofilaments (NF-200)
The staining of neurofilament NF-200 in the groups NGF (4.60 × 106 ± 2.05 × 105), CAPE (6.11 × 106 ± 2.25 × 105), and CAPE + NGF was significantly higher than that in controls (3.02 × 106 ± 1.56 × 105). No significant difference was observed between the groups CAPE + NGF (6.13 × 106 ± 1.64 × 105) and CAPE alone (6.11 × 106 ± 2.25 × 105). In the group CIS + NGF (3.56 × 106 ± 1.95 × 105), the staining of NF-200 was significantly lower than that in the group NGF (4.60 × 106 ± 2.05 × 105). The staining of NF-200 was significantly higher in the group CIS + NGF + CAPE (4.43 × 106 ± 1.56 × 105) than that in the group CIS + NGF (3.56 × 106 ± 1.95 × 105) (Fig. 9a). Neurofilaments are stained in green and nuclei are stained in blue (Fig. 9b).

Effect of CAPE on neurofilament NF-200 of PC12 cells in the presence/absence of NGF and cisplatin. Additions: NGF 100 ng/mL, CIS 5 μM, CAPE (10 μM). a Quantification of the fluorescence of immunostained cells by using ImageJ. b Fluorescence photomicrographs showing neurofilaments stained with anti-neurofilament 200 kD labeled with FITC and nuclei stained with Hoechst 33342 (×40). Additions: NGF 100 ng/mL, CAPE 10 μM, and CIS 5 μM. In the bar graph, data are presented as mean ± SEM of three different experiments performed in triplicates. *Significantly different from the control group (p < 0.05); #significantly different from NGF (p < 0.05); &significantly different from CIS + NGF (p < 0.05)
Discussion
Cisplatin-induced neurotoxicity affects axons and cell bodies of sensory neurons in the peripheral nervous system. Neurite retraction is one of the most important events in the early stages of neuronal apoptosis (Sadri et al. 2010). We have recently demonstrated the early effects of cisplatin on the neuritogenesis of PC12 cells (Ferreira et al. 2016a), a neuronal model largely employed to evaluate neurite outgrowth and cell differentiation. In that study, we used a low concentration (5 μM) of cisplatin, which we demonstrated did not affect cell viability. We have now used the same model to assess the neuroprotective effect of CAPE against the inhibitory effects of cisplatin on neurite outgrowth and neuroplasticity-related proteins. Studies have suggested that CAPE protects against different toxicities of cisplatin in rats mainly due to its antioxidant potential (Ozen et al. 2004; Kizilay et al. 2004). Accordingly, the neuroprotective potential of CAPE has also been demonstrated in experimental models of spinal cord injury induced by (ii) ischemia-reperfusion (Ilhan et al. 1999), by (ii) methotrexate (Uzar et al. 2006), or yet, (iii) in a rat model of immune encephalomyelitis induced by myelin basic protein (MBP) from guinea pigs (Ilhan et al. 2004). In these models, the damage is associated with oxidative stress. Altogether, these studies provide a body of evidence of the involvement of antioxidant mechanisms in the neuroprotective action of CAPE; however, there is little information on the involvement of neurotrophic mechanisms in the neuroprotective effect of CAPE. We have previously demonstrated that the neurotrophic activity of CAPE is an important mechanism of neuroprotection in both in vivo and in vitro models exposed to dopaminergic neurotoxins associated with Parkinsonism (dos Santos et al. 2014; Barros Silva et al. 2013). In the present study, we have evaluated the protective effect of CAPE on neurite outgrowth and on the expression of plasticity-related proteins (GAP-43, synapsin I, and synaptophysin) in PC12 cells exposed to 5 μM cisplatin. Additionally, we investigated the involvement of both NGF itself and NGF-high-affinity receptor (trkA) in the neurotrophic mechanism of CAPE. To investigate the involvement of other neurotrophin receptors besides trkA, we evaluated the effects of CAPE on the differentiation of another neuronal model (SH-SY5Y cells) with a different phenotype for trk receptors (trkB).
In order to select an effective concentration of CAPE, we evaluated the effect of several concentrations of CAPE (1–50 μM) on the differentiation and the viability of PC12 cells exposed to cisplatin. The concentration of cisplatin used in the differentiation assays was 5 μM, which is the lowest concentration of cisplatin that significantly impairs neurite outgrowth without affecting cell viability (previously determined in Ferreira et al. 2016a). In the viability assays, we used 32 μM cisplatin (IC50, previously determined in Ferreira et al. 2016a). Altogether, the results show that the most effective concentration of CAPE in this model of neurotoxicity is 10 μM, because it is the lowest concentration that (i) protects against the cell death induced by cisplatin (IC50) and (ii) induces the highest level of cell differentiation. In our previous study, the same concentration of CAPE protected against the inhibition of neuritogenesis induced by the dopaminergic neurotoxin MPP+ iodide (1-methyl-4-phenylpyridinium iodide), the active metabolite of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), known to induce Parkinsonism in vivo (dos Santos et al. 2014).
We had previously demonstrated that 5 μM CIS reduces the expression of neuronal proteins associated with axonal growth (GAP-43) and synaptogenesis (synapsin I and synaptophysin), which might be related to the axonal damage found in the peripheral neuropathy induced by cisplatin. To investigate if CAPE interferes in this pathway, we evaluated its effects on the same proteins in PC12 cells exposed to 5 μM cisplatin. GAP-43 is a major constituent of the growth cone, which is responsible for guiding the growth of axons and modulating the formation of new connections (Benowitz and Routtenberg 1997). Synaptophysin and synapsin I regulate the synaptic vesicle fusion and neurotransmitter release (Das et al. 2004; Theil 1993). CAPE (10 μM) minimized the downregulation of these neuronal proteins induced by cisplatin. This result is in line with our previous finding that CAPE alone induces neuritogenesis and increases the expression of neuron-typical proteins in PC12 cells (dos Santos et al. 2014).
To better understand the neurotrophic mechanism of CAPE, we investigated the involvement of NGF/trkA pathway by using K252a, an antagonist of the NGF-high-affinity receptor (trkA) (Phan et al. 2014). PC12 cells express NGF-high-affinity receptors (trkA) and respond to NGF stimulation by stopping division, extending neurites, and differentiating (Calabrese 2008; Huang and Reichardt 2001; Vaudry et al. 2002). K252a reduced the effect of NGF after 72 h of incubation and the effect of CAPE after 96 h of incubation, which suggests different affinities with the receptor trkA. It also suggests the participation of trkA-dependent signaling pathway in the neuritogenic effect of CAPE. However, this finding does not exclude the participation of other neurotrophin receptors. To investigate this possibility, we used SH-SY5Y cells, a neuronal cell line that does not express trkA receptors (Edsjo et al. 2001); instead, these cells respond to retinoic acid treatment by expressing trkB/trkC receptors, of which the high-affinity ligands are the neurotrophins BDNF, NT-3, and NT-4/5 (Kaplan et al. 1993). According to our findings, CAPE did not induce differentiation in SH-SY5Y cells; on the contrary, CAPE reduced the neuritogenesis induced by retinoic acid. It suggests that CAPE inhibits the activity of retinoic acid on trkB. One possibility is that CAPE occupies an inactive site of trkB or even the active site without activating it and preventing the binding of RA on the active site. Accordingly, a study showed that propolis, which contains CAPE, decreases GAP-43 and inhibits neurite outgrowth of RA-stimulated SH-SY5Y cells (Kim and Yoo 2016). These two findings support our hypothesis that CAPE might inhibit trkB in SH-SY5Y cells, preventing the differentiation induced by retinoic acid. On the other hand, another study reported that CAPE protects against the death of dopaminergic neurons in lipopolysaccharide (LPS) and 6-hydroxydopamine (6-OHDA) models of Parkinson’s disease by increasing the expression of BDNF (Kurauchi et al. 2012), the trkB receptor ligand. That study, however, does not access the induction of neurite outgrowth or neuritogenesis/synaptogenesis modulators; therefore, the neuroprotective effect of CAPE observed in those models might not be specifically related to the neurotrophic action of BDNF on trkB receptors. Instead, it might be elicited by the action of BDNF on p75NTR receptors, which are known to elicit cell death but also cell survival. Binding of neurotrophins to p75NTR receptor promote neuronal survival through the nuclear factor-κB (NF-κB) pathway (Longo and Massa 2013; Dechant and Barde 2002).
Besides the involvement of NGF-high-affinity receptors (trkA), we also investigated the role of NGF itself in the protective mechanism of CAPE. The association of CAPE and NGF did not induce a significant increase in the number of differentiated cells nor increase the expression of neuronal proteins as compared to the effect of CAPE alone. These results show that the neuritogenic effect of CAPE is not additive to the effect of NGF, which suggests a common pathway. Results also show that there is no need to add NGF to observe the neuritogenic action of CAPE. This finding alone suggests that CAPE activates trkA pathway but does not reveal if CAPE itself activates trkA or if it increases the expression of NGF, which in turn, activates trkA. To answer this question, we determined the levels of NGF by enzyme-linked immunosorbent assay (ELISA). No significant difference was observed in cells treated with CAPE as compared to controls. Altogether, the findings indicate that the neuroprotective mechanism of CAPE involves induction of neuroplasticity by the activation of the trkA-dependent signaling pathway, regardless of the absence of NGF.
Finally, we confirmed the neuronal differentiation induced by CAPE by analyzing the density of 200-kD neurofilaments (NF-200), a useful immunocytochemical marker of axons (Wu et al. 1998). Neurofilaments are the major components of the cytoskeleton of neuronal cells and they provide specific support for developing neurites (Murphy et al. 1993; Schimmelpfeng et al. 2004). There is a direct relationship between neurite outgrowth and neurofilament density, i.e., neurofilament proteins increase with differentiation of neuronal cell lines (Flaskos et al. 1999). In our study, cisplatin decreased the density of neurofilaments and CAPE minimized this effect. Additionally, CAPE increased the density of neurofilaments in PC12 cells regardless of the absence of NGF.
Conclusion
CAPE attenuates the inhibitory effects of cisplatin on markers of neurite outgrowth and synaptogenesis, such as GAP-43, synapsin I, synaptophysin, and 200-kD neurofilament. The same concentration of CAPE increases the viability of PC12 cells exposed to the IC50 of cisplatin. The neuroprotective mechanism of CAPE is not dependent on NGF and does not potentiate the effect of NGF, but might involve the activation of the NGF-high-affinity receptors, trkA. The participation of other neurotrophin receptors such as trkB (BDNF/NT-4-selective) and trkC (NT-3-selective) is unlikely. This is the first study to show the participation of neuroplasticity in the neuroprotective effect of CAPE against the neurotoxicity of cisplatin. The beneficial effect of CAPE on cisplatin-induced peripheral neuropathy should be further investigated.
References
Albers JW, Chaudhry V, Cavaletti G , Donehower RC (2014). Interventions for preventing neuropathy caused by cisplatin and related compounds Cochrane Database Syst Rev. CD005228
Avan A, Postma TJ, Ceresa C, Avan A, Cavaletti G, Giovannetti E, Peters GJ (2015) Platinum-induced neurotoxicity and preventive strategies: past, present, and future. Oncologist 20(4):411–432. https://doi.org/10.1634/theoncologist.2014-0044
Bak J, Kim HJ, Kim SY, Choi YS (2016) Neuroprotective effect of caffeic acid phenethyl ester in 3-nitropropionic acid-induced striatal neurotoxicity. Korean J Physiol Pharmacol 20(3):279–286. https://doi.org/10.4196/kjpp.2016.20.3.279
Barros Silva R, Santos NA, Martins NM, Ferreira DA, Barbosa F Jr, Oliveira Souza VC, Kinoshita A, Baffa O, Del-Bel E, Santos AC (2013) Caffeic acid phenethyl ester protects against the dopaminergic neuronal loss induced by 6-hydroxydopamine in rats. Neuroscience 233:86–94. https://doi.org/10.1016/j.neuroscience.2012.12.041
Benowitz LI, Routtenberg A (1997) GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci 20(2):84–91. https://doi.org/10.1016/S0166-2236(96)10072-2
Burton AW, Fanciullo GJ, Beasley RD, Fisch MJ (2007) Chronic pain in the cancer survivor: a new frontier. Pain Med 8(2):189–198. https://doi.org/10.1111/j.1526-4637.2006.00220.x
Calabrese EJ (2008) Enhancing and regulating neurite outgrowth. Crit Rev Toxicol 38(4):391–418. https://doi.org/10.1080/10408440801981981
Cascinu S, Cordella L, del Ferro E, Fronzoni M, Catalano G (1995) Neuroprotective effect of reduced glutathione on cisplatin-based chemotherapy in advanced gastric cancer: a randomized double-blind placebo-controlled trial. J Clin Oncol 13(1):26–32. https://doi.org/10.1200/JCO.1995.13.1.26
Cascinu S, Catalano V, Cordella L, Labianca R, Giordani P, Baldelli AM, Beretta GD, Ubiali E, Catalano G (2002) Neuroprotective effect of reduced glutathione on oxaliplatin-based chemotherapy in advanced colorectal cancer: a randomized, double-blind, placebo-controlled trial. J Clin Oncol 20(16):3478–3483. https://doi.org/10.1200/JCO.2002.07.061
Das KP, Freudenrich TM, Mundy WR (2004) Assessment of PC12 cell differentiation and neuritic growth: a comparison of morphological and neurochemical measures. Neurotoxicol Teratol 26(3):397–406. https://doi.org/10.1016/j.ntt.2004.02.006
Dechant G, Barde YA (2002) The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci 5(11):1131–1136. https://doi.org/10.1038/nn1102-1131
dos Santos NA, Martins NM, Silva RDEB, Ferreira RS, Sisti FM, dos Santos AC (2014) Caffeic acid phenethyl ester (CAPE) protects PC12 cells from MPP+ toxicity by inducing the expression of neuron-typical proteins. Neurotoxicology 45:131–138. https://doi.org/10.1016/j.neuro.2014.09.007
Edsjo A, Hallberg B, Fagerstrom S, Larsson C, Axelson H, Pahlman S (2001) Differences in early and late responses between neurotrophin-stimulated trkA- and trkC-transfected SH-SY5Y neuroblastoma cells. Cell Growth Differ 12(1):39–50
Ferreira RS, dos Santos NA, Martins NM, Fernandes LS, dos Santos AC (2016a) Non-cytotoxic concentration of cisplatin decreases neuroplasticity-related proteins and neurite outgrowth without affecting the expression of NGF in PC12 cells. Neurochem Res 41(11):2993–3003. https://doi.org/10.1007/s11064-016-2019-5
Ferreira RS, dos Santos NA, Martins NM, Fernandes LS, dos Santos AC (2016b) Non-cytotoxic concentration of cisplatin decreases neuroplasticity-related proteins and neurite outgrowth without affecting the expression of NGF in PC12 cells. Neurochem Res
Flaskos J, Fowler MJ, Teurtrie C, Hargreaves AJ (1999) The effects of carbaryl and trichlorphon on differentiating mouse N2a neuroblastoma cells. Toxicol Lett 110(1-2):79–84. https://doi.org/10.1016/S0378-4274(99)00142-3
HAUSHEER FH, SCHILSKY RL, BAIN S, BERGHORN EJ, LIEBERMAN F (2006) Diagnosis, management, and evaluation of chemotherapy-induced peripheral neuropathy. Semin Oncol 33(1):15–49. https://doi.org/10.1053/j.seminoncol.2005.12.010
HUANG EJ, REICHARDT LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24(1):677–736. https://doi.org/10.1146/annurev.neuro.24.1.677
Huang Y, Jin M, Pi R, Zhang J, Chen M, Ouyang Y, Liu A, Chao X, Liu P, Liu J, Ramassamy C, Qin J (2013) Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neurosci Lett 535:146–151. https://doi.org/10.1016/j.neulet.2012.12.051
Ilhan A, Koltuksuz U, Ozen S, Uz E, Ciralik H, Akyol O (1999) The effects of caffeic acid phenethyl ester (CAPE) on spinal cord ischemia/reperfusion injury in rabbits. Eur J Cardiothorac Surg 16(4):458–463. https://doi.org/10.1016/S1010-7940(99)00246-8
Ilhan A, Akyol O, Gurel A, Armutcu F, Iraz M, Oztas E (2004) Protective effects of caffeic acid phenethyl ester against experimental allergic encephalomyelitis-induced oxidative stress in rats. Free Radic Biol Med 37(3):386–394. https://doi.org/10.1016/j.freeradbiomed.2004.04.022
Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ (1993) Induction of TrkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Eukaryotic Signal Transduction Group. Neuron 11(2):321–331
Kim HB, Yoo BS (2016) Propolis inhibits neurite outgrowth in differentiating SH-SY5Y human neuroblastoma cells. Toxicol Res 32(3):239–243. https://doi.org/10.5487/TR.2016.32.3.239
Kizilay A, Kalcioglu MT, Ozerol E, Iraz M, Gulec M, Akyol O, Ozturan O (2004) Caffeic acid phenethyl ester ameliorated ototoxicity induced by cisplatin in rats. J Chemother 16(4):381–387. https://doi.org/10.1179/joc.2004.16.4.381
Kurauchi Y, Hisatsune A, Isohama Y, Mishima S, Katsuki H (2012) Caffeic acid phenethyl ester protects nigral dopaminergic neurons via dual mechanisms involving haem oxygenase-1 and brain-derived neurotrophic factor. Br J Pharmacol 166(3):1151–1168. https://doi.org/10.1111/j.1476-5381.2012.01833.x
L'episcopo F, Serapide MF, Tirolo C, Testa N, Caniglia S, Morale MC, Pluchino S, Marchetti B (2011) A Wnt1 regulated Frizzled-1/beta-Catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: therapeutical relevance for neuron survival and neuroprotection. Mol Neurodegener 6(1):49. https://doi.org/10.1186/1750-1326-6-49
Longo FM, Massa SM (2013) Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat Rev Drug Discov 12(7):507–525. https://doi.org/10.1038/nrd4024
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65(1-2):55–63. https://doi.org/10.1016/0022-1759(83)90303-4
Murphy A, Breen KC, Long A, Feighery C, Casey EB, Kelleher D (1993) Neurofilament expression in human T lymphocytes. Immunology 79(1):167–170
Murtaza G, Karim S, Akram MR, Khan SA, Azhar S, Mumtaz A, Bin Asad MH (2014) Caffeic acid phenethyl ester and therapeutic potentials. Biomed Res Int 2014:145342
Natarajan K, Singh S, Burke TR Jr, Grunberger D, Aggarwal BB (1996) Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci U S A 93(17):9090–9095. https://doi.org/10.1073/pnas.93.17.9090
Ozen S, Akyol O, Iraz M, Sogut S, Ozugurlu F, Ozyurt H, Odaci E, Yildirim Z (2004) Role of caffeic acid phenethyl ester, an active component of propolis, against cisplatin-induced nephrotoxicity in rats. J Appl Toxicol 24(1):27–35. https://doi.org/10.1002/jat.941
Pace A, Savarese A, Picardo M, Maresca V, Pacetti U, del Monte G, Biroccio A, Leonetti C, Jandolo B, Cognetti F, Bove L (2003) Neuroprotective effect of vitamin E supplementation in patients treated with cisplatin chemotherapy. J Clin Oncol 21(5):927–931. https://doi.org/10.1200/JCO.2003.05.139
Phan CW, Lee GS, Hong SL, Wong YT, Brkljaca R, Urban SABD, Malek SN, Sabaratnam V (2014) Hericium erinaceus (Bull.: Fr) Pers. cultivated under tropical conditions: isolation of hericenones and demonstration of NGF-mediated neurite outgrowth in PC12 cells via MEK/ERK and PI3K-Akt signaling pathways. Food Funct 5(12):3160–3169. https://doi.org/10.1039/C4FO00452C
Planting AS, Catimel G, de Mulder PH, de Graeff A, Hoppener F, Verweij J, Oster W, Vermorken JB (1999) Randomized study of a short course of weekly cisplatin with or without amifostine in advanced head and neck cancer. EORTC Head and Neck Cooperative Group. Ann Oncol 10(6):693–700. https://doi.org/10.1023/A:1008353505916
Rasband WS (1997–2014) ImageJ. U. S. National Institutes of Health, Bethesda, Maryland, USA http://imagej.nih.gov/ij/
Sadri S, Bahrami F, Khazaei M, Hashemi M, Asgari A (2010) Cannabinoid receptor agonist WIN-55,212-2 protects differentiated PC12 cells from organophosphorus-induced apoptosis. Int J Toxicol 29(2):201–208. https://doi.org/10.1177/1091581809359708
Santos NAGD, Martins NM, Silva RDB, Ferreira RS, Sisti FM, Santos ACD (2014) Caffeic acid phenethyl ester (CAPE) protects PC12 cells from MPP+ toxicity by inducing the expression of neuron-typical proteins. Neurotoxicology 45:131–138. https://doi.org/10.1016/j.neuro.2014.09.007
Schimmelpfeng J, Weibezahn KF, Dertinger H (2004) Quantification of NGF-dependent neuronal differentiation of PC-12 cells by means of neurofilament-L mRNA expression and neuronal outgrowth. J Neurosci Methods 139(2):299–306. https://doi.org/10.1016/j.jneumeth.2004.05.010
Tapley P, Lamballe F, Barbacid M (1992) K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene 7(2):371–381
Theil G (1993) Synapsin I, synapsin II, and synaptophysin: marker proteins for synaptic vesicle. Brain Pathol 3(1):87–95. https://doi.org/10.1111/j.1750-3639.1993.tb00729.x
Tolba MF, Azab SS, Khalifa AE, Abdel-RAHMAN SZ, Abdel-NAIM AB (2013) Caffeic acid phenethyl ester, a promising component of propolis with a plethora of biological activities: a review on its anti-inflammatory, neuroprotective, hepatoprotective, and cardioprotective effects. IUBMB Life 65(8):699–709. https://doi.org/10.1002/iub.1189
Uzar E, Sahin O, Koyuncuoglu HR, Uz E, Bas O, Kilbas S, Yilmaz HR, Yurekli VA, Kucuker H, Songur A (2006) The activity of adenosine deaminase and the level of nitric oxide in spinal cord of methotrexate administered rats: protective effect of caffeic acid phenethyl ester. Toxicology 218(2-3):125–133. https://doi.org/10.1016/j.tox.2005.10.014
van den Berg R, Haenen GRMM, van den Berg H, BAST A (2001) Transcription factor NF-κB as a potential biomarker for oxidative stress. Br J Nutr 86(S1):S121–S127. https://doi.org/10.1079/BJN2001340
Vaudry D, Stork PJ, lazarovici P, Eiden LE (2002) Signaling pathways for PC12 cell differentiation: making the right connections. Science 296(5573):1648–1649. https://doi.org/10.1126/science.1071552
Wu G, Fang Y, Lu ZH, Ledeen RW (1998) Induction of axon-like and dendrite-like processes in neuroblastoma cells. J Neurocytol 27:1–14
Acknowledgments
The authors would like to thank CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, grant number 140106/2015-4, Rafaela Scalco Ferreira) and FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, processo 2017/09332-7) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ferreira, R.S., dos Santos, N.A.G., Martins, N.M. et al. Caffeic Acid Phenethyl Ester (CAPE) Protects PC12 Cells from Cisplatin-Induced Neurotoxicity by Activating the NGF-Signaling Pathway. Neurotox Res 34, 32–46 (2018). https://doi.org/10.1007/s12640-017-9849-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-017-9849-z