
Abstract
Reactive oxygen species (ROS) readily oxidize the sulfur-containing amino acids cysteine and methionine (Met). The impact of Met oxidation on the fast inactivation of the skeletal muscle sodium channel NaV1.4 expressed in mammalian cells was studied by applying the Met-preferring oxidant chloramine-T or by irradiating the ROS-producing dye Lucifer Yellow in the patch pipettes. Both interventions dramatically slowed down inactivation of the sodium channels. Replacement of Met in the Ile–Phe–Met inactivation motif with Leu (M1305L) strongly attenuated the oxidizing effect on inactivation but did not eliminate it completely. Mutagenesis of Met1470 in the putative receptor of the inactivation lid also markedly diminished the oxidation sensitivity of the channel, while that of other conserved Met residues in intracellular linkers connecting the membrane-spanning segments (442, 1139, 1154, 1316, 1469) were of minor importance. The results of mutagenesis, assays of other NaV channel isoforms (NaV1.2, NaV1.5, NaV1.7), and the kinetics of the oxidation-induced removal of inactivation collectively indicate that multiple Met residues need to be oxidized to completely impair inactivation. This arrangement using multiple Met residues confers a finely graded oxidative modulation of NaV channels and allows organisms to adapt to a variety of oxidative stress conditions, such as ischemic reperfusion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Excess reactive oxygen species (ROS), as found in oxidation stress situations, affect lipids, nucleic acids, and proteins in cells. In proteins, the sulfur-containing amino acids cysteine (Cys) and methionine (Met) are readily oxidized by ROS [1]. Oxidation of Met generates the two Met sulfoxide (MetO) epimers Met–S–O and Met–R–O, which are reduced by the methionine sulfoxide reductases (MSRs) A and B, respectively [2]. Oxidation and reduction of methionines may serve different functions [3]: (1) Intracellular ROS can be scavenged by reversible oxidation of Met residues that are not critical for the function of a protein. Such residues may prevent other critical Met residues from becoming oxidized, as was postulated for the Escherichia coli glutamine synthetase enzyme complex [4]. (2) MSRs may reduce oxidized Met residues that are critical for protein function, thus, serving a role as repair enzymes. For example, oxidative loss of calmodulin functions, such as activation of plasma membrane Ca2+-ATPase, may be restored by MSRs [5]. (3) Reversible Met oxidation may regulate specific oxidation-sensitive processes. Coexpression of Drosophila Shaker C/B potassium channels in Xenopus oocytes with MSRA or MSRB protects fast inactivation of the channel against oxidation, an effect that could be attributed to a Met residue in the N-terminal ball domain, which is responsible for fast inactivation [6–8].
Several lines of evidence argue that oxidative modification of voltage-gated sodium channels (NaV channels) with pathophysiological consequences also occurs (e.g., [9–12]) but the underlying molecular mechanisms remain elusive. NaV channels rapidly open upon membrane depolarization to allow Na+ influx. The resulting current is transient because the channels inactivate quickly. In this inactivation process, a very well-conserved hydrophobic triad consisting of Ile–Phe–Met (IFM) in the linker between domains 3 and 4 (D3–D4) interacts with moieties on the channels’ inner pore entries (e.g., [13, 14]). Because MetO is more hydrophilic than Met [15], the hydrophobic interaction between the linker and its receptor on the channel may be disturbed if MetO is present. In fact, several studies using oxidants, such as chloramine-T (ChT) and H2O2, indicated that oxidation of Met may impair fast inactivation in both neuronal and muscle Nav channels [16–19]. Similar effects are evoked by irradiation of HEK 293 cells expressing the human isoforms of NaV1.4 or NaV1.5 with UV-A (320–380 nm wavelength) light, which triggers the production of intracellular ROS [20]. However, a mutant of the rat NaV1.4 channel with the inactivating IFM motif mutated to IFI remained sensitive to both UV-A and H2O2 exposure [20], thus, suggesting that the Met in the inactivation motif is not the only target.
We have examined the oxidation sensitivity of NaV channel inactivation by replacing conserved Met residues in the IFM motif and other intracellular linkers of the rat NaV1.4 channel and subjecting the expressed channels to oxidation. Mutation of Met1305 in the IFM motif in the D3–D4 linker drastically decreased oxidation sensitivity. Essentially the same effect was observed for residue Met1470 in the S4–S5 linker of domain 4 and also for a combination of Met1305 and Met1470 mutants. The mutagenesis results and the kinetics of oxidation-induced modification of channel gating suggest that at least two Met residues are oxidized to impair inactivation. Because the mutation of other Met residues conserved among mammalian NaV channel types had only minor effects, we postulate that the Met residues in the IFM motif and in its receptor are primarily responsible for the oxidation sensitivity of NaV1.4 channel inactivation.
Materials and methods
Expression plasmids and mutagenesis
The α-subunit-encoding rat NaV channel gene Na V 1.4 (P15390; [21]) in the plasmid vector pcDNA3 was used as a background for mutagenesis. Site-specific mutagenesis was performed to replace methionine with leucine at positions 442, 1139, 1154, 1305, 1316, 1469, 1470. Mutant nomenclature is as follows: IFL: M1305L; IFM_LL: M1469L·M1470L; IFM_LM: M1469L; IFM_ML: M1470L; IFL_LL: M1305L·M1469L·M1470L; IFM_4L: M442L·M1139L·M1154L·M1316L; IFM_6L: IFM_LL combined with IFM_4L; IFL_6L: IFL combined with IFM_6L. As a control, the following wild-type channels were used: rat NaV1.2 (P04775; [22]), human NaV1.7 (NP002968; [23]), and human NaV1.5 (Q14524; [24]). Because the cardiac NaV1.5 channels harbor a cysteine residue in the pore region, it is sensitive to extracellular cysteine-modifying agents. We, therefore, constructed the mutant NaV1.5_C373Y to generate a pore region in domain-1 similar to that in NaV1.4 channels. All channel types expressed well, and typically cells with 1–10 nA of maximal inward current, were included for analysis.
Cell culture
HEK 293 cells (CAMR, Porton Down, Salisbury, UK) were maintained in Dulbecco’s Modified Eagle’s Medium mixed 1:1 with Ham’s F12 medium and supplemented with 10% fetal calf serum in a 5% CO2 incubator. Cells were trypsinized, diluted with culture medium, and grown in 35-mm dishes. Electrophysiological experiments were performed 1–5 days after plating. HEK 293 cells were transfected with the respective plasmids using the Rotifect® (Roth, Karlsruhe, Germany) transfection reagent following the vendor’s instructions.
Electrophysiological measurements
Whole-cell, voltage-clamp experiments were performed as described previously [25]. Briefly, patch pipettes with resistances of 0.7–2.0 MΩ were used, and the series resistance was compensated for >70% to minimize voltage errors. A patch-clamp amplifier EPC9 was operated by PatchMaster software (both HEKA Elektronik, Lambrecht, Germany). Holding potential was −120 mV. Leak and capacitive currents were corrected with a p/n method with a leak holding voltage of −120 mV. Currents were low-pass filtered at 5 kHz and sampled at a rate of 25 kHz. All experiments were performed at 19–21°C. The patch pipettes contained (mM) 35 NaCl, 105 cesium fluoride, 10 ethylene glycol tetraacetic acid, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.4 with CsOH). The bath solution contained (mM) 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES (pH 7.4 with NaOH).
Chloramine-T-induced oxidation
ChT was diluted in the respective bath solution immediately before application. It was applied by total exchange of the bath solution within about 10 s. For most experiments, 200 μM ChT were applied. Higher ChT concentrations markedly reduce the success rate of the experiments and, more importantly, also cause a “loss of channels”, presumably by an irreversible destruction of the protein.
Lucifer Yellow-induced oxidation
Cells were loaded with the fluorescent dye Lucifer Yellow (LY; 1 mM; Sigma) via the patch pipette. After recording control currents, cells were illuminated with 436-nm light from a PolyChrome-1 light source (XBO lamp, TILL Photonics) using a 40× dry objective.
Current–voltage relationships
From a holding potential of –120 mV, cells were depolarized to −80 through +60 mV in steps of 10 mV for 40 ms every 3 s. The peak current–voltage relationships were fit according to a Hodgkin-Huxley formalism with m = 3 activation gates and a single-channel conductance according to the Goldman–Hodgkin–Katz equation.
V m is the voltage of half-maximal gate activation and k m the corresponding slope factor. Γ is the maximal conductance of all channels and E rev the reversal potential.
Voltage dependence of fast inactivation
From a holding potential of −120 mV, cells were conditioned for 500 ms at voltages ranging from −140 to 0 mV in steps of 5 mV. Subsequently, peak current was determined at −10 mV and normalized to a control peak current measured before conditioning. The repetition interval was 3 s. The normalized peak current plotted versus the conditioning voltage was described with a Boltzmann function:
with the half-maximal inactivation voltage V h and the corresponding slope factor k h that indicates the voltage dependence of inactivation.
Time course of inactivation removal
The time course of oxidation-induced loss of inactivation was monitored by measuring the ratio of current after 10 ms and the peak current from pulses elicited to −10 mV at an interval of 5 s (r). The inactivation index, 1-r(t), was plotted as a function of time (t) and fit with the following function:
with the ratio before oxidation, r 0 , the time of oxidation start, t 0 , the time constant, τ, and an exponent, n. The ratio after infinite oxidation is \(r_\infty \), which was set to 1 throughout.
Data analysis and statistics
Data were analyzed with FitMaster (HEKA Elektronik) and IgorPro (WaveMetrics, Lake Oswego, OR, USA). Averaged data are presented as mean ± SEM (n = number of independent measurements). Groups of data were compared with a two-sided Student’s t-test or analysis of variance followed by a post hoc Bonferroni test when appropriate.
Results
Removal of fast inactivation by chloramine-T
Rat skeletal muscle sodium channels (NaV1.4) were expressed in HEK 293 cells, and Na+ currents were recorded in the whole-cell configuration in response to depolarizations to −10 mV applied every 5 s. The influence of various concentrations of the membrane permeant ChT, applied extracellullarly, was assayed. Superposition of current traces measured before and 200 s after ChT application (Fig. 1a) shows that increasing ChT concentrations remove inactivation in a progressive manner. Similar removal of inactivation was observed at other voltages (not shown). Current activation was not markedly affected, but at high ChT (1 mM) concentration, a peak current reduction could be observed after long waiting periods (not shown).

a–c Effect of ChT on the inactivation of NaV1.4 channels in HEK 293 cells. a Current responses to depolarizations to −10 mV before (black) and 200 s after (gray) application of the indicated ChT concentration. b Superposition of inactivation index data from representative experiments in which the indicated concentration of ChT was applied at time zero. The superimposed curves are fits according to Eq. 3 with an exponent set to 2. The vertical gray bar indicates 200 s, i.e., the time from which the current traces in (a) were selected. c The time constants determined from fits as in panel (b) were transformed to rate constants (k = 1/τ) and plotted as a function of the ChT concentration. n = 4–11. The straight line is the result of a linear fit with a slope of 11.7 ± 0.5 (sM)−1
The inactivation index (see “Materials and methods”), an operational descriptor proportional to the extent of inactivation, was plotted as a function of time (Fig. 1b). Increasing concentrations of ChT resulted in a loss of inactivation as indicated by a marked decrease in the inactivation index being about half-maximal after 100 s when 1 mM ChT was applied. The time course of inactivation removal could not be adequately described with a single exponential because the onset of the effect showed a noticeable degree of sigmoidicity. Therefore, as an operational data descriptor, we chose to fit the results with Eq. 3, assuming an exponent of n = 2. This approach resulted in reasonable data descriptions (see Fig. 1b), but it should be noted that deviations from an exponent of 2 would improve the fit considerably in some cases (see below).
The rate of ChT-induced loss of inactivation was estimated from the time constants of data fits as shown in Fig. 1b and is plotted in Fig. 1c as a function of ChT concentration. The rate roughly depends on the ChT concentration in a linear manner indicating that the approach to fit the time course with n = 2 extracted a rate constant describing the oxidation of a target in a first-order reaction.
As shown in Fig. 2a, progressive loss of inactivation induced by 200 μM ChT did not continue when ChT was washed away with external solutions containing the reducing agent dithiothreitol (DTT; 1 mM). DTT is expected to inactivate any residual ChT in the bath chamber and should restore thiol modifications at cysteine residues. Even prolonged exposure to DTT did not restore inactivation (verified in each of the four independent experiments for the wild type and mutant M1316L); neither the time course of current inactivation nor the current magnitude changed upon DTT wash after ChT application (Fig. 2b). This result suggests that ChT either did not alter the redox state of cysteine residues or cysteine modification did not result in noticeable changes in channel function.

a, b Removal of inactivation is irreversible. a Inactivation index as a function of time; application of 200 μM ChT and wash with 1 mM DTT is indicated. b Current responses to depolarizations to −10 mV at the times during the experiment indicated by the white data points in (a)
To ensure that the inactivation removal effect of ChT is caused by its ability to oxidize proteins, we used p-toluenesulfonamide (p-TSA). p-TSA is similar to ChT but has an amide moiety instead of the N-NaCl group in ChT and, thus, is incapable of generating reactive species. When applied to NaV1.4 channels at a concentration of 2 mM for 1,000 s, no alteration of channel inactivation was observed (n = 3; data not shown).
Effect of chloramine-T on various NaV channel types
The time course of inactivation was also observed to be sensitive to oxidation in other NaV channel types (e.g., [19, 20, 26]). To compare the sensitivity quantitatively, we expressed NaV1.2, NaV1.5, and NaV1.7 channels in HEK 293 cells and subjected them to ChT. Application of 200 μM extracellular ChT potently and rapidly blocked wild-type NaV1.5 channels (more than 50% in 30 s), probably because of a cysteine residue in the tetrodotoxin binding site in the pore loop of domain-1. Therefore, we constructed and analyzed mutant NaV1.5_C373Y in these experiments. This mutant has inactivation properties indistinguishable from those of the wild type, but the peak inward current is less sensitive to oxidation caused by ChT (only about 20% rapid current reduction upon application of 200 μM ChT). The inactivation of all channel types examined was similarly sensitive to ChT (Fig. 3).

a, b Removal of inactivation in NaV isoforms NaV1.2, NaV1.4, NaV1.5_C373Y, and NaV1.7. a Superimposed current traces for depolarization from −120 mV to −10 mV before (black) and 500 s after application of 200 μM ChT (gray) for the indicated channel types. b Statistics on inactivation index after 500 s
Influence of conserved methionine residues in the core domain of NaV1.4 channels
Given the lack of reversibility under DTT application and the previously shown preference of ChT to oxidize Met residues [6, 27], the loss of NaV channel inactivation is probably mediated by oxidation of one or more methionines in the NaV1.4 protein. A previous report has indeed suggested that Met oxidation plays a role in oxidation-induced removal of inactivation of NaV channels in toad skeletal muscle fibers [19].
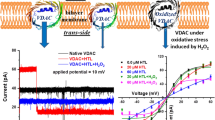
There is a conserved Met residue in the inactivation domain (IFM motif) between domain-3 and domain-4 (Fig. 4a). Thus, we generated a mutant of NaV1.4 in which this Met was replaced with leucine (M1305L, generating an IFL motif) and examined the sensitivity to ChT (Fig. 4b, top). As indicated by Wang and Wang [20], elimination of the Met from the IFM-motif fails to completely remove the oxidation sensitivity of the channel inactivation. However, the time course of ChT-induced loss of inactivation is much slower than in the wild type. Owing to this slow time course with 0.2 mM ChT, we could not accurately conclude if the slowing of inactivation reaches a steady state within a typical lifetime of a recording situation (30 min). However, treatment with 1 mM ChT resulted in complete loss of inactivation. Therefore, for kinetic analysis, we assumed that ultimately all channels lose inactivation. Superposition of current traces from several cells recorded 300 s after start of ChT application (Fig. 4b, bottom) clearly shows that the IFL mutant is less sensitive than the wild type. In Fig. 4c, the inactivation index after 500 s ChT (0.2 and 1.0 mM) application is presented. For the wild type, this is 0.474 ± 0.035 (n = 10) and for mutant, IFL 0.727 ± 0.017 (n = 5; P = 0.007) in 0.2 mM ChT; for 1.0 mM ChT, the numbers are 0.019 ± 0.007 (n = 8) and 0.449 ± 0.050 (n = 4; P < 0.001). The finding that mutation of the Met residue in the IFM-motif alters but does not eliminate the oxidation sensitivity of the inactivation process suggests that other Met residues are involved.

a–c Influence of methionine mutations on the effect of ChT. a Schematic diagram illustrating the topology of a NaV channel α-subunit. Conserved Met residues in the core domain excluding the N- and C-terminal regions are indicated as circles. The numbers refer to the positions in rat NaV1.4. b Examples of ChT-induced (200 μM) loss of inactivation (gray) with superimposed fits (black) according to Eq. 3 for the wild type and the indicated mutants. Bottom: Superposition of current traces obtained from different cells exposed to 200 μM ChT for 300 s. c Mean inactivation index obtained 500 s after application of 0.2 mM (white) or 1.0 mM ChT (gray). n = 4–12
The rat NaV1.4 channel protein in total contains 66 Met residues, making it impractical mutating all of them individually and in combination with the one in the inactivation domain. Because oxidation-induced loss of inactivation was also observed in other NaV channel types (Fig. 3), one can speculate that the relevant methionines might be conserved. In addition, if such methionines were subject to cellular regulation, such as reduction by methionine sulfoxide reductases, they are expected to be accessible from the cytosolic side. A multiple sequence alignment of all nine mammalian NaV channels revealed that there are seven conserved Met residues in the core domain excluding the N- and C-terminal domains that face the cytosol (Fig. 4a). Most noticeably, there is a double-Met motif in the putative receptor for the inactivation domain in the S4–S5 linker of domain-4 (M1469, M1470) and two methionines in the corresponding linker of domain-3 (M1138, M1154). The remaining two methionines are in S6 of domain-1 (M442) and in the inactivation linker connecting domain-3 and domain-4 (1316). The documented relevance of the residues M1469 and M1470 for rapid inactivation [28] prompted us to generate a double mutant M1469L·M1470L, both in the background of the wild type (IFM) and in the background of the IFL mutant (termed IFM_LL and IFL_LL, respectively).
Both mutants, when exposed to 200 μM ChT, retained oxidation sensitivity, but the time course of inactivation removal was noticeably slower than in the wild-type channel (Fig. 4b). Using the inactivation index after 500 s of 0.2 and 1.0 mM ChT application as a measure (Fig. 4c), mutants IFM_LL (0.779 ± 0.015, n = 6; 0.394 ± 0.055, n = 5) and IFL_LL (0.863 ± 0.010, n = 6; 0.644 ± 0.076, n = 5) were less sensitive than the wild type (P < 0.05; P < 0.001), also shown in the superpositions of individual current traces 300 s after ChT application (Fig. 4b). In constructs lacking the methionine(s) in the linker (IFL) or receptor (LL), removal of the other respective Met significantly slowed down the effect of ChT even further: IFL vs. IFL_LL, P < 0.001; IFM_LL vs. IFL_LL, P < 0.01. Thus, the Met in the IFM-motif as well as the Met diad in the putative receptor contribute to the oxidation sensitivity of NaV channels in a somewhat additive manner. Individual mutations of methionines in the putative receptor revealed that M1470 is much more important for the channel’s sensitivity than M1469 (Fig. 4c): Inactivation index after 500 s in 1 mM ChT was 0.058 ± 0.025 (n = 6) for IFM_LM and 0.364 ± 0.044 (n = 5) for IFM_ML (P > 0.001). Nevertheless, even if all these methionines are replaced with leucines, the channels still have some ChT sensitivity suggesting the involvement of additional residues.
We, therefore, introduced further mutations altering M442, M1130, M1154, and M1316 to leucines into the mutants IFM_LL and IFL_LL, termed IFM_6L and IFL_6L, respectively. Unfortunately, both mutants exhibited a strongly left-shifted voltage dependence of steady-state inactivation; the half-maximal inactivation voltage (V h ) was −72.9 ± 0.7 mV (n = 22) for the wild type and −111.0 ± 2.5 mV (n = 10) for mutant IFM_6L and for −112.5 ± 1.2 mV (n = 21) IFL_6L. As a consequence, even small left-shifts in the steady-state inactivation induced by ChT resulted in a substantial reduction of peak current even if the holding voltage was lowered from −120 to −140 mV. The determined loss of inactivation is, therefore, an overestimation, as properly inactivating channels will be selectively eliminated from the statistics owing to a shift in the steady-state inactivation. Nevertheless, mutant IFM_6L was significantly less sensitive to ChT than the wild type and mutants IFL and IFM_LL (inactivation index after 500 s: 0.904 ± 0.014, n = 6; 0.394 ± 0.040, n = 9 P < 0.001). The comparison of IFM_LL and IFM_6L shows that the additional replacement of the four methionines significantly diminishes the sensitivity of the channel further. This is corroborated by a small but significant decrease of sensitivity in mutant IFM_4L compared with the wild type (P < 0.01; Fig. 4c). Likewise, mutant IFL_6L was significantly less sensitive than mutant IFL_LL (P < 0.01; Fig. 4c).
Time course of ChT-induced loss of inactivation
In the experiments shown in Fig. 4, the time course of the loss of inactivation mediated by application of 200 μM ChT was described with Eq. 3, allowing the exponent n to adjust, i.e., providing a variable sigmoidicity. This exponent was, then, plotted versus the respective time constant, τ, in Fig. 5a for the wild type and several mutants. Two trends are clearly observed: (1) there is a strong negative correlation between n and τ. (2) While the wild-type data require an n between 2 and 3, n approaches unity as the mutants lose their sensitivity towards ChT such that the sigmoidicity in the modification kinetics is diminished. The correlation between n and τ is consistent with the idea that the loss of inactivation by ChT is mediated by oxidative modification of multiple Met residues in the wild-type channel. As the target Met residues are removed, the exponent n approaches unity, but the modification of kinetics slows.

a, b Sigmoidicity of ChT-induced loss of inactivation. a Results of the fits according to Eq. 3 to data as shown in Fig. 4b are plotted with their individual error estimates: exponent, n, indicating the degree of sigmoidicity, versus the time constant, τ. b Time course of the inactivation index of NaV1.4 channels upon repeated application of 500 μM ChT. Between the ChT applications, the cells were bathed in 1-mM DTT-containing solution. The continuous curve is a data fit assuming that the loss of inactivation proceeds under the second ChT application with the same kinetics as at the end of the first application. Exponent n = 2.01; time constant τ = 205 s
The idea that multiple target residues account for the sigmoidal time course reflected in the greater-than-unity exponent value in the wild-type channel suggests that pretreatment of the channel with ChT should arrest them in a certain state. Loss of inactivation should resume without sigmoidicity after long interruptions of ChT application. In Fig. 5b, an experiment is shown illustrating that this is indeed the case. ChT was applied, and inactivation loss starts with an exponent of 2. Then, ChT was washed away with DTT solutions to stop the reaction immediately. After about 200 s, ChT was applied again and the loss of inactivation proceeded without any noticeable lag or sigmoidicity. This result also rules out that the sigmoidicity arises from slow accumulation of ChT inside the cell.
Alternative oxidation of NaV1.4 channels by irradiated Lucifer Yellow
To test whether the effect of oxidation on NaV channel inactivation is specific for ChT, we also applied H2O2 to HEK 293 cells expressing wild-type channels. As previously shown by Wang and Wang [20], H2O2 also removed inactivation (data not shown). Another means of generating reactive oxygen species in living cells is to load cells with the dye Lucifer Yellow (LY) via the patch pipette and to illuminate it with blue light (436 nm). This method was shown to be effective in removing inactivation of NaV channels in mouse hippocampal neurons [26]. It has the great advantage that no mechanical agitation by changing of external bath solutions is required. Light-exposed LY (1 mM) effectively removed inactivation of the wild-type NaV1.4 channel (Fig. 6a,b). The M-to-L mutants were much less sensitive than the wild type. The inactivation index after 500 s light exposure was 0.570 ± 0.076 (n = 7) for the wild type, 0.951 ± 0.009 (n = 6) for mutant IFL, and 0.971 ± 0.005 (n = 6) for mutant IFM_LL (P < 0.01). Similar to what was found for ChT, removal of methionines in the IFM domain and in the receptor in domain-4 strongly reduced the oxidation sensitivity of the channel. For LY, we did not observe an additional effect of the extra methionines eliminated in the IFM_6L mutation because the protection obtained with mutation IFM_LL was already almost complete.

a–c Removal of inactivation with light-exposed Lucifer Yellow (LY). a Patch pipettes contained 1 mM LY and light (436 nm) was turned on at time zero. The left panels show examples of LY-induced loss of inactivation (gray) with superimposed fits (black) according to Eq. 3 for the wild type and the indicated mutants. Right: Mean inactivation index obtained 500 s after start of illumination. n values are indicated in parentheses. b Superposition of current traces obtained from different cells exposed to LY and light for 300 s. c Results of the fits according to Eq. 3 to data as shown in panel (a) are plotted with their individual error estimates: exponent, n, indicating the degree of sigmoidicity, versus the time constant, τ
The time course of LY-induced loss of inactivation of the wild type and the mutants was also described with Eq. 3. Similar to the experiments with ChT (Fig. 5a), we found a strong negative correlation between the estimated exponent (n) and the time constant (τ; Fig. 6c). In addition, this analysis shows that the M-to-L mutations have a stronger protective effect when modification is mediated by LY irradiation as compared to ChT application: The time constant of wild-type modification was 388 ± 37 s for ChT and 415 ± 91 s for LY; mutation IFL slowed down the modification rate by a factor of 2.5 ± 0.3 and 8.5 ± 3.3, mutant IFM_LL by a factor of 2.1 ± 0.3 and 9.3 ± 4.7 for ChT and LY, respectively.
Discussion
Regulation of proteins by oxidation is a common cellular mechanism, relevant to various physiological and pathophysiological phenomena and the process of aging (e.g., [3, 29, 30]). Therefore, the detailed investigation of molecular mechanisms involved in oxidative protein modification is of general interest. However, investigation of oxidative protein modification is not always straightforward, in part, because of limited amino acid specificity of the oxidizing agents. In case of thiol modifications of cysteine moieties, specific protocols are available, but oxidation of methionines requires harsher and potentially less specific treatments. Thus, unequivocal molecular characterization of oxidative protein modification requires the identification of target residues to exclude unspecific oxidation events.
In this study, we investigated the sensitivity of voltage-gated sodium channels towards Met-directed oxidation caused by two independent oxidative treatments and identified multiple target Met residues by mutagenesis. We expressed various NaV channel types in HEK 293 cells and either applied the mild oxidant ChT or irradiated LY, which was loaded into the cells via the patch pipettes. In both cases, oxidation impaired the fast inactivation of NaV channels in a time- and dose-dependent manner. This effect was almost identical in four different NaV channel isoforms (NaV1.2 (brain), NaV1.4 (skeletal muscle), NaV1.5_C373Y (heart), and NaV1.7 (peripheral nervous system)). For the heart channel, we used the single-point mutant C373Y in order to avoid direct oxidation of the tetrodotoxin binding site in the pore.
The reducing agent DTT failed to restore inactivation following treatment with ChT or LY irradiation, suggesting that oxidation of Met residues might be involved in the removal of fast inactivation of the channel. Because all NaV channel isoforms tested showed similar oxidation sensitivity, we reasoned that the relevant Met residues might be conserved. The most obvious Met residue (Met1305) involved in channel inactivation resides in the D3–D4 inactivation linker and is part of the “IFM motif”. When mutated to leucine, channel modification by ChT and irradiated LY was considerably slower, suggesting that Met1305 in the IFM motif is a potential target of oxidation.
This result is compatible with a previous report on the oxidation sensitivity of NaV channel inactivation in toad skeletal muscle fibers, in which an involvement of Met residues was hypothesized [19]. Eaholtz and colleagues [31] provided more direct evidence by studying the potency of small peptides corresponding to the amino acid sequence of this inactivation domain to “inactivate” or block NaV channels. When in such peptides Ile–Phe–Met was replaced with Ile–Phe–MetO, the inactivating potency of the peptides vanished, suggesting that a Met-oxidized inactivation linker does not function properly. However, a report by Wang and Wang [20] suggests that the Met in the inactivation domain is not responsible for the loss of inactivation induced by either ultraviolet light or H2O2, as both interventions removed inactivation similarly in the NaV1.4 wild-type channels and in a mutant in which the inactivation domain contained Ile–Phe–Ile. We saw that IFL channels retain some degree of oxidation sensitivity, but the time course of inactivation removal in the mutant was much slower than in the wild type. One likely explanation for this discrepancy could be that application of ChT or irradiation of LY more specifically attacks Met residues as compared to channel exposure to UV light or H2O2, which may more readily modify other amino acids.
Further mutagenesis revealed that methionines in the receptor of the inactivation lid, Met1469/1470 in the S4–S5 linker of domain 4, also contribute to the channels’ oxidation sensitivity with M1470 playing a particularly important role. Combining leucine replacements at Met1305 and Met1469/1470 resulted in channels that were even less sensitive to oxidation. These results clearly show that inactivation of NaV channels is sensitive to oxidative events. Loss of fast inactivation appears to be a multi-step process involving the oxidation of the IFM motif as well as its receptor site in domain 4. This was furthermore demonstrated by analyzing the time course of inactivation removal upon oxidation. While the wild-type channels showed a sigmoidicity with an exponent of two or greater, the mutated channels exhibited an exponent close to unity. The molecular interpretation could be that both, the inactivation lid and its receptor, must be oxidized to effectively impair inactivation, essentially acting as a coincidence detector. When one target is removed by an M-to-L mutation, a single oxidation event is much less likely to occur such that only minor effects on channel inactivation are observed. This was particularly well observed when modification was elicited with irradiated LY in the patch pipette.
Additional conserved Met residues are present in intracellular regions of the channel protein (Fig. 4a). Most of them are located at the interface between membrane and cytosol to form a ring or cluster of residues at the intracellular entry of the channel. One could, therefore, speculate that these methionines may act as radical scavengers. If this was true, a channel with an intact IFM motif, but with all other methionines absent, might be more sensitive to oxidative stress than a channel with all native methionines in place. This scenario was tested with mutant IFM_6L, which, however, was not more oxidation-sensitive than mutant IFM_LL. Thus, our experiments do not provide direct evidence for a scavenging mechanism inside NaV channel proteins, but we cannot completely rule out this possibility for milder long-term oxidative stress situations.
Several examples of oxidation by ChT having a marked impact on ion channel function have been reported. Shaker potassium channels with prominent C-type inactivation [32] and hERG1 potassium channels [33] become nonfunctional when exposed to ChT. Calcium-activated potassium channels of the BK-type exhibit an increased activity on treatment with ChT [27], and this oxidative regulation requires a triad of Met residues in the C-terminal domain [34]. In Shaker C/B channels, treatment with ChT disrupts rapid N-type inactivation by a selective oxidation of a Met residue in the N-terminal inactivation ball domain [6, 7]. The loss of N-type inactivation in Shaker C/B by ChT somewhat resembles the oxidative regulation of fast inactivation in NaV channels reported here. However, the mechanism of the oxidative regulation in NaV channels is more complex because our mutagenesis results show that multiple Met residues in the IFM inactivation motif and its receptor site are involved. This conclusion is further strengthened by the presence of sigmoidicity in the onset of loss of inactivation.
Oxidation of Met residues by ChT leads to formation of methionine sulfoxide, and such posttranslational modification should, in principle, be reversible in the presence of methionine sulfoxide reductases (MSRs). Su et al. [33] report that the hERG1 channel is less sensitive to extracellularly applied ChT when MSRA is present in the patch pipette. We failed to observe such a protecting or repairing effect of MSRA on the inactivation of NaV channels (data not shown). The reasons for that are not clear, but most likely, a limited accessibility of the oxidized Met residues to the reducing enzyme or the relatively low catalytic turnover rate of MSRA plays a role.
The importance of proper NaV channel inactivation for neuronal and muscle functions is manifested by several inherited diseases (e.g., LQT-3 syndrome and myotonia; for an overview, see [35]) in which the pathomechanism is postulated to involve an incomplete NaV channel inactivation. Hence, under oxidative stress situations, incomplete NaV channel inactivation may result in dramatic changes of electrical activity. A prominent example is cardiac arrhythmia occurring as a consequence of an ischemia or reperfusion insult; oxidation of cardiac NaV channels (e.g., by application of H2O2) results in an increase in slowly inactivating “late” sodium currents, which may cause fatal prolongations of cardiac action potentials [11, 12]. Because the critical Met residues identified for NaV1.4 channels are conserved in cardiac NaV1.5 channels, similar molecular mechanisms of oxidation-induced loss of NaV channel inactivation may also apply to the electrical properties of cardiac tissue. In fact, cardiac arrhythmia is part of the ischemia or reperfusion complications [36–38], and an impaired inactivation of cardiac NaV channels would contribute an arrhythmogenic potential. A possible intervention could be a specific blockade of oxidized, non-inactivating channels as demonstrated for the beneficial effects of ranolazine for the postischemic heart [39].
Considering the potential pathophysiological consequences implicated in a loss of NaV channel inactivation, it is surprising that oxidation-sensitive residues are conserved in the inactivation machinery, although alternative residues (e.g., leucines) would produce NaV channels with similarly rapid and complete inactivation. It, thus, appears as if NaV channels carry an unnecessary “risk” of becoming oxidatively damaged. One speculation is that oxidation-regulated inactivation may offer an evolutionary advantage, allowing organisms to regulate firing frequency in neurons or contraction in cardiac tissue when cells are exposed to oxidative stress. The arrangement using multiple Met residues thereby confers a finely graded oxidative modulation of NaV channels and allows the organisms to adapt to a variety of oxidative stress conditions. In this respect, it is interesting to note that NaV channels of insects (e.g., the Para channel of the fruit fly Drosophila melanogaster) harbor an “MFM” inactivation motif, which is expected to be even more sensitive to oxidation than their mammalian relatives. In addition, at the putative receptor site for the inactivation domain, Para channels harbor an “AM” rather than an “MM” motif, i.e., residue M1470 shown to be important for the oxidation sensitivity of the channels is conserved. Although the physiological relevance still needs to be investigated, NaV channels are prone of being oxidatively damaged if the oxidative stress exceeds a critical level, a phenomenon that may be relevant in various pathophysiological conditions and during the process of aging.
References
Vogt W (1995) Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic Biol Med 18:93–105
Weissbach H, Etienne F, Hoshi T, Heinemann SH, Lowther LW, Matthews B, John GS, Nathan C, Brot N (2002) Peptide methionine sulfoxide reductase: structure, mechanism of action and biological function. Arch Biochem Biophys 397:172–178
Hoshi T, Heinemann SH (2001) Regulation of cell function by methionine oxidation and reduction. J Physiol 531:1–11
Levine RL, Mosoni L, Berlett BS, Stadtman ER (1996) Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci U S A 93:15036–15040
Bigelow DJ, Squier TC (2005) Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim Biophys Acta 1703:121–134
Ciorba MA, Heinemann SH, Weissbach H, Brot N, Hoshi T (1997) Regulation of potassium channel function by methionine oxidation and reduction. Proc Natl Acad Sci U S A 94:9932–9937
Kuschel L, Hansel A, Schönherr R, Weissbach H, Brot N, Hoshi T, Heinemann SH (1999) Molecular cloning and functional characterization of a human peptide methionine reductase (hMsrA). FEBS Lett 456:17–21
Jung S, Hansel A, Kasperczyk H, Hoshi T, Heinemann SH (2002) Activity, tissue distribution and site-directed mutagenesis of a human peptide methionine sulfoxide reductase of type B: hCBS1. FEBS Lett 527:91–94
Desaphy J-F, De Luca A, Imbrici P, Camerino DC (1998) Modification by ageing of the tetrodotoxin-sensitive sodium channels in rat skeletal muscle fibres. Biochim Biophys Acta 1373:37–46
Lin CS-Y, Grosskreutz J, Burke D (2002) Sodium channel function and the excitability of human cutaneous afferents during ischaemia. J Physiol 538:435–446
Ma JH, Luo AT, Zhang PH (2005) Effect of hydrogen peroxide on persistent sodium current in guinea-pig ventricular myocytes. Acta Pharmacol Sin 26:828–834
Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L (2006) Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharm Exp Therap 318:214–222
Eaholtz G, Scheuer T, Catterall WA (1994) Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron 12:1041–1048
Tang L, Kallen RG, Horn R (1996) Role of an S4–S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. J Gen Physiol 108:89–104
Black SD, Mould DR (1991) Development of hydrophobicity parameters to analyze proteins which bear post- or cotranslational modifications. Anal Biochem 193:72–82
Wang GK (1984) Irreversible modification of sodium channel inactivation in toad myelinated nerve fibres by the oxidant chloramine-T. J Physiol 346:127–141
Wang GK, Brodwick MS, Eaton DC (1985) Removal of sodium channel inactivation in squid axon by the oxidant chloramine-T. J Gen Physiol 86:289–302
Huang JM, Tanguy J, Yeh JZ (1987) Removal of sodium channel inactivation and block of sodium channels by chloramine-T in crayfish and squid giant axons. Biophys J 52:155–163
Quiñonez M, DiFranco M, González F (1999) Involvement of methionine residues in the fast inactivation mechanism of the sodium current from toad skeletal muscle fibers. J Membr Biol 169:83–90
Wang GK, Wang SY (2002) Modifications of human cardiac sodium channel gating by UVA light. J Membr Biol 189:153–165
Trimmer JS, Cooperman SS, Tomiko SA, Zhou JY, Crean SM, Boyle MB, Kallen RG, Sheng ZH, Barchi RL, Sigworth FJ, Goodman RH, Agnew WS, Mandel G (1989) Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron 3:33–49
Noda M, Ikeda T, Suzuki H, Takeshima H, Takahashi T, Kuno M, Numa S (1986) Expression of functional sodium channels from cloned cDNA. Nature 322:826–828
Klugbauer N, Lacinova L, Flockerzi V, Hofmann F (1995) Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendokrine cells. EMBO J 14:1084–1090
Gellens ME, George Jr AL, Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG (1992) Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A 89:554–558
Chen H, Gordon D, Heinemann SH (2000) Modulation of cloned skeletal muscle sodium channels by the scorpion toxins Lqh II, Lqh III, and LqhαIT. Pflügers Arch 439:423–432
Higure Y, Katayama Y, Takeuchi K, Ohtubo Y, Yoshii K (2003) Lucifer Yellow slows voltage-gated Na+ current inactivation in a light-dependent manner in mice. J Physiol 550:159–167
Tang XD, Daggett H, Hanner M, Garcia ML, McManus O, Brot N, Weissbach H, Heinemann SH, Hoshi T (2001) Oxidative regulation of large conductance calcium-activated potassium channels. J Gen Physiol 117:253–273
McPhee JC, Ragsdale DS, Scheuer T, Catterall WA (1998) A critical role for the S4–S5 intracellular loop in domain IV of the sodium channel α-subunit in fast inactivation. J Biol Chem 273:1121–1129
Stadtman ER (2006) Protein oxidation and aging. Free Radic Res 40:1250–1258
Chakravarti B, Chakravarti DN (2007) Oxidative modification of proteins: age-related changes. Gerontology 53:128–139
Eaholtz G, Colvin A, Leonard D, Taylor C, Catterall WA (1999) Block of brain sodium channels by peptide mimetics of the isoleucine, phenylalanine, and methionine (IFM) motif from the inactivation gate. J Gen Physiol 113:279–293
Schlief T, Schönherr R, Heinemann SH (1996) Modification of C-type inactivating shaker potassium channels by chloramine-T. Pflügers Arch 431:483–493
Su Z, Limberis J, Martin RL, Xu R, Kolbe K, Heinemann SH, Hoshi T, Cox BF, Gintant B (2007) Functional consequences of methionine oxidation of hERG potassium channels. Biochem Pharm 74:702–707
Ciali Santarelli L, Wassef R, Heinemann SH, Hoshi T (2006) Three methionine residues located within the RCK domains confer oxidative sensitivity to large-conductance calcium-activated K+ channels. J Physiol 571:329–348
Ashcroft FM (2000) Ion Channels and Disease. Academic, San Diego
Birnbaum Y, Leor J, Kloner RA (1997) Pathobiology and clinical impact of reperfusion injury. J Thromb Thrombolysis 4:185–195
Gazmuri RJ, Ayoub IM, Kolarova J (2003) Myocardial protection during resuscitation from cardiac arrest. Curr Opin Crit Care 9:199–204
Kevin LG, Novalija E, Stowe DF (2005) Reactive oxygen species as mediators of cardiac injury and protection: the relevance to anesthesia practice. Anesth Analg 101:1275–1287
Hale SL, Kloner RA (2006) Ranolazine, an inhibitor of the late sodium channel current, reduces postischemic myocardial dysfunction in the rabbit. J Cardiovasc Pharmacol Ther 11:249–255
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (HE2993/7-1, S.H.H.), IZKF/TMWFK (B307-04004, S.H.H.), and National Institutes of Health (T.H.). We thank J. Trimmer for providing cDNA coding for rNaV1.4, N. Klugbauer for hNaV1.7, S. Noda for rNaV1.2, A. George for hNaV1.5, and S. Arend and A. Rossner for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kassmann, M., Hansel, A., Leipold, E. et al. Oxidation of multiple methionine residues impairs rapid sodium channel inactivation. Pflugers Arch - Eur J Physiol 456, 1085–1095 (2008). https://doi.org/10.1007/s00424-008-0477-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0477-6